
Abstract
Sepsis and the acute respiratory distress syndrome (ARDS) each cause substantial morbidity and mortality. In contrast to other lung diseases, the entire course of disease in these syndromes is measured in days to weeks rather than months to years, which raises unique challenges in achieving precision medicine. We review advances in sepsis and ARDS resulting from omics studies, including those involving genome-wide association, gene expression, targeted proteomics, and metabolomics approaches. We focus on promising evidence of biological subtypes in both sepsis and ARDS that consistently display high risk for death. In sepsis, a gene expression signature with dysregulated adaptive immune signaling has evidence for a differential response to systemic steroid therapy, whereas in ARDS, a hyperinflammatory pattern identified in plasma using targeted proteomics responded more favorably to randomized interventions including high positive end-expiratory pressure, volume conservative fluid therapy, and simvastatin therapy. These early examples suggest heterogeneous biology that may be challenging to detect by clinical factors alone and speak to the promise of a precision approach that targets the right treatment at the right time to the right patient.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Point Summary-
Disease heterogeneity in ARDS and sepsis has challenged the conduct of omics studies involving these syndromes, yet precision medicine approaches are sorely needed to improve outcomes.
-
Genomics studies of ARDS and sepsis are challenged by small sample sizes and the difficulty in identifying appropriate controls.
-
Gene expression studies have identified biologically distinct expression signatures that retroactively identify differential response to routine treatments applied in the ICU. In sepsis, a signature of dysregulated adaptive immune signaling has evidence to stratify patients according to a differential response to systemic steroid therapy. In ARDS, patients with a hyperinflammatory pattern identified in plasma using targeted proteomics responded more favorably to randomized interventions including high positive end-expiratory pressure, volume conservative fluid therapy, and simvastatin therapy.
-
Fewer ARDS and sepsis metabolomics and unbiased proteomics studies exist, but as these approaches become more standardized, additional biomarkers may be identified.
Introduction
Sepsis and the acute respiratory distress syndrome (ARDS) are two syndromes causing a sobering proportion of deaths in the intensive care unit (ICU). By one estimate, sepsis was responsible for between one third and one half of inpatient deaths [1], whereas ARDS has been observed to complicate almost one quarter of critical care admissions requiring mechanical ventilation, with a mortality rate exceeding 35% [2]. Despite significant advances in our understanding of the pathologic mechanisms contributing to each of these syndromes, neither sepsis nor ARDS can boast specific pharmacologic therapy with a consistently proven effect. Numerous trials in sepsis [3,4,5] and ARDS [6,7,8] have failed to establish drug therapy for these deadly syndromes. One major factor that may contribute to this treatment gap is the profound heterogeneity encompassed by patients meeting criteria for each syndrome. A valid concern is that our syndromic definitions [9,10,11], although useful in identifying patients who share clinical factors and who may benefit from standardized care [12,13,14,15], may have almost no utility in predicting a patient’s biologic subclassification nor in predicting mortality, expected complications, or response to therapy. Precision medicine options for these syndromes, whereby the correct drug could be targeted to the patients most likely to be helped and least likely to be harmed, are sorely needed.
For complex traits such as asthma or cystic fibrosis, the knowledge of a patient’s biologic endotype provides clues about a patient’s prognosis, pathophysiology, and expected response to therapy [16,17,18,19,20], yet such a breakthrough has yet to arrive for sepsis or ARDS. In this chapter, we review the contributions of genomic medicine to identifying potential biologic subgroups in sepsis and ARDS, a necessary first step for precision medicine. In addition, we consider specific challenges to pursuing omics approaches in each of these traits, as well as potential opportunities we envision in the near future.
Targeted Proteomics: Laying the Foundation for Precision Medicine of Sepsis and ARDS
Increasingly, we have objective evidence that response to therapy is nonuniform and potentially predictable by factors beyond clinical features. In ARDS, this has been consistently demonstrated among clinical trial populations using an analytic method known as latent class analysis (LCA) to uncover potentially unobserved subpopulations while remaining agnostic to outcomes. In analyses considering clinical variables including vital signs, ventilator data, and laboratory values in addition to exploratory plasma biomarkers representing inflammation, vascular dysfunction, or alveolar injury, a latent class model consistently identified two classes of ARDS trial subjects that differed in their plasma expression of inflammatory biomarkers epitomized by interleukin (IL-) 8 or IL-6, and by their degree of systemic illness, characterized by low blood pressure and low serum bicarbonate [21,22,23,24].
Not only were subjects in the “hyperinflammatory” subphenotype group more likely to die, but the LCA group assignment (hyperinflammatory versus non-hyperinflammatory) exhibited significant statistical interaction with randomized treatment effects (Table 18.1) [21, 22, 24]. Thus, when the randomized interventions of higher positive end-expiratory pressure (PEEP), conservative fluid strategy, or simvastatin therapy were analyzed in groups stratified by LCA assignment, each therapy seemed to have a mortality benefit only observed in the hyperinflammatory group, with no signal for improvement in the non-hyperinflamed group [21, 22, 24]. In each trial, there was no evidence for heterogeneity in treatment effect by baseline severity of illness, as defined by the Acute Physiology and Chronic Health Evaluation (APACHE) score, nor by the severity of ARDS, as defined by the ratio of arterial partial pressure of oxygen to fraction of inspired oxygen (PaO2:FiO2).
Reproducible biologically defined ARDS subgroups based on plasma protein expression patterns were also reported in a population of sepsis-associated ARDS subjects from a prospective sepsis cohort applying a Ward clustering algorithm [25]. Showing remarkable similarity to the LCA-derived subgroups, the clustering algorithm detected two classes of ARDS, one “reactive” defined by high plasma concentrations of markers of inflammation, coagulation, and endothelial activation compared to the “noninflamed” group, and a significantly higher mortality was observed for the reactive subgroup. Although the overlap in plasma protein signatures between the Calfee “hyperinflamed” and Bos “reactive” subgroups is striking [21,22,23, 25], both studies sampled fairly similar candidate biomarkers that have previously performed well in human and animal studies of sepsis-associated ARDS [26, 27], so the overlap is less surprising than if the authors had used unbiased discovery or untargeted proteomics approaches.
Similarly, in sepsis, it is conceivable that heterogeneity of treatment effect may underlie some of the negative overall findings for such drugs as recombinant human interleukin-1 receptor antagonist (rhIL1RA) [28,29,30], antitumor necrosis factor [31, 32], or activated protein C [4, 33, 34]. In a subgroup reanalysis of a randomized trial of rhIL1RA for sepsis [29], the mortality benefit of rhIL1RA differed significantly between subjects with high baseline endogenous plasma interleukin-1 receptor antagonist (IL1RA) , who seemed to benefit from the drug, and those without elevated plasma IL1RA in whom there was no effect [35]. A separate subgroup reanalysis of the same rhIL1RA trial demonstrated a very strong signal for benefit among subjects with clinically defined macrophage activation syndrome [36]. Although subgroup analyses must be viewed with caution due to underpowering and the risk of unstable effect estimates [37,38,39], these reports nonetheless highlight the potential for precision application of sepsis therapy if replicated in prospectively defined studies.
What Can Be Learned from Genetic Approaches in Complex, Non-Mendelian Traits?
Neither sepsis nor ARDS is considered a classic monogenic or “Mendelian” trait, whereby the expression of the trait is easily predictable by parsing the inheritance of one genetic locus through several generations. However, multiple lines of evidence support a major interplay between genetic variation and patterned responses to injury and infection. Primary, or inherited, immunodeficiency diseases (PIDD) include over 330 specific disorders caused by at least 320 monogenetic changes [40, 41] and span broad subgroups that include defects in just one aspect of the immune system (e.g., antibody, innate, T-cell, natural killer cell, neutrophil) to combined immunodeficiencies, autoimmune diseases, or autoinflammatory disorders. Some syndromes present as an inherited susceptibility to one particular type of infection, such as mycobacteria, fungi, or certain bacteria, and thus, elucidation of the respective genetic underpinnings has helped to pinpoint critical host responses to specific pathogens [42,43,44,45,46]. Further, the identification of monogenic conditions causing auto-inflammatory conditions that mimic the clinical features of sepsis while remaining culture-negative [47,48,49] highlight the primacy of host response in driving shock and organ failure.
Further, there is ample evidence that historic infectious threats likely shaped genetic architecture through natural selection [50, 51]. The single missense variant in the beta-globin gene responsible for sickle cell anemia (rs334) persists at a frequency of 5–10% in genotyped African populations [52, 53], a frequency much higher than expected for such a deleterious mutation. However, because individuals who carry only one copy of rs334 seem to be protected from malarial infection [51, 54], this variant is common in populations where malaria has been, or continues to be, a threat. Similar examples may explain the striking variation in genes encoding cytokines [55], or genes that control activation of the complement syndrome, some of which have also been strongly implicated in inflammatory traits like age-related macular degeneration [56,57,58].
Accepting that our genes influence response to infection or injury, and that such historic threats have in turn shaped genetic architecture, it remains true that most patients with sepsis do not harbor a single genetic variant that explains their risk for sepsis or sepsis death. Nonetheless, there exists strong evidence that sepsis death exhibits significant heritability. In a classic study merging genealogic records and population health information, biologic parents and their children displayed a much stronger concordance for premature death from infection than did adopted parents and their children, suggesting that genes play a stronger role in response to infection than does environment [59]. The relative risk (RR) of dying prematurely from infection when one parent had also died from infection was almost 6 (95% CI 2.47–13.7), a larger risk than was observed for cancer or even vascular disease [59]. Just as unraveling the monogenic PIDD have suggested precision treatment options that sometimes obviate the need for bone marrow transplantation [47], it may be that better recognition of dysregulated genes contributing to sepsis outcomes suggests novel treatment paradigms for this deadly disease.
Similar data do not exist to support the inherited susceptibility to ARDS, in part because ARDS was only described with the advent of modern ICU care [60], but one might consider the syndrome of acute hypoxia and bilateral lung opacities following a potential insult – ARDS [10] – to be a patterned response to injury or infection. Although to our knowledge no pedigrees exist of ARDS, genetic investigations have suggested novel pathophysiologic processes. Recognizing that we are unlikely to explain a large proportion of the variance in the risk for sepsis or ARDS by one or even a small handful of genes, the dissection of trait-associated pathways may still suggest individuals predisposed to these syndromes via specific mechanisms and, thus, suggest groups who are likely to respond to specific interventions.
Knowledge- and Discovery-Based Genomics Studies
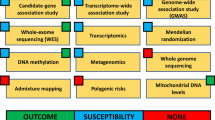
In general terms, there are two major approaches to identify inherited variation that may influence a trait (Fig. 18.1). Knowledge-based approaches, sometimes referred to as candidate gene studies , select specific genes or pathways already hypothesized to contribute to a disease and test for a higher frequency of genetic variants in disease-positive subjects compared to subjects without the disease. Advantages of the candidate approach include the straightforward design, typically as either a case-control or cohort study, low cost, and the fact that next steps after a positive finding are relatively clear. However, precisely because candidate gene studies are predicated on existing knowledge of sepsis or ARDS pathophysiology, these studies have a high risk of failure. For complex traits like sepsis or ARDS, the expected effect size of any given genetic variant is modest, with an odds ratio less than 1.4, and consequently most studies are statistically underpowered to detect an effect even if one exists. Further, even if the selected candidate gene does play a central role in ARDS pathophysiology, researchers still need to genotype the causal part of the gene responsible for the trait or, leveraging linkage disequilibrium [61], a variant in linkage with the causal variant. We are only beginning to understand the complexities of genetic regulation beyond the traditional paradigm of cis regulation, whereby local DNA sequence dictates local messenger RNA (mRNA) sequence, which in turn explains protein sequence. With the application of next-generation sequencing techniques to better understand DNA-protein binding, noncoding RNA regulatory elements, epigenetic changes that may silence or activate gene expression, and the impact of three-dimensional chromatin organization [62,63,64,65,66], it is now apparent that early genotyping strategies may have been too simplistic. Thus, the failure to detect associations does not exonerate a gene from playing a significant role. Finally, the candidate gene approach now seems highly inefficient in the era of next generation sequencing approaches.

Both knowledge-based and discovery-based methods can identify new candidate markers that span all aspects of biology and clinical features. Novel analytic techniques can then combine clinical, genetic, transcriptomic, metabolomic, and proteomic data to achieve the goals of precision medicine: identifying new therapies via refined mechanistic insight and unpacking clinical heterogeneity into biologically meaningful subgroups. PCR polymerase chain reaction, GWAS genome-wide association study, RNA-Seq RNA sequencing, mRNA messenger RNA
Genome-wide association studies (GWAS) assay single nucleotide polymorphisms (SNPs) at over 500,000 loci across the genome using nanofabricated arrays of oligonucleotide probes specific for individual SNPs. Then, using knowledge of linkage disequilibrium between SNPs based on large-scale genotyping of multiple populations [52, 67, 68], investigators can impute genotypes at loci that were not genotyped, allowing dense characterization of genetic variation for less than 100 US dollars per sample. However, though the array-based GWAS does characterize DNA variation “across the genome” and is considered a discovery approach, it is not truly bias-free, as the arrays are built using oligonucleotide probes for known SNPs and imputation steps rely upon preexisting knowledge of LD relationships between SNPs. The technology that yields data closest to truly bias-free genome sequences is next-generation sequencing (NGS) , in which massively parallel sequencing of fragmented input DNA occurs. NGS is the preferred method to discover new variants or private variants that occur in only one family, or only one individual, as well as to study rare variants [69].
Factors beyond the inherited genomic DNA sequence influence the expression of genes. Profiling of messenger RNA (mRNA) or, more broadly, entire transcriptomes (i.e., transcriptomics) has enabled major advances in the understanding of cancer and complex traits like asthma [16, 70, 71]. Gene expression studies also can be thought of as following either knowledge-based or discovery-based paths. To understand the mRNA abundance or expression pattern of a specific transcript, one could use traditional polymerase chain reaction (PCR) methods using an oligonucleotide probe or probes complementary to the sequence(s) of interest. When roughly 20–30 thousand probes are arrayed onto a single nanofabricated platform to assess global gene expression, we term this a whole genome microarray , which is a discovery method, albeit based on probes and, thus, not bias-free. For broader unbiased characterization of transcriptomes, investigators can use RNA-sequencing (RNA-Seq), a NGS approach that sequences a complementary DNA (cDNA) library prepared from input RNA. Gene expression studies have unique challenges compared to genomics in that the former are cell type and context specific and highly dynamic. Beyond mRNA, multiple noncoding RNA species have been identified and demonstrated to influence transcription, translation, message stability, splicing, enhancing/silencing, epigenetic regulation, and even molecular scaffolding [72]. Bias-free sequencing has elucidated the breadth of the noncoding RNA landscape, and the study of noncoding RNA in critical illness remains in its infancy. Epigenetic changes, which describe inherited but modifiable DNA-protein interactions, such as histone modifications or DNA methylation patterns, also modify gene expression and can be assessed in targeted, high-throughput, or bias-free applications.
Unique Challenges to Achieving Precision Medicine in Critical Care
Although the promise of omics techniques to contribute to precision medicine options is undeniable, specific challenges complicate the application of these methods to sepsis and ARDS. Both syndromes are complex genetic traits, requiring both an extreme environmental insult like infection or exposure to a ventilator and host susceptibility. This gene-by-environment interaction can be complex to study and poses unique barriers to identifying omics signals even when present.
First, the genomic signature of sepsis or ARDS will never occur in isolation. Sepsis by definition is a systemic disease, and multiple organ dysfunction is often central to its diagnosis. Dissecting out the signature of sepsis from that of secondary kidney, lung, brain, or liver injury requires unique analytic tools as well as potentially arbitrary decisions classifying changes as sepsis related or not. With genomic material from the infecting microbe potentially circulating in blood, sequencing techniques might amplify bacterial, viral, parasitic, or fungal genomes rather than the patient’s cells. ARDS is also frequently complicated by coincident non-lung organ failure and frequently occurs on the background of sepsis, such that identifying the specific ARDS signature may be difficult. Further, liver and kidney dysfunction can alter the clearance of proteins and metabolites. When metabolic or proteomic changes seem to distinguish ARDS cases from non-cases, these could represent an important feature of the causal pathway in sepsis or may simply reflect end-organ dysfunction with impaired clearance.
Second is the problem of identifying a suitable control population and appropriately designing the study. In many diseases, large convenience cohorts of healthy adults can be used as controls, and large-scale genomic resources such as the UK Biobank can be very powerful to detect a genetic signal [73]. For ICU diseases, however, the use of healthy controls may be problematic, precisely because of the gene-by-environment interaction that requires a severe environmental insult to manifest sepsis or ARDS. A person’s genome may contain multiple risk variants for ARDS, but if she never develops sepsis, exposure to a ventilator, or another ARDS precipitant, she may never exhibit lung flooding. The presence of such a subject in the control group would attenuate any signal for ARDS risk, even if multiple ARDS cases carry the same variant. Similar issues arise when designing a sepsis study. In a case-control design, should controls be a group of patients who never had pneumonia or, instead, a group infected with pneumonia and thus at risk to develop sepsis, but who remained relatively well? For this reason, many critical care investigators choose a cohort design, which alleviates the concern for selection bias, but comes at increased cost and lower efficiency. Design issues may complicate the analytic phase as well. A frequent criticism of a potential new prognostic marker for sepsis or ARDS is that it merely reflects severity of illness, and investigators are asked to confirm that associations remain independent of illness severity characterized by simplified acute physiology or APACHE scores [74,75,76]. While appropriate adjusting for potential confounders has face validity for any analysis, there is a counterargument that the very processes driving acute physiologic derangement and captured by such scores may be in the causal path influencing sepsis or ARDS risk and outcome. Observing that an association persists across multiple levels of illness severity or predicted mortality can sometimes mitigate this concern [77].
Third, timing of biospecimen sampling is both critically important and yet challenging to enact. For both sepsis and ARDS, genomic, proteomic, and metabolomic signatures change substantially within hours to days, and the timeframe to collect samples is highly compressed. Whereas genomic DNA samples should be stable over time and could be collected after the acute event, RNA, protein, or metabolite profiling often requires specific collection strategies and is only relevant if collected during the illness itself. At the same time, critically ill patients are frequently unable to consent for themselves, suffer from anemia [78], and have multiple competing clinical needs that may limit the ability to conduct observational research in the early hours of ICU admission. Furthermore, it is challenging to define “time zero” for sepsis; is it when the patient presents to the emergency room, the first low blood pressure, or the first fever? The importance of time-course analysis in sepsis was highlighted by gene expression work by Sweeney et al. [79], in which a clear sepsis gene expression signature emerged in early sepsis, but was later swamped by recovery signals. For ARDS, does the clock start when exogenous oxygen exceeds 4 liters per minute, when the chest radiograph is first abnormal, or when the patient is intubated and meeting all consensus criteria [10, 80]? For each omics study, these issues should be carefully considered and protocolized to ensure the highest possible scientific rigor.
Finally, there is the issue of which tissue warrants profiling. Peripheral blood – easily obtained by a blood draw and either left whole or segmented into constituent blood cells – is convenient, widely available, and relevant, as a potential snapshot of circulating host response. Buffy coat gene expression signatures reproducibly separate sepsis cases from controls [81, 82], and circulating inflammatory cells may be the critical actor in sepsis pathology. However, peripheral blood has numerous limitations. Genes that are expressed exclusively by endothelium, epithelium, stromal tissue, or tissue specific to the infected organ will not be captured by whole blood or leukocyte gene expression profiling. Though there may be strong interest to evaluate vascular mRNA, a vessel biopsy will remain highly unlikely and circulating endothelial cells are difficult to collect and may be fundamentally distinct compared to intact vasculature [83]. For ARDS, there is general consensus that lung tissue would provide the maximal utility for gene, protein, and metabolite expression. However, patients with ARDS are rarely subjected to lung biopsy due to their tenuous stability and the risk of the procedure [84]. Easily obtained peripheral blood is not a consistent surrogate for omics states in the lung. In ARDS, peripheral blood gene expression may be swamped by sepsis severity rather than lung injury per se [85]. Even when limiting analysis to only the mononuclear cell fraction and carefully timing blood draws to coincide with bronchoalveolar lavage (BAL) , the signature of alveolar macrophages in ARDS is markedly distinct from synchronous peripheral blood monocytes [86]. Despite these formidable challenges, there are numerous examples of incremental and occasionally transformational progress toward precision medicine that speak to tremendous potential of omics approaches in sepsis and ARDS (Table 18.1).
Sepsis Genetics: Hints at Heterogeneous Treatment Effects
Although the promise of genetics to contribute to precision medicine options for sepsis remains strong, progress to date has been relatively modest. Candidate gene association studies have yielded a number of variants that reliably associate with increased susceptibility to specific infections and occasionally with a higher frequency of hypotension or death [87,88,89]. As candidate gene studies extend from our preexisting paradigm of sepsis, most of the interrogated genes have been those influencing host response, immune regulation, or vascular regulation. Genome-wide studies of sepsis outcome – a highly heritable trait [59] – have also been published and suggest novel pathways that merit consideration.
In one of the first published GWAS for sepsis survival, investigators from the Genetics of Sepsis and Septic Shock in Europe (GenOSept) consortium used a discovery population of approximately 1000 subjects with community-acquired pneumonia and replicated findings in an additional 1000 individuals from clinical trials or an ongoing pneumonia cohort. The GenOSept authors reported a fairly convincing LD peak on chromosome 5 in the FER gene encoding Fps/Fes-related tyrosine kinase that associated with lower risk of death (meta-analysis odds ratio 0.56, 95% confidence interval 0.41–0.66) with a p-value robust to multiple comparison testing (p = 5.6 × 10−8) [90]. Interestingly, the association with death was attenuated by expanding the population to include septic subjects with abdominal infections, suggesting that the protective association may be relevant only to pulmonary sepsis. The FER gene encodes a non-receptor protein tyrosine kinase implicated in actin cytoskeleton regulation as well as chemotaxis and leukocyte migration – areas already of interest in sepsis pathophysiology [91,92,93] – thus, it is an attractive sepsis candidate gene. Mortality was 25% for homozygous carriers of the dominant allele compared to 10% in homozygous recessive carriers [90], suggesting that genotype might act as a prognostic enrichment tool to help select a high-risk population [11]. However, as is often the case with genomic findings, replication of this variant has been inconsistent, and it was not associated with mortality in a smaller population of septic subjects enrolled in clinical trials in Germany [94]. A second GWAS found that a rare missense variant in gene VSP13A was associated with very high risk for mortality, and this VSP13A SNP was associated with higher sequential organ failure assessment score in a separate pneumonia study, providing possible replication. VSP13A encodes for vacuolar protein sorting 13 homolog A and has been implicated in autophagy, another pathway relevant to sepsis [95].
Examples of genetic studies contributing to precision therapy in sepsis are indirect, but a few do exist. Meyer et al. identified a synonymous coding SNP in the gene IL1RN encoding interleukin-1 receptor antagonist (IL1RA) that associated with reduced ARDS risk in both trauma and sepsis populations, as well as with reduced sepsis mortality in the VASST septic shock trial [96,97,98]. The SNP seemed to be functional, associating with increased plasma IL1RA among patients at risk for ARDS, lower plasma interleukin-1 beta (IL1β) during septic shock, and as a site of allelic imbalance with more efficient IL1RN gene expression following endotoxin challenge. As these data suggested that more efficient plasma IL1RA generation might be protective in sepsis, Meyer and colleagues used plasma from a completed clinical trial of recombinant human IL1RA (rhIL1RA) for sepsis to phenotype sepsis patients for pre-randomization plasma IL1RA and IL1β expression. They detected a differential effect of rhIL1RA on mortality based on plasma IL1RA expression, such that rhIL1RA seemed to reduce mortality among “IL1RA-high” subjects [adjusted risk difference (ARD) −12%, 95% CI −23% to −1%], p = 0.044, but not among “IL1RA-low” subjects (ARD +7%, 95% CI −4% to +17%), resulting in a statistically significant interaction term [35]. As a subgroup analysis, the observation of lower mortality was insufficient evidence to change practice [77, 99] yet it demonstrates that individualized sepsis treatment based on plasma biomarker expression is possible. Given numerous potential genetic associations with sepsis in pathways associated with drug targets (Table 18.2), testing for heterogeneous treatment effect by genotype or plasma protein expression as a routine addition to interventional trials is an approach that, if adequately powered, could be promising to identify precision targets.
ARDS Genetics and the Search for Causal Intermediates
Numerous candidate gene studies have been undertaken in ARDS populations to elucidate key factors associated with either risk of ARDS or ARDS mortality [100, 101]. Among the best replicated loci, genes contributing to inflammatory response (IL6, IL10, IL1RN, PI3, MBL2, NFKB1, TLR1), vascular regulation (ACE, VEGFA, MYLK, ANGPT2, SERPINE1), oxidant stress (NFE2L2, HMOX1), and lung epithelial function (SFTPB) are overrepresented. Discovery approaches have also been published [102] and have contributed new candidate genes such as PPFIA1, which encodes for liprin alpha 1, a gene expressed in lung and numerous tissues that plays a role in regulating focal adhesions and cell-matrix interactions. As previously mentioned, medium-throughput candidate gene DNA array studies identified risk variants in ARDS that may have potential therapeutic implications, as for IL1RN, the gene encoding IL1RA, or ANGPT2, the angiopoietin-2 gene that contributes to vascular permeability [98, 103].
Given results of GWAS over the past decade, it is usually unreasonable to expect that common variants will be associated with complex diseases with effect sizes large enough to be statistically significant in studies involving fewer than 2000 cases. In complex traits where numerous relatively frequent SNPs are hypothesized to alter risk with modest effect sizes (odds ratio 1.1–1.5) [104], a well-powered study should include many thousands of cases and non-cases, and no such ARDS population yet exists. Nonetheless, an approach by which genetics may help advance a precision medicine platform is by integrating genetic association results with those of other omics studies to prioritize candidate biomarkers. For example, plasma markers could be prioritized based on genetic results to identify those with the most direct relationships with ARDS risk or mortality. To borrow from a cardiology example, genetics provided strong inferential evidence that plasma concentration of low-density lipoprotein cholesterol (LDL) was the major risk factor for coronary artery disease (CAD) risk and mortality. Plasma LDL is strongly genetically regulated and variants that influence plasma LDL strongly associate with CAD in a consonant fashion; genetic variants that lower LDL associate with reduced lifetime CAD risk and those that elevate LDL associate with high CAD and mortality [105,106,107]. Thus, drugs targeting LDL are a mainstay of CAD prevention and treatment, and we term LDL a “causal” marker for CAD. Causal markers are lacking for ARDS, but if a causal marker were identified, it could speed drug development via the design of high-throughput screens to identify compounds that alter the marker.
A few examples of leveraging genetics to infer causal ARDS intermediates are worth highlighting. Recognizing that platelets contribute to both microvascular and immune system activation during ARDS and that the lung is a major site of platelet biogenesis [108,109,110], Wei and colleagues focused on genes shown to strongly associate with platelet counts in healthy subjects and verified that variants in the gene LRRC16A also associate with platelet count in a critically ill population. Further, the same platelet-associated variant is also associated with ARDS risk, and a small but significant portion of the ARDS risk was mediated through platelet count, implicating thrombocytopenia as a causal intermediate for ARDS risk [111]. The same group then identified an independent locus in the LRRC16A gene associated with both a falling platelet trajectory in the ICU and ARDS mortality [112] and statistically demonstrated that declining platelet count mediated the association between LRRC16A and death. These examples of genetic mediation analysis are one demonstration of using genetic data to adapt causal inference methodology for the identification of causal disease intermediates. By mathematically disassembling an association between an explanatory variable (gene variant) and outcome (ARDS) into direct (gene-ARDS) and indirect (gene-platelet and platelet-ARDS) effects, one can infer the relative proportion of effect for the candidate mediator. The concept of using drugs to target platelet abundance or platelet trajectory to modify ARDS risk or mortality may seem unfamiliar, yet there was strong rationale for the LIPS-A study that tested whether aspirin reduced ARDS risk and found that it reduced ARDS risk though with a smaller effect size than anticipated [113]. Future work in this area may be fruitful.
A complementary approach termed Mendelian randomization (MR) leverages the association between genetic variants and intermediates such as plasma biomarkers to infer a biomarker’s causality. Each individual’s genetic variants are independently assigned by random assortment of parental alleles, according to the law of independent assortment, and alleles are distributed independently of any potential confounder. MR leverages this independence and applies the instrumental variable method to reduce a potential predictor variable to the portion that is least confounded, least susceptible to measurement error, and least vulnerable to reverse causation. Reilly and colleagues used MR to infer a potential causal role for plasma angiopoietin-2 (ANG2) and ARDS risk following sepsis; ANG2 was selected as a marker based on a genetic association between the angiopoietin-2 gene (ANGPT2) and ARDS they had previously identified [114]. Further, via mediation analysis, they found that plasma ANG2 mediated a substantial proportion (>34%) of the association between ANGPT2 variants and ARDS risk [114], whereas no direct effect between ANGPT2 and ARDS was observed. Together, these data highlight the potential for drugs that block ANG2 signaling to improve ARDS outcomes and the promise of applying a similar study design to prioritize interventions in future trials.
Sepsis Gene Expression Studies: Ready for Clinical Launch?
Gene expression studies are close to yielding findings that can be clinically translated into prognostic and predictive biomarkers in sepsis. Much of the early work demonstrating the power of whole blood, or peripheral leukocyte, gene expression signatures to discriminate biologically meaningful sepsis subgroups originated with Hector Wong’s work in pediatric populations. Using unsupervised hierarchical clustering, his group consistently identified three patterns of gene expression among pediatric patients with septic shock [82, 115, 116]: “subclass A” patients, characterized by repression of adaptive immunity genes and glucocorticoid receptor signaling, who exhibited higher severity of illness, fewer ICU-free days, and higher mortality.
Given that glucocorticoids are frequently administered to patients with septic shock, the same group then asked whether the effect of glucocorticoids on sepsis mortality was associated with baseline gene expression patterns. They reported a potential interaction (p = 0.089) with steroids increasing mortality in subclass A patients with an odds ratio of 4.1 (95% CI: 1.4–12.0), but not in subclass B patients [117]. Further, by using a plasma biomarker risk stratification tool as a prognostic marker that reliably identified high risk for mortality [11], along with the glucocorticoid-response gene expression subclassification signature, Wong and colleagues established preliminary proof that the gene expression signature could be used to identify subjects who respond favorably to glucocorticoids among those with high predicted mortality [118].
This work set the stage for a precision clinical trial of corticosteroids leveraging the PERSEVERE pediatric biomarker risk model [119], which is based on plasma expression of five biomarkers (C-C chemokine ligand 3, interleukin 8, heat shock protein 70 kDa 1B, granzyme B, and matrix metallopeptidase 8), to identify high-risk subjects. Subsequently, a 100-gene mRNA classifier was used to identify sepsis subclass and limit enrollment to subclass B patients. By focusing on high-risk individuals, the prognostic approach seeks to improve trial efficiency, whereas the predictive approach, in theory, will limit the potential for the intervention to harm patients predicted to do worse with corticosteroid treatment. The Stress Hydrocortisone in Pediatric Septic Shock (SHIPSS) is a phase III randomized double-blind placebo-controlled trial to test whether steroids are beneficial in refractory septic shock; the trial will use plasma and gene expression classifiers to determine whether the above approach is ready for clinical use.
Similar efforts have been undertaken in adult sepsis, with the results of several large, highly cited adult sepsis trials highlighted in Table 18.3. In every case, authors were able to identify a subset of patients at increased risk of death, though the precise method of detecting classes and the number of clusters varied. The UK Genomic Advances in Sepsis (GAiNS) group performed whole blood gene expression profiling by microarray on a discovery cohort of 265 subjects with severe pneumonia, with replication in a second pneumonia population of 106 [120]. Using unsupervised hierarchical cluster analysis of the most variable 10% of transcript probes, they identified two dominant clusters which they termed “sepsis response signatures 1 and 2” (SRS1, SRS2). The SRS1 subtype had a 27% mortality at 28 days, while SRS2 had 17% mortality. Although SRS1 subjects were more likely to require vasoactive medications and had higher Sequential Organ Failure Assessment (SOFA) scores at baseline, there was no statistically significant difference in baseline APACHE II score, need for mechanical ventilation, or use of renal replacement therapy. Combinations of clinical covariates performed poorly at predicting SRS membership with misclassification rates of 20–40%. Thus, SRS grouping seemed to add prognostic value beyond typical clinical scoring systems [120]. Investigators were able to reduce their classifier to seven transcripts that predicted SRS classification in both discovery and validation cohorts. In pathway analysis annotating the genes that were most differentially expressed between SRS1 and SRS2 groups, the high-mortality SRS1 group did not exhibit increased expression of cytokine or inflammatory genes, but, rather, exhibited dysregulation of genes related to T-cell activation, cell death, apoptosis, necrosis, cytotoxicity, and phagocyte movement, in addition to upregulation of genes that characterize endotoxin tolerance [121, 122], suggesting a defective adaptive immune signature characterized SRS1 subjects.
In consonant fashion, a group from the Netherlands created the observational Molecular Diagnosis and Risk Stratification of Sepsis (MARS) cohort and applied slightly different clustering methodology to identify four clusters of sepsis subjects [121]. A 140-gene classifier reliably identified cluster membership when applied to two additional adult sepsis populations, and it identified three of four clusters in a pediatric sepsis population. Although the particular genes that best discriminated the high-risk cohort varied by study, this may be due to high correlation among many dysregulated genes, rather than a failure to replicate. Indeed, a meta-analysis by Sweeney et al. [123] that included mortality data from 14 diverse datasets of bacterial infection, including both adult and child cohorts, identified three clusters (a high-mortality “inflammopathic” cluster, a low-mortality “adaptive” cluster, and an additional “coagulopathic” cluster characterized by abnormal coagulation profiles) that could be distinguished with an 11-gene signature. They assessed overlap of their signature-based clusters with the high-mortality clusters identified in the MARS and Wong et al. pediatric cohorts, and they found substantial overlap in patients identified by each cohort’s high-mortality/high-inflammation endotype.
Thus, while the specific genes selected for gene expression signatures vary, across numerous populations, a high-mortality subset of septic patients with dysregulated adaptive immunity can be identified, and “high-risk” gene expression status enhances mortality prediction over the APACHE score [121]. As mentioned above, the PERSEVERE trial is using these methods for targeted enrollment into a personalized trial of corticosteroid therapy for high-risk patients, demonstrating the potential such signatures have for personalized medicine trials. Adults with sepsis may also exhibit heterogeneous response to glucocorticoids, and one study has suggested that response may be predicted by whole blood gene expression patterns. Using a parsimonious classifier based on the expression of seven transcripts that distinguished the high-mortality, adaptive immune dysregulated SRS1 group [120], Antcliffe et al. retrospectively assigned SRS classification to subjects with septic shock in the VANISH trial [124, 125]. Although there was no mortality benefit observed for corticosteroids in the overall trial, a statistical interaction was detected between steroid allocation and SRS grouping, such that SRS2 subjects exhibited a higher risk of death from sepsis when randomized to steroids [124]. As SRS2 is classically the low-mortality group of sepsis characterized by higher levels of adaptive immune signaling, it may be that corticosteroids disrupt an otherwise favorable host response to infection in some patients.
Gene Expression Studies in ARDS
Four groups have published whole blood gene expression studies in ARDS to date (including a pediatric cohort of acute respiratory failure), and an additional two publicly available datasets from the GLUE grant of trauma also include ARDS phenotyping. Each cohort is individually small (13–67 cases) and heterogeneous in terms of timing of sampling. In each case, a unique set of ARDS-associated genes has been identified, and the ability to rigorously replicate the genes identified in other cohorts has been modest [126]. Sweeney et al. performed a meta-analysis study of all six publicly available datasets to attempt to find a common gene expression signature across the disparate cohorts, but in contrast to the clear signal found with these methods in septic cohorts [79, 127], no expression signature could robustly distinguish ARDS cases from controls [85]. Limiting the ARDS cohorts to more homogenous groups (e.g., only adults with sepsis, excluding those with trauma) did not enhance the classifier signal. Interestingly, the top ARDS-associated genes in the meta-analysis are related to sepsis according to Gene Ontology classifiers, suggesting a potential overwhelming signal from sepsis and severity of illness may have obscured any lung-specific ARDS signals.
Metabolomics Studies in Sepsis and ARDS
Metabolomics is the study of all small molecules in an organism or tissue, including peptides, lipids, nucleic acids, and carbohydrates. Metabolite levels can change rapidly in response to cellular perturbations, such as the switch to anaerobic metabolism in exercise or sepsis that leads to lactate production. Thus, metabolites are particularly promising targets for personalized medicine because they reflect dynamic changes in the host. In contrast to well-established profiling methods for gene expression or SNP assessment, metabolomic profiling methods are rapidly evolving. The number of detected and quantified human metabolites included in the human metabolome database (HMDB) has increased from 8000 in 2010 to 18,000 in 2019, and the presumed number of actual metabolites is likely >100,000 [128]. This growth in the number of identifiable metabolites, and the parallel growth of metabolomic analytic and statistical methods, makes it difficult to compare studies across years and cohorts.
Metabolic changes in sepsis are widely recognized, as lactate, the end product of anaerobic metabolism, is the most widely used sepsis biomarker. Lactate measurement is a Centers for Medicare & Medicaid Services quality measure for patients with sepsis. Elevated lactate (>4 mmol/L) defines septic shock in the most recent sepsis guidelines, and serial lactate measurement and clearance can be used to assess adequacy of resuscitation [9, 129]. In addition to lactate, numerous metabolites are measured in basic chemistry (e.g., serum bilirubin, creatinine) that are followed as part of standard ICU care and comprise critical elements of our ICU scoring systems [74, 76].
Broad profiling of the plasma metabolome in sepsis has shown promise for biomarker identification. Langley et al. performed nontargeted profiling of plasma in 63 sepsis survivors and 31 non-survivors and identified widespread metabolic abnormalities between the two groups, involving pathways such as fatty acid transport, β-oxidation, gluconeogenesis, and the citric acid cycle. This group identified a biomarker panel of five metabolites, along with age and hematocrit, that outperformed lactate and APACHE score in mortality prediction in several studies [130]. Interestingly, when Rogers et al. examined the same populations with different metabolomics analytic strategies, a separate predictive biomarker panel was identified, likely a result of the high correlation among many metabolites [131] and emphasizing the lack of consistency among different metabolomics analysis strategies.
ARDS metabolomics studies are summarized in Table 18.4. These studies vary widely in terms of fluid studied (plasma, free edema, exhaled breath condensate, bronchoalveolar lavage), control population, and metabolic profiling techniques. All are fairly small, involving fewer than 50 ARDS patients in any individual study. Although an ideal control population might have respiratory failure and a condition that mimics the hypoxia of ARDS, such as hydrostatic pulmonary edema or pneumonia, in practice, most studies used convenience or noncritically ill controls. Not surprisingly, the ARDS-associated metabolites in such disparate populations are far from conclusive in terms of either pathway or individual metabolite. Further, given the extent of heterogeneity in sample type and control population, these data are not amenable to meta-analysis. Larger ARDS metabolomics studies are needed, with a focus on careful phenotyping and consistent sample preparation methods.
Conclusion
Despite unique challenges in sepsis and ARDS, the past 10 years have shown substantial advances in the prospect of precision medicine for these deadly diseases. Large-scale gene expression and targeted proteomics plasma studies are identifying biologically distinct patterns of expression that at least retroactively identify a differential response to routine treatments applied in the ICU. Once metabolomics and proteomics approaches become more standardized, investigators may identify additional biomarkers for use in clinical trials that serve either as enrollment criteria to enrich for high-risk subgroups or for potential predictive enrichment to select a population for whom an intervention is more likely to have a positive effect. Prospective randomized trials based on biologic classification will be a reality in the near future, and the era of critical illness precision medicine might thus begin.
References
Liu V, Escobar GJ, Greene JD, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA. 2014;312(1):90–2.
Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800.
Zeni F, Freeman B, Natanson C. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med. 1997;25(7):1095–100. PubMed PMID: 9233726. Epub 1997/07/01. eng.
Ranieri VM, Thompson BT, Barie PS, Dhainaut J-F, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366(22):2055–64. PubMed PMID: 22616830.
Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309(11):1154–62. PubMed PMID: 23512062.
Rice TW, Wheeler AP, Thompson BT, de Boisblanc BP, Steingrub J, Rock P. Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA. 2011;306(14):1574–81. PubMed PMID: 21976613. Epub 2011/10/07. eng.
Smith FG, Perkins GD, Gates S, Young D, McAuley DF, Tunnicliffe W, et al. Effect of intravenous β-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet. 2012;379(9812):229–35.
National Heart, Lung, and Blood Institute ARDS Clinical Trials Network, Truwit JD, Bernard GR, Steingrub J, Matthay MA, et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med. 2014;370(23):2191–200. PubMed PMID: 24835849. Pubmed Central PMCID: 4241052.
Singer M, Deutschman CS, Seymour C, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–10.
Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526–33. PubMed PMID: 22797452.
Prescott HC, Calfee CS, Thompson BT, Angus DC, Liu VX. Toward smarter lumping and smarter splitting: rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am J Respir Crit Care Med. 2016;194(2):147–55.
The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342(18):1301–8. PubMed PMID: 10793162.
Ouellette DR, Patel S, Girard TD, Morris PE, Schmidt GA, Truwit JD, et al. Liberation from mechanical ventilation in critically ill adults: an official American College of Chest Physicians/American Thoracic Society clinical practice guideline: inspiratory pressure augmentation during spontaneous breathing trials, protocols minimizing sedation, and noninvasive ventilation immediately after extubation. Chest. 2017;151(1):166–80.
Girard TD, Alhazzani W, Kress JP, Ouellette DR, Schmidt GA, Truwit JD, et al. An official American Thoracic Society/American College of Chest Physicians clinical practice guideline: liberation from mechanical ventilation in critically ill adults. Rehabilitation protocols, ventilator liberation protocols, and cuff leak tests. Am J Respir Crit Care Med. 2017;195(1):120–33. PubMed PMID: 27762595.
Girard TD, Kress JP, Fuchs BD, Thomason JWW, Schweickert WD, Pun BT, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet. 2008;371(9607):126–34.
Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci. 2007;104(40):15858–63.
Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198–207. PubMed PMID: 25199059.
Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455–66. PubMed PMID: 23688323.
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. PubMed PMID: 25981758.
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR Potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. PubMed PMID: 22047557.
Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2(8):611–20. Pubmed Central PMCID: PMC4154544.
Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. 2017;195(3):331–8. PubMed PMID: 27513822.
Calfee CS, Delucchi KL, Sinha P, Matthay MA, Hackett J, Shankar-Hari M, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med. 2018;6(9):691–8. PubMed PMID: 30078618. Pubmed Central PMCID: 6201750.
Sinha P, Delucchi KL, Thompson BT, McAuley DF, Matthay MA, Calfee CS, et al. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med. 2018;44(11):1859–69. PubMed PMID: 30291376.
Bos LD, Schouten LR, van Vught LA, Wiewel MA, Ong DSY, Cremer O, et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax. 2017;72(10):876–83.
Ware LB, Matthay MA, Parsons PE, Thompson BT, Januzzi JL, Eisner MD. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome∗. Crit Care Med. 2007;35:1821–8. PubMed PMID: 17581482. Epub 2007/06/22. Eng.
Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med. 2005;33(1):1–6; discussion 230–2. PubMed PMID: 15644641. Epub 2005/01/13. eng.
Opal SM, Fisher CJ, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. Crit Care Med. 1997;25(7):1115–24.
Fisher CJ Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271(23):1836–43. PubMed PMID: 8196140. Epub 1994/06/15. eng.
Fisher CJ Jr, Slotman GJ, Opal SM, Pribble JP, Bone RC, Emmanuel G, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22(1):12–21. PubMed PMID: 8124953. Epub 1994/01/01. eng.
Panacek EA, Marshall JC, Albertson TE, Johnson DH, Johnson S, MacArthur RD, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab’)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004;32(11):2173–82. PubMed PMID: 15640628. Epub 2005/01/11. eng.
Reinhart K, Menges T, Gardlund B, Harm Zwaveling J, Smithes M, Vincent JL, et al. Randomized, placebo-controlled trial of the anti-tumor necrosis factor antibody fragment afelimomab in hyperinflammatory response during severe sepsis: the RAMSES Study. Crit Care Med. 2001;29(4):765–9. PubMed PMID: 11373466.
Bernard GR, Vincent J-L, Laterre P-F, LaRosa SP, Dhainaut J-F, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709. PubMed PMID: 11236773.
Man M, Close SL, Shaw AD, Bernard GR, Douglas IS, Kaner RJ, et al. Beyond single-marker analyses: mining whole genome scans for insights into treatment responses in severe sepsis. Pharmacogenomics J. 2012;13(3):218–26.
Meyer NJ, Reilly JP, Anderson BJ, Palakshappa JA, Jones TK, Dunn TG, et al. Mortality benefit of recombinant human interleukin-1 receptor antagonist for sepsis varies by initial interleukin-1 receptor antagonist plasma concentration. Crit Care Med. 2018;46:21–8. PubMed PMID: 28991823. Pubmed Central PMCID: PMC5734955. Epub 2017/10/11. eng.
Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 Receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial∗. Crit Care Med. 2016;44(2):275–81. PubMed PMID: 00003246-201602000-00005.
Wang R, Lagakos SW, Ware JH, Hunter DJ, Drazen JM. Statistics in medicine--reporting of subgroup analyses in clinical trials. N Engl J Med. 2007;357:2189.
Hernandez AV, Boersma E, Murray GD, Habbema JD, Steyerberg EW. Subgroup analyses in therapeutic cardiovascular clinical trials: are most of them misleading? Am Heart J. 2006;151:257.
Rothwell PM. Treating individuals 2. Subgroup analysis in randomised controlled trials: importance, indications, and interpretation. Lancet. 2005;365:176–86.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128.
Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232–45.
Wang Z, Sun Y, Fu X, Yu G, Wang C, Bao F, et al. A large-scale genome-wide association and meta-analysis identified four novel susceptibility loci for leprosy. Nature Commun. 2016;7:13760.
Rowe PC, McLean RH, Wood RA, Leggiadro RJ, Winkelstein JA. Association of homozygous C4B deficiency with bacterial meningitis. J Infect Dis. 1989;160(3):448–51. PubMed PMID: 2788199.
Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26(6):454–70.
Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 2015;212(5):619–31.
von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321(5889):691–6. PubMed PMID: 18669862. Pubmed Central PMCID: 2688396. Epub 2008/08/02. eng.
Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, Wittkowski H, Bialkowski A, Tzaribachev N. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011;63:840.
Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–44.
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1 receptor antagonist. N Engl J Med. 2009;360(23):2426–37.
Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, et al. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2(10):e286. PubMed PMID: 15361935. Pubmed Central PMCID: 515367. Epub 2004/09/14. eng.
Elguero E, Délicat-Loembet LM, Rougeron V, Arnathau C, Roche B, Becquart P, et al. Malaria continues to select for sickle cell trait in Central Africa. Proc Natl Acad Sci. 2015;112(22):7051–4.
Consortium TGP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285.
Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104.
Van Dyke AL, Cote ML, Wenzlaff AS, Land S, Schwartz AG. Cytokine SNPs: comparison of allele frequencies by race and implications for future studies. Cytokine. 2009;46(2):236–44.
de Vries RR, Meera Khan P, Bernini LF, van Loghem E, van Rood JJ. Genetic control of survival in epidemics. J Immunogenet. 1979;6(4):271–87. PubMed PMID: 521665. Epub 1979/08/01. eng.
Maller J, George S, Purcell S, Fagerness J, Altshuler D, Daly MJ, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38(9):1055–9. PubMed PMID: 16936732. Epub 2006/08/29. eng.
Klein R, Zeiss C, Chew E, Tsai J, Sackler R, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–9 . PubMed PMID. https://doi.org/10.1126/science.1109557.
Sorensen TI, Nielsen GG, Andersen PK, Teasdale TW. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med. 1988;318(12):727–32. PubMed PMID: 3347221. Epub 1988/03/24. eng.
Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2(7511):319–23. PubMed PMID: 4143721.
Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10(4):241–51. PubMed PMID: 19293820. Epub 2009/03/19. eng.
ENCODE_Project_Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74.
ENCODE_Project_Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011;9(4):e1001046.
Berdasco M, Esteller M. Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet. 2019;20(2):109–27.
Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19(12):789–800.
Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R, Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet. 2018;19(9):535–48.
Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. PubMed PMID: 20981092. Pubmed Central PMCID: 3042601.
The International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–8.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333.
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–7. PubMed PMID: 10521349.
Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–5.
Strobel EJ, Yu AM, Lucks JB. High-throughput determination of RNA structures. Nat Rev Genet. 2018;19(10):615–34.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9.
Knaus WA, Wagner DP, Draper EA, Zimmerman JE, Bergner M, Bastos PG, et al. The APACHE III prognostic system. Risk prediction of hospital mortality for critically ill hospitalized adults. Chest. 1991;100(6):1619–36. PubMed PMID: 1959406. Epub 1991/12/01. eng.
Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818.
Le Gall J-R, Lemeshow S, Saulnier F. A new simplified acute physiology score (SAPS II) based on a European/North American Multicenter Study. JAMA. 1993;270(24):2957–63.
Kent DM, Rothwell PM, Ioannidis JP, Altman DG, Hayward RA. Assessing and reporting heterogeneity in treatment effects in clinical trials: a proposal. Trials. 2010;11(1):1–11.
Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340(6):409–17. PubMed PMID: 9971864.
Sweeney TE, Shidham A, Wong HR, Khatri P. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci Transl Med. 2015;7(287):287ra71. PubMed PMID: 25972003. Pubmed Central PMCID: 4734362.
Levitt JE, Bedi H, Calfee CS, Gould MK, Matthay MA. Identification of early acute lung injury at initial evaluation in an acute care setting prior to the onset of respiratory failure. Chest. 2009;135(4):936–43. PubMed PMID: 19188549. Pubmed Central PMCID: 2758305. Epub 2009/02/04. eng.
Cazalis M-A, Lepape A, Venet F, Frager F, Mougin B, Vallin H, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. 2014;2(1):20. PubMed PMID: 26215705.
Wong HR, Cvijanovich N, Allen GL, Lin R, Anas N, Meyer K, et al. Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med. 2009;37(5):1558–66. PubMed PMID: 19325468. Pubmed Central PMCID: 2747356.
Dolan JM, Meng H, Sim FJ, Kolega J. Differential gene expression by endothelial cells under positive and negative streamwise gradients of high wall shear stress. Am J Physiol Cell Physiol. 2013;305(8):C854–66. PubMed PMID: 23885059. Epub 07/24.
Palakshappa JA, Meyer NJ. Which patients with ARDS benefit from lung biopsy? Chest. 2015;148(4):1073–82. PubMed PMID: 25950989. Epub 2015/05/08. eng.
Sweeney TE, Thomas NJ, Howrylak JA, Wong HR, Rogers AJ, Khatri P. Multicohort analysis of whole-blood gene expression data does not form a robust diagnostic for acute respiratory distress syndrome. Crit Care Med. 2018;46(2):244–51. PubMed PMID: 29337789. Pubmed Central PMCID: PMC5774019. Epub 2018/01/18.
Morrell ED, Radella F 2nd, Manicone AM, Mikacenic C, Stapleton RD, Gharib SA, et al. Peripheral and alveolar cell transcriptional programs are distinct in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2018;197(4):528–32. PubMed PMID: 28708019. Pubmed Central PMCID: PMC5821902. Epub 2017/07/15.
Sutherland AM, Walley KR. Bench-to-bedside review: association of genetic variation with sepsis. Crit Care. 2009;13(2):210.
Villar J, Maca-Meyer N, Pérez-Méndez L, Flores C. Bench-to-bedside review: understanding genetic predisposition to sepsis. Critical Care Lond Engl. 2004;8(3):180–9. PubMed PMID: 15153236. Epub 04/29.
Lu H, Wen D, Wang X, Gan L, Du J, Sun J, et al. Host genetic variants in sepsis risk: a field synopsis and meta-analysis. Crit Care. 2019;23(1):26.
Rautanen A, Mills TC, Gordon AC, Hutton P, Steffens M, Nuamah R, et al. Genome-wide association study of survival from sepsis due to pneumonia: an observational cohort study. Lancet Respir Med. 2015;3(1):53–60.
Jacobson JR, Dudek SM, Birukov KG, Ye SQ, Grigoryev DN, Girgis RE, et al. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am J Respir Cell Mol Biol. 2004;30(5):662–70. PubMed PMID: 14630613.
Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1168–78. PubMed PMID: 11350795.
Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163(3):510–22. PubMed PMID: 7775594.
Schöneweck F, Kuhnt E, Scholz M, Brunkhorst FM, Scherag A. Common genomic variation in the FER gene: useful to stratify patients with sepsis due to pneumonia? Intensive Care Med. 2015;41(7):1379–81.
Lee S, Lee SJ, Coronata AA, Fredenburgh LE, Chung SW, Perrella MA, Nakahira K, Ryter SW, Choi AM. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014;20(3):432–42. PubMed PMID: 23971531.
Russell JA, Walley KR, Singer J, Gordon AC, Hebert PC, Cooper DJ, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877–87. PubMed PMID: 18305265. Epub 2008/02/29. eng.
Meyer NJ, Ferguson JF, Feng R, Wang F, Patel PN, Li M, et al. A functional synonymous coding variant in the IL1RN gene is associated with survival in septic shock. Am J Respir Crit Care Med. 2014;190(6):656–64. PubMed PMID: 25089931. Pubmed Central PMCID: PMC4214110. Epub 2014/08/05. eng.
Meyer NJ, Feng R, Li M, Zhao Y, Sheu CC, Tejera P, et al. IL1RN coding variant is associated with lower risk of acute respiratory distress syndrome and increased plasma IL-1 receptor antagonist. Am J Respir Crit Care Med. 2013;187(9):950–9. PubMed PMID: 23449693. Epub 2013/03/02. Eng.
Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol. 2014;11(2):81–90.
Reilly JP, Christie JD, Meyer NJ. Fifty years of research in ARDS. Genomic contributions and opportunities. Am J Respir Crit Care Med. 2017;196(9):1113–21. PubMed PMID: 28481621. Pubmed Central PMCID: PMC5694838. Epub 2017/05/10. eng.
Acosta-Herrera M, Pino-Yanes M, Perez-Mendez L, Villar J, Flores C. Assessing the quality of studies supporting genetic susceptibility and outcomes of ARDS. Front Genet. 2014;5:20. PubMed PMID: 24567738. Pubmed Central PMCID: 3915143.
Christie JD, Wurfel MM, Feng R, O’Keefe GE, Bradfield J, Ware LB, et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS One. 2012;7(1):e28268.
Meyer NJ, Li M, Feng R, Bradfield J, Gallop R, Bellamy S, et al. ANGPT2 genetic variant is associated with trauma-associated acute lung injury and altered plasma angiopoietin-2 isoform ratio. Am J Respir Crit Care Med. 2011;183:1344–53. PubMed PMID: 21257790. Epub 2011/01/25. eng.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53. PubMed PMID: 19812666. Pubmed Central PMCID: 2831613. Epub 2009/10/09. eng.
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72. PubMed PMID: 16554528.
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–5.
Linsel-Nitschke P, Götz A, Erdmann J, Braenne I, Braund P, Hengstenberg C, et al. Lifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease—a Mendelian Randomisation study. PLoS One. 2008;3(8):e2986.
Wu J, Sheng L, Wang S, Li Q, Zhang M, Xu S, et al. Analysis of clinical risk factors associated with the prognosis of severe multiple-trauma patients with acute lung injury. J Emerg Med. 2012;43(3):407–12.
Wang T, Liu Z, Wang Z, Duan M, Li G, Wang S, et al. Thrombocytopenia is associated with acute respiratory distress syndrome mortality: an international study. PLoS One. 2014;9(4):e94124.
Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105.
Wei Y, Wang Z, Su L, Chen F, Tejera P, Bajwa EK, et al. Platelet count mediates the contribution of a genetic variant in LRRC 16A to ARDS risk. Chest. 2015;147(3):607–17.
Wei Y, Tejera P, Wang Z, Zhang R, Chen F, Su L, et al. A missense genetic variant in LRRC16A/CARMIL1 improves acute respiratory distress syndrome survival by attenuating platelet count decline. Am J Respir Crit Care Med. 2017;195(10):1353–61. PubMed PMID: 27768389. Pubmed Central PMCID: 5443896.
Kor DJ, Carter RE, Park PK, et al. Effect of aspirin on development of ards in at-risk patients presenting to the emergency department: the lips-a randomized clinical trial. JAMA. 2016;315(22):2406–14.
Reilly JP, Wang F, Jones TK, Palakshappa JA, Anderson BJ, Shashaty MGS, et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med. 2018;44(11):1849–58.
Wong HR, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Willson DF, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med. 2009;7:34. PubMed PMID: 19624809. Pubmed Central PMCID: 2720987. Epub 2009/07/25. eng.
Wong HR, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, et al. Validation of a gene expression-based subclassification strategy for pediatric septic shock. Crit Care Med. 2011;39(11):2511–7. PubMed PMID: 21705885. Pubmed Central PMCID: 3196776. Epub 2011/06/28. eng.
Wong HR, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, Bigham MT, et al. Developing a clinically feasible personalized medicine approach to pediatric septic shock. Am J Respir Crit Care Med. 2015;191(3):309–15. PubMed PMID: 25489881.
Wong HR, Atkinson SJ, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, et al. Combining prognostic and predictive enrichment strategies to identify children with septic shock responsive to corticosteroids. Crit Care Med. 2016;44(10):e1000–3. PubMed PMID: 27270179. Pubmed Central PMCID: 5026540.
Wong H, Salisbury S, Xiao Q, Cvijanovich N, Hall M, Allen G, et al. The pediatric sepsis biomarker risk model. Crit Care. 2012;16(5):R174. PubMed PMID. https://doi.org/10.1186/cc11652.
Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4:259.
Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. 2017;5(10):816–26. PubMed PMID: 28864056. Epub 2017/09/03.
Pena OM, Pistolic J, Raj D, Fjell CD, Hancock REW. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011;186(12):7243–54.
Sweeney TE, Azad TD, Donato M, Haynes WA, Perumal TM, Henao R, et al. Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit Care Med. 2018;46(6):915–25. PubMed PMID: 29537985. Pubmed Central PMCID: PMC5953807. Epub 2018/03/15.
Antcliffe DB, Burnham KL, Al-Beidh F, Santhakumaran S, Brett SJ, Hinds CJ, et al. transcriptomic signatures in sepsis and a differential response to steroids: from the VANISH Randomized Trial. Am J Respir Crit Care Med. 2019;199:980–6. PubMed PMID: 30365341.
Gordon AC, Mason AJ, Thirunavukkarasu N, Perkins GD, Cecconi M, Cepkova M, et al. Effect of early vasopressin vs norepinephrine on kidney failure in patients with septic shock: the VANISH randomized clinical trial early vasopressin vs norepinephrine on kidney failure in septic shock patients early vasopressin vs norepinephrine on kidney failure in septic shock patients. JAMA. 2016;316(5):509–18.
Kangelaris KN, Prakash A, Liu KD, Aouizerat B, Woodruff PG, Erle DJ, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol. 2015;308(11):L1102–13. PubMed PMID: 25795726. Pubmed Central PMCID: 4451399.
Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med. 2016;8(346):346ra91. PubMed PMID: 27384347.
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D17. PubMed PMID: 29140435. Pubmed Central PMCID: PMC5753273. Epub 2017/11/16.
Nguyen HB, Rivers EP, Knoblich BP, Jacobsen G, Muzzin A, Ressler JA, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med. 2004;32(8):1637–42. PubMed PMID: 15286537.
Langley RJ, Tsalik EL, van Velkinburgh JC, Glickman SW, Rice BJ, Wang C, et al. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5(195):195ra95. PubMed PMID: 23884467.
Rogers AJ, McGeachie M, Baron RM, Gazourian L, Haspel JA, Nakahira K, et al. Metabolomic derangements are associated with mortality in critically ill adult patients. PLoS One. 2014;9(1):e87538. PubMed PMID: 24498130. eng.
Nakada T-A, Russell JA, Wellman H, Boyd JH, Nakada E, Thain KR, et al. Leucyl/cystinyl aminopeptidase gene variants in septic shock. Chest J. 2011;139(5):1042–9.
Chen QX, Wu SJ, Wang HH, Lv C, Cheng BL, Xie GH, et al. Protein C -1641A/-1654C haplotype is associated with organ dysfunction and the fatal outcome of severe sepsis in Chinese Han population. Hum Genet. 2008;123(3):281–7.
Walley KR, Russell JA. Protein C -1641 AA is associated with decreased survival and more organ dysfunction in severe sepsis. Crit Care Med. 2007;35(1):12–7. PubMed PMID: 17080006. Epub 2006/11/03. eng.
Tejera P, Wang Z, Zhai R, Su L, Sheu C-C, Taylor DM, et al. Genetic polymorphisms of peptidase inhibitor 3 (elafin) are associated with acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2009;41(6):696–704.
Wang Z, Beach D, Su L, Zhai R, Christiani DC. A genome-wide expression analysis in blood identifies pre-elafin as a biomarker in ARDS. Am J Respir Cell Mol Biol. 2008;38(6):724–32. PubMed PMID: 18203972. Pubmed Central PMCID: 2396250. Epub 2008/01/22. eng.
Marshall RP, Webb S, Bellingan GJ, Montgomery HE, Chaudhari B, McAnulty RJ, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166(5):646–50. PubMed PMID: 12204859.
Marshall RP, Webb S, Hill MR, Humphries SE, Laurent GJ. Genetic polymorphisms associated with susceptibility and outcome in ARDS. Chest. 2002;121(3 Suppl):68S–9S. PubMed PMID: 11893690.
Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, et al. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet. 2000;58(3):181–91. PubMed PMID: 11076040.
Dahmer MK, O'Cain P, Patwari PP, Simpson P, Li SH, Halligan N, et al. The influence of genetic variation in surfactant protein B on severe lung injury in African American children. Crit Care Med. 2011;39(5):1138–44. PubMed PMID: 21283003. Epub 2011/02/02. eng.
Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, Hutton P, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. 2017;196(3):328–39. PubMed PMID: 28036233. Pubmed Central PMCID: PMC5549866. Epub 2016/12/31.
Schubert JK, Muller WP, Benzing A, Geiger K. Application of a new method for analysis of exhaled gas in critically ill patients. Intensive Care Med. 1998;24(5):415–21. PubMed PMID: 9660254.
Stringer KA, Serkova NJ, Karnovsky A, Guire K, Paine R 3rd, Standiford TJ. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma (1)H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol. 2011;300(1):L4–L11. PubMed PMID: 20889676. Pubmed Central PMCID: 3023293.
Rai RK, Azim A, Sinha N, Sahoo JN, Singh C, Ahmed A, et al. Metabolic profiling in human lung injuries by high-resolution nuclear magnetic resonance spectroscopy of bronchoalveolar lavage fluid (BALF). Metabolomics. 2013;9(3):667–76.
Evans CR, Karnovsky A, Kovach MA, Standiford TJ, Burant CF, Stringer KA. Untargeted LC–MS metabolomics of bronchoalveolar lavage fluid differentiates acute respiratory distress syndrome from health. J Proteome Res. 2014;13(2):640–9.
Bos LD, Weda H, Wang Y, Knobel HH, Nijsen TM, Vink TJ, et al. Exhaled breath metabolomics as a noninvasive diagnostic tool for acute respiratory distress syndrome. Eur Respir J. 2014;44(1):188–97. PubMed PMID: 24743964. Epub 2014/04/20.
Singh C, Rai RK, Azim A, Sinha N, Ahmed A, Singh K, et al. Metabolic profiling of human lung injury by H-1 high-resolution nuclear magnetic resonance spectroscopy of blood serum. Metabolomics. 2015;11(1):166–74. PubMed PMID: WOS:000348343300016. English.
Rogers AJ, Contrepois K, Wu M, Zheng M, Peltz G, Ware LB, et al. Profiling of ARDS pulmonary edema fluid identifies a metabolically distinct subset. Am J Physiol Lung Cell Mol Physiol. 2017;312(5):L703–L9. PubMed PMID: 28258106. Pubmed Central PMCID: PMC5451591. Epub 2017/03/05.
Izquierdo-Garcia JL, Nin N, Jimenez-Clemente J, Horcajada JP, Arenas-Miras MDM, Gea J, et al. Metabolomic profile of ARDS by nuclear magnetic resonance spectroscopy in patients with H1N1 influenza virus pneumonia. Shock. 2018;50(5):504–10. PubMed PMID: 29293175. Epub 2018/01/03.
Viswan A, Singh C, Rai RK, Azim A, Sinha N, Baronia AK. Metabolomics based predictive biomarker model of ARDS: a systemic measure of clinical hypoxemia. PLoS One. 2017;12(11):e0187545. PubMed PMID: 29095932. Pubmed Central PMCID: PMC5667881. Epub 2017/11/03.
Maile MD, Standiford TJ, Engoren MC, Stringer KA, Jewell ES, Rajendiran TM, et al. Associations of the plasma lipidome with mortality in the acute respiratory distress syndrome: a longitudinal cohort study. Respir Res. 2018;19(1):60. PubMed PMID: 29636049. Pubmed Central PMCID: PMC5894233. Epub 2018/04/11.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rogers, A.J., Meyer, N.J. (2020). Precision Medicine in Critical Illness: Sepsis and Acute Respiratory Distress Syndrome. In: Gomez, J., Himes, B., Kaminski, N. (eds) Precision in Pulmonary, Critical Care, and Sleep Medicine. Respiratory Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-31507-8_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-31507-8_18
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-31506-1
Online ISBN: 978-3-030-31507-8
eBook Packages: MedicineMedicine (R0)