
Abstract
Sickle cell disease is characterized by vaso-occlusive and hemolysis events that may occur in a variable range of clinical presentations. Patients with this disease, particularly with acute chest syndrome, suffer acute episodes of chest pain caused by vaso-occlusive crisis, that tend to repeat, especially under the influence of lower respiratory infections, dehydration, and any cause of hypoxemia. Children and adolescents with sickle cell disease must be periodically evaluated through oximetry, lung function tests, sleep study, and echocardiograms to detect obstructive and restrictive diseases that frequently worsens the lung condition. Asthma and sleep disorders contribute to morbidity and must be controlled.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
Definition
Sickle cell disease (SCD) is a type of recessive autosomal hemoglobinopathy. It is caused by a specific mutation in the gene that codifies the globin beta-chain in the hemoglobin (Hb) molecule, which results in a substitution of glutamic acid by a valine in the 6th position of the chain, thus creating HbS. This Hb is polymerized in the red blood cell, causing an alteration in its shape and hemolysis, which causes acute events and progressive damage in different organs. In 1910 James Herrick was the first to describe the characteristic shape of the red blood cell (drepanocytes, sickle cell, from the Latin falx, falcis: “sickle shape”). Subsequently, Pauling et al. identified electrophoresis anomalies in HbS, and proposed the term “molecular disease” in 1949, thus demonstrating for the first time that the production of an abnormal protein could be the cause of a genetic disorder.
There are several genotypes of the disease, causing different clinical manifestations. The concept of sickle cell anemia refers to the most usual presentation, in which the patient inherits HbS from both parents, thus being homozygote for the disease (HbSS); those who inherent an S-gene and another causing alteration in the β-chain will have other presentations of sickle cell disease, such as HbSC or HbS/β thalassemia (β0 when there is no production of β chains; β+ when there is partial production). In all of these, HbS represents the greatest proportion of Hb in the erythrocyte. Heterozygous patients inherent only one allele from their parents: the other allele is normal (HbA), and they are named carriers (HbAS), and have what is known as “sickle cell trait.” Carriers are usually asymptomatic.
Epidemiology
The distribution of HbS in the world indicates two factors: migration and the selection of individuals who are HbS carriers because of their resistance to malaria in endemic regions. The greatest prevalence of SCD is in tropical Africa. It has been estimated that every year 230,000 children affected by this disease are born in this region (0.74% of all live births), in comparison to the 2,600 children born in the United States, and 1,300 in Europe. Migrations have spread the disease in America, not only in the African population, but also in their descendants. Diagnosis is confirmed with Hb analysis through electrophoresis or chromatography.
Etiology and Physiopathology
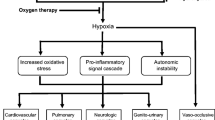
The physiopathological manifestations of SCD are hemolysis and vaso-occlusion (Fig. 52.1). When red blood cells flow in microcirculation, Hb deoxygenation is caused, which involves a change in the composition of the molecule. HbS is less soluble in a deoxygenated environment (which can also be caused by stress, hypoxia, or acidosis), and therefore Hb is polymerized within the red blood cell, and its membrane is distorted, originating a rigid cell with poor deformability. Hb polymers trigger a cascade of events within the cell:
-
a.
Activation of transport channels, with potassium loss and cellular dehydration.
-
b.
Disruption of the red blood cell membrane, thus exposing it to membrane components, such as band 3 protein, which binds to specific antibodies, promoting erythrophagocytosis caused by macrophages.
-
c.
Liberation of hemoglobin in the plasma, with Fe3+, which promotes oxidative damage in the microenvironment.

Physiopathology of sickle cell anemia. (Source: Adapted from Rees DC et al. Lancet. 2010;376:218–222)
Vaso-occlusive crises are caused by erythrocyte and leukocyte entrapment in the microcirculation, originating vascular obstruction and tissue ischemia. Although this process requires HbS polymerization, the trigger event for vascular obstruction is a type of inflammation. This inflammation results from an interaction between the erythrocyte and vascular endothelium, causing obstruction and ischemia episodes, which are followed by a restitution of the vascular flow, causing tissue damage mediated by reperfusion. Then, oxidative stress is triggered, which causes adhesion molecule overexpression, increasing inflammatory cytokines synthesis and leukocytosis. Hemolysis also contributes to vaso-occlusion. Hemoglobin liberation in plasma, caused by intravascular hemolysis, generates superoxide radicals and hydroxyl, which are potent inhibitors of nitric oxide (NO). This compound is produced under normal conditions in the endothelium and regulates the basal vasodilator tone, inhibits platelets, hemostatic activation, and the expression of adhesion molecules dependent on the nuclear factor kβ (FNkβ). Hb release into the plasma also causes endothelial dysfunction and NO resistance. Hemolysis also liberates arginase-1 in the erythrocyte, which metabolizes arginine into ornithine, exhausting the substrate required to synthesize NO. All of this helps to maintain hypercoagulability, with an increase in the platelet activation and the levels of procoagulant factors in the blood.
It is important to note that acute and chronic inflammatory events happen in the lung because erythrocytes are exposed to relatively low O2 tensions, as well as the slow flow of the cells. The airway and vascular system are in close connection, which eases the transference of inflammatory mediators among each other.
Clinical Manifestation of Sickle Cell Disease
SCD has considerable phenotypical heterogenicity, influenced by genetic and environmental factors. Hb of fetal concentration (HbF), coexistence of other hemoglobinopathies, and certain types of polymorphism in simple nucleotides modulate the risk of certain complications. Among environmental factors, environmental humidity, cold, and pollution negatively influence the patient, and particularly by increasing vaso-occlusive events.
Complications worsen with age. In infants, dactylitis (painful inflammation of the fingers and toes), anemia, hyperbilirubinemia, splenomegaly, and infections in the respiratory tract are common. Among other complications, children may present growth and puberty delay, cognitive alterations, and cerebrovascular accidents. Adults tend to have articular pain, chronic ulcers in the legs, kidney failure, and neurocognitive disorders.
Sickle cell anemia complications can appear in any organ, and some of them can be very serious. In this chapter we only present the pulmonary complications (Table 52.1).
Respiratory Clinical Manifestations, Diagnostic Approach and Treatment
Acute Chest Syndrome
Acute chest syndrome (ACS) is a symptom of sudden pulmonary damage, defined as an infiltration of new consolidated alveoli in chest X-rays, with no evidence of atelectasis, and which involves at least a whole lung segment. Generally, the patient presents with chest pain, fever, tachypnea, wheezing, cough, and hypoxemia. The Cooperative Study of Sickle Cell Disease (CSSCD) reported an incidence of 29% (12.8 episodes for 100 patient-years) in patients with sickle cell anemia type SS. Almost half the patients with sickle cell anemia will present with one episode of acute chest syndrome, which is the second cause of hospitalization, after vaso-occlusive crisis (VOC). This may be the initial presentation, although it can also appear after the first 3 days, in 10% to 20% of the cases during their hospital stay. Children between 2 and 4 years of age have the greatest incidence (25.3 years per patient).
Risk factors for this complication involving having HbSS or HbS/β0, thalassemia, asthma, chronic hypoxemia, low HbF, tobacco smoke exposure, general anesthesia, and surgery, mainly abdominal, and during the winter season. There are multiple causes for ACS. The National Acute Chest Syndrome Study Group (NACSSG) studied the causes in 671 episodes presented in 538 patients. Infections were the main cause in 29% of the cases. It is thought that respiratory infections promote an inflammatory response in the lung. Pneumonia caused by Chlamydia was the most common cause, followed by the pneumonia caused by Mycoplasma, viral pneumonia, and bacterial infections last.
Another cause for the acute chest syndrome is fat embolism. During a bone ischemic event, a piece of the marrow or bone that is detached because of a bone marrow stroke migrates through the blood flow to the lungs. As a consequence, sudden inflammation appears. This syndrome caused by fat embolism tends to have a severe clinical presentation, and it tends to appear with pain, neurological symptoms, thrombocytopenia, and increase in transaminases.
Activation of the secretory phospholipase A2 hydrolyzes the fat emboly in sn-2 position, creating free fat acids and lysophospholipids, which cause lung damage. When arachidonic acid is caused by this hydrolysis, a series of inflammation mediators appear, such as thromboxane, leukotrienes, and prostaglandins. Secretory phospholipase A2 increases (336 ± 209 ng/ml; basal level 10 ± 8.4 ng/ml) during an acute chest syndrome presentation, and this increase can predict the disease within the following 24–48 h. Nevertheless, this analysis is still not commercially available, and it has to be validated as a diagnostic test.
A third mechanism of the ACS is pulmonary strokes. This phenomenon has not been carefully studied, and it could be an exclusion diagnosis. Recently, it has been observed that patients may have pulmonary artery thrombosis. This etiology was discovered in 17% of the cases using multidetector computed tomography (CT). Most of the positive findings (81%) in the study by Mekontso Dessap et al. were partial defects in blood vessels. Patients received anticoagulation treatment. Risk factors involved were thrombocytosis and less evidence of hemolysis.
The clinical presentation of the ACS fluctuates in severity. The physician must be alert to detect the beginning of this process. The typical clinical presentation is characterized by chest or limb pain, and fever, followed by hypoxemia and dyspnea. Sometimes the patient has yellow sputum, probably related to fat embolism. Lung examination may reveal crackles, wheezing, and reduction of lung sounds. It is common to find a reduction of hemoglobin levels of 0.7 g/dl on average, and sometimes thrombocytopenia is also present. Thrombocytopenia is an independent risk factor to suffer from multilobe ACS, as well as for needing assisted ventilation. Although most patients can be treated without problems, 13% may require assisted ventilation. CSSCD investigators reported a mortality of 1.1% of children and 4.3% in adults. Other studies have reported a rate of almost 9% in adults.
During an episode of vaso-occlusive crisis, it is recommended to constantly monitor the oximetry. If the patient develops hypoxemia (oximetry <95%), or if during auscultation symptoms or signs compatible with acute chest syndrome appear, a chest X-ray will be required. The diagnosis is confirmed by the finding of a new consolidation in the chest X-ray. Consolidations are more common in lower and medial lung fields (Fig. 52.2). There may be pleural effusion, usually sterile. This infiltration is long lived, usually continuing between 10 and 12 days, especially if an infection is involved. Computerized tomography gives a better view of hyperfusion, reduced number of arterioles and venules, and areas with ground glass presentation.

Acute chest syndrome (ACS) shown in chest X-ray in 12-year-old schoolchild with ACS (a) at admittance caused by coronavirus (CoV), and (b) after ACS has been established
It is recommended to repeat the hemogram to diagnose anemia and thrombocytopenia, as well as obtaining a crossed histocompatibility test, in case a transfusion is needed. Obtaining a gasometric analysis will be of use during the treatment, particularly if the patient presents with a case of respiratory distress. If fever is present, a blood culture is required, to identify the bacterial agent in a reduced number of patients (3.5%). The following are not routine tests: sputum culture, D-dimer exam, and computerized tomography.
Bronchoscopy is not necessary for diagnosis and it is not recommended. Nevertheless, in special cases, if the patient’s condition worsens considerably in spite of the treatment, it could help to find a cause such as plastic bronchitis. This complication consists of a mucus blockage caused by bronchial secretions that worsens the hypoxemic state and lung condensations. In this case, the procedure improves the patient’s condition, both clinically and radiographically. Secretion blockages can be removed with saline washing and continuous suction of the blockage, which is then attached to the tip of the bronchoscope; after this, the bronchoscope is removed, with the blockage of the airway attached to it.
Figure 52.3 summarizes the algorithm of diagnosis and management of ACS. One of the most important measures to avoid in this syndrome is directed spirometry with 10 maximum inspirations every 1–2 h when the patient is awake. Small children who do not know how to use the equipment may blow soap bubbles. Another important measure is to adequately manage pain, without oversedation. Maintenance hydration is important during the vaso-occlusive process, because patients with SCD do not concentrate urine correctly and tend to dehydrate. Nevertheless, excessive liquids should be avoided, as this promotes lung edema.

Algorithm of diagnosis and management of ACS
A combination of an IV cephalosporine, such as cefuroxime, with a macrolide such as oral azithromycin, is the standard care. As an alternate regime, in case of allergies, a quinolone or a macrolide with a beta-lactam antibiotic is recommended. Oxygen must be supplied to keep the saturation over 95%. Blood transfusion must not surpass a maximum of Hb 10–11 g/dl to avoid increasing viscosity. This precaution may prevent ACS worsening, and is are recommended for severe anemia or persistent hypoxemia. Transfused red blood cells must be S negative. Blood replacement is recommended for the cases when the initial hemoglobin is high (Hb ≥ 10 g/dl) and the patient requires transfusion. Other indications for blood replacement are severe hypoxemia in spite of oxygen therapy, patients who need an artificial respirator, multilobe ACS, and also if there is no improvement after a simple transfusion.
An echocardiography is recommended for patients who need intensive care to detect lung hypertension. If this is the case, patients must receive intensive treatment with blood replacement.
Because of airway hyperreactivity in SCD, some investigators propose the routine use of bronchodilators, although others only use it for treating wheezing or prolonged expiration.
Use of steroids such as IV dexamethasone 0.3 mg/kg every 12 h at four doses may shorten hospitalization time, but it is associated to relapses in up to 25% of the patients once the treatment is interrupted, and therefore its use is controversial. Although NO seems to be related to the pathology of the hemolysis, as well as improving ACS in preliminary trials, it did not reduce the duration of the vaso-occlusive crisis. Therefore, it had no impact on the development of this syndrome in clinical trials done afterward.
ACS morbidity is wide, both in the short and long term. This syndrome may be conditioned to the development of chronic lung disease, acute lung hypertension during the episode (specially in adults), and respiratory failure (13% of NACSSG patients). Wide lobal affectation, platelets <200,000/mm3, and a history of heart disease are independent prognosis factors for respiratory failure. For difficult cases of respiratory failure that do not respond to conventional treatment, high-frequency oscillator and extracorporeal membrane oxygenation (ECMO) have been used.
Neurological events are other complications during ACS, appearing in 11% of the patients. The most common events are alterations of the mental state, convulsions, and neuromuscular abnormalities. A platelet count under 200,000 mm3 is a risk factor for neurological complications.
The prevalence of acute kidney damage increases with the severity of the chest syndrome. Patients with this syndrome may develop multiple organ failure. Hydroxyurea , a drug that has the capacity to increase HbF, or a chronic regime of monthly transfusions could reduce the recurrence or severity of the episodes. Patients who continue to experiencing episodes even when treated with hydroxyurea or transfusions are candidates for bone marrow transplant, especially if they have a histocompatible sibling.
Pulmonary Function Tests
Spirometry, lung volume, diffusing capacity, or t transfer factor of the lung for carbon monoxide (DLCO), corrected for hemoglobin, and reactivity measures of the reversible airway with bronchodilator are routine lung function tests for patients with SCD. The methacholine parasympathetic agonist challenge test is not done routinely, because of the low risk to induce vasoconstriction and vaso-occlusion.
These patients suffer from lung dysfunction (Table 52.2). During childhood, obstructive ventilatory dysfunction may appear in 35% to 37% of the cases, and restrictive ventilatory dysfunction may appear in 8%. The Wedderburn study, which included children with SCA who were 5–16 years old, concluded that functional residual capacity (FRC), forced expiratory volume in 1 s (FEV1), vital forced capacity (VFC), and peak expiratory flow (PEF) are lower in comparison to healthy children, but the FEV1/VFC proportion is the same. These differences persist after receiving a bronchodilator, which suggests a mild restrictive disease. After the 10 years of age, 14% of the patients had restrictive or obstructive alterations. Children with sickle cell anemia may lose 3% of FEV1 and 2% of total capacity per year.
Besides this, patients with SCA have a reduced percentage of peak expiratory pressure (PEP), in comparison to peak inspiratory pressure (PIP), which is the inverse proportion of normal adults and children. This PEP deficiency may contribute to a reduction in lung volume, as well as atelectasis. Changes in expiratory muscle force are caused by the fact that abdominal and internal intercostal muscles are supplied by epigastric and intercostal arteries, which do not have extensive collaterals. On the contrary, the diaphragm contributes to inspiration, and it is supplied by several arteries, and therefore is more resistant to ischemic processes.
As patients age, lung function worsens. Only 10% of the adults with sickle cell disease have a normal lung examination according to the CSSCD analysis, which was conducted before the general use of hydroxyurea. Generally, adult patients showed a reduced lung cavity of about 70 ± 15% on average in relationship to the expected value, with restrictive pattern and a DLCO adjusted for 64 ± 20% expected value of hemoglobin. Spirometry was within normal, with FEV1 and FVC values about 83% and 84% of what was expected, respectively.
Therefore, a yearly evaluation of lung function in children who can cooperate and adults is important to detect changes. The exam must be preferentially done when the patient is stable, and at least 2 weeks after a painful episode. Although hydroxyurea reduces the number of ACS events in both adults and children, there is currently not enough information to know if it will modify the changes in lung function. The study BABY HUG Follow Up II, sponsored by the National Institute of Health in the United States, will evaluate if this drug preserved lung function after 10 years, in comparison to those children who have not received hydroxyurea.
Airway Hyperreactivity and Asthma
Airway hyperreactivity, measured after bronchodilator use or subfreezing air hyperventilation, is more common in children with SCD than in control patients, particularly for younger children. Prevalence of such hyperreactivity may be as high as 85% in children with SCD and asthma, and 64% for SCD patients without asthma. Hyperreactivity detected after methacholine is 78% in patients with SCD, in comparison to 18% in children in the general population.
As a comparison, Valdivia et al. pointed out that there was a 9% prevalence of asthma, and 20% of wheezing, during the last 12 months before the study in Chilean schoolchildren. In the United States, 16.8% of 291 children with SS hemoglobin, who were followed as part of the multi-centric observational study of the CSSCD, had bronchial asthma. This prevalence is similar to the asthma prevalence among Afro-Americans. It is important to point out that asthma patients have a two- to four fold risk to develop acute chest syndrome during a hospitalization for a vaso-occlusive crisis, as well as having long hospitalizations, and to be readmitted 3 days after they were discharged. Also, asthma is associated to a double death risk among patients with SCD.
It is not clear why asthma may complicate SCD. The explanation may lie in common mechanisms, such as leukotrienes. Leukotrienes are lipids derived from arachidonic acid that mediate inflammation. Phospholipase A2 releases arachidonic acid from the cellular membranes and causes the production of A4 leukotriene. Leukotriene B4, which is produced in this process, is involved in the activation and chemiotaxis of neutrophils and cysteine leukotrienes, causing bronchoconstriction, smooth muscle proliferation, mucus production, and airway edema; these may be associated to pain pathogeny.
The clinical record must include questions about the onset of respiratory distress and wheezing, exercise intolerance, and family history of asthma. Patients can be evaluated at the same time with questionnaires to detect asthma. Controlling environmental factors and the possibility of other conditions, such as gastroesophageal reflux, sinusitis, and obstructive sleep apnea, must be considered.
A patient with asthma and SCD must be treated as any other, following the recommendations for asthma diagnosis and treatment summarized by a group of experts at the National Institute of Health (NIH) in the United States, which are also available on the Internet. Although there is some evidence that systemic steroids may exacerbate a vaso-occlusive crisis, the benefit of asthma control exceeds the risks. Therefore, inhaled or systemic steroids should be administered if they are needed. If a beta-3 agonist is needed, it may be necessary to have an electrocardiogram to evaluate the QTc interval, because such agonists may increase morbidity in patients who have a prolonged QTc.
Hypoxemia
Hypoxemia is defined as a hemoglobin saturation in the oximetry under 95%. Hemoglobin desaturation in sickle cell anemia is in part caused by the movement to the right of the hemoglobin dissociation curve, due to HbS properties and the effects of chronic anemia through biphosphoglycerate 2.3. A retrospective analysis reported by Quinn, which involved 585 children under 20 years old, with an average age of 9 years old, found that patients with SS or S/β0 thalassemia had an average pulse oximetry saturation (Sat O2) of 96.3%; 33% presented with a saturation <96%, and 2.8% presented a saturation under 90%. When compared to the patients who had HbSC or S/β+ thalassemia, they had an average O2 saturation of 98.7%; 3.6% had a saturation level <96%, and 0.5% at <90%. A good saturation was correlated with higher Hb, reticulocyte count, and lower age in the patients with SS/Sβ0.
It is recommended to control O2 saturation during the follow-up of these patients. It is also important to check those patients who received IV opioids during pain exacerbations to prevent a prolonged hypoxemia. Oxygen administration in hypoxemic patients with sickle cell disease is a safe treatment that does not increase anemia.
Sleep Obstructive Apnea
Snoring prevalence during sleep tends to be increased in patients with sickle cell disease (37%), and the same happens for sleep obstructive apnea (19%), because of the increase in the lymphatic tissue of amygdale, adenoids, and retropharyngeal nodes, which may increase because of the functional asplenia. At the same time, apnea/hypopnea and night hypoxemia are related to priapism, pain exacerbations, and cerebrovascular events. Clinical history must include questions about sleep disorders, and if there is a suspicion of such, a polysomnography must be requested.
Obstructive sleep apnea can be treated through amygdalotomy or adenoidectomy, if there is hyperplasia in these structures. For resistant cases, oxygen should be administered, with or without a BiPAP machine.
Thromboembolism
Patients with sickle cell disease are not free from suffering from venous thrombosis and lung embolism, because anemia is a procoagulant condition, and if this is added to the bedridden condition of pain exacerbations, the risk increases. Doppler ultrasound tests of the limbs and chest helicoidal computerized tomography are useful to detect lung embolism. The management of these complications is the same as for any other type of patients.
Chronic Pulmonary Disease
Although advanced chronic pulmonary disease is rare in children with sickle cell disease, this disease is related to acute and recurrent events of vascular etiology, such as acute chest syndrome, lung edema, and chronic fibrosis. Fibrosis can be observed mainly in the bases of the lung, and it can be detected through high-resolution tomography or chest X-ray. Chronic lung disease tends to be progressive, worsening hypoxemia, dyspnea, and reducing the parameters for lung function. A patient with a corrected DLCO for Hb under 40% has a severe lung disease. Sometimes, different levels of chronic lung disease are referenced in sickle cell anemia (Table 52.3).
Lung Hypertension
Lung hypertension is defined as media lung arterial pressure (mPAP) ≥25 mm Hg, determined through heart catheterization, and it is responsible for the death of 30% of adults with SS:
It is thought that lung hypertension is caused by hemolysis and NO reduction, as was discussed in the physiopathology section. Both NO and endothelin-1 are opposing vaso-active factors that regulate lung vascular tone. Endothelin-1 and the plasminogen activator inhibitor-1 (PAI-1) are increased by the effect of hypoxia-inducible factor 1-alpha (HIF-1α) subunit in endothelial cells of lung microvasculature. At the same time, HIF-1α is produced by the placental growth factor, which is a pro-angiogenic factor, produced by sickle cells. Plasma hemin may also contribute to the activation of the immune system and inflammation. Other clinical factors that contribute are chronic lung disease, hypercoagulability condition of the sickle cell anemia, such as low levels of C and S protein, and a previous history of splenectomy.
The American College of Cardiology Foundation and the American Heart Association recommend a yearly evaluation for the patients under risk of developing lung hypertension, such as patients with sickle cell anemia. Echocardiograms may be used as a test of adverse prognostic if tricuspid regurgitation rate (TRR) is ≥2.5 m/s, which is the value of two standard deviations above the median. At the same time, TRR is used to calculate mPAP. mPAP is estimated using a modified Bernoulli equation: mPAP = 0.61sPAP + 2, where sPAP is the systolic pressure of the lung artery, which is estimated through the formula sPAP = 4 × TRR(Kanter and Kruse-Jarres 2013) + right atrium pressure. TRR prevalence at ≥2.6 m/s is present in 11% of the children. In adults, 30% will have a TRR ≥ 2.5 m/s, but only 6–10% have lung hypertension because of catheterization.
Besides the echocardiography evaluation, N-terminal pro-b-type natriuretic peptide (NT-proBNP), the 6-min walk test, and use of a catheter are steps in the diagnostic process (Fig. 52.4).

Diagnostic algorithm of lung hypertension
Lung hypertension can be arterial, venous, or mixed: 50% of patients with lung hypertension have the arterial presentation (mPAP ≥25 mm Hg) and lung capillary pressure (PCP ≤15 mm Hg), which means that the vasculopathy is mainly located in the arterial lung system. Further, 50% of patients have venous-lung hypertension with mPAP ≥25 mm Hg, and besides this, the diastole final pressure of the left ventricle is >15 mm Hg, or there is evidence of diastolic failure. If the transpulmonary pressure gradient is <12 mm Hg, lung hypertension is venous; and if it is ≥12 mm Hg, it is mixed.
Lung hypertension therapy in SCD is divided into therapies aimed to anemia, such as hydroxyurea and blood transfusions to keep HbS under 20%. Relative to therapies directed to lung hypertension, the Walk PHaSST double-blind clinical study “Treatment: Progression of pulmonary hypertension and sickle cell anemia with sildenafil treatment” was designed to treat those patients with a tricuspid regurgitation rate >2.7 m/s with sildenafil. There were 483 participants, 52 of them (10.8%) were 12–20 years old, and most of them were adults. Unfortunately, the study ended early, because the adverse events reported for the patients treated with sildenafil had a twofold increase, particularly in relationship to pain, in comparison to those who received placebo. No differences in efficiency were found.
Another alternative for lung hypertension is to use endotelin-1 receptor antagonists, such as bosentan and ambrisetan. Nevertheless, double-blind studies ASSET-1 and -2, which studied bosentan in patients with sickle cell anemia and lung hypertension, were closed because of lack of enrollment. Therefore, information about these drugs in relation to these patients is limited.
In patients with systemic arterial hypertension or diastolic disorder, pressure should be controlled, and antidiuretic treatment should be started if there is heart failure. Prostanoid analogues are effective to treat lung hypertension. Epoprostenol administration reduces lung arterial pressure and lung vascular resistance, as well as increasing the cardiac output in patients with sickle cell anemia and lung hypertension. However, the chronic treatment of these patients has not been reported. An adolescent with sickle cell anemia and lung hypertension, which was resistant to sildenafil, bosentan, and treprostinil, who continued with severe symptoms, underwent a bilateral lung transplant successfully after 1-year follow-up.
Sources
Adekile AD. What’s new in the pathophysiology of sickle cell disease? Med Princ Pract. 2013;22:311–2.
Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Rev. 2013;27:279–87.
Klings ES, Wyszynski DF, Nolan VG, Steinberg MH. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med. 2006;173:1264–9.
Knight-Madden J, Forrester TS, Lewis NA, Greenough A. Asthma in children with sickle cell disease and the association with acute chest syndrome. Thorax. 2005;60:206–10.
Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118:855–64.
MacLean JE, Atenafu E, Kirby-Allen M, MacLusky IB, Stephens D, Grasemann H, et al. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am J Respir Crit Care Med. 2008;178:1055–9.
Mekontso Dessap A, Deux JF, Abidi N, Lavenu-Bombled C, Melica G, Renaud B, et al. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 2011;184:1022–9.
Miller AC, Gladwin MT. Pulmonary complications of sickle cell disease. Am J Respir Crit Care Med. 2012;185(11):1154–65.
Minniti CP, Sable C, Campbell A, Rana S, Ensing G, Dham N, et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: association with hemolysis and hemoglobin oxygen desaturation. Haematologica. 2009;94(3):340–7.
Morris CR. Asthma management: reinventing the wheel in sickle cell disease. Am J Hematol. 2009;84:234–41.
Odievre MH, Verger E, Silva-Pinto AC, Elion J. Pathophysiology insights in sickle cell disease. Indian J Med Res. 2011;134:532–7.
Ong BA, Caboot J, Jawad A, McDonough J, Jackson T, Arens R, et al. Respiratory muscle force and lung volume changes in a population of children with sickle cell disease. Br J Haematol. 2013;163:112–7.
Paul RN, Castro OL, Aggarwal A, Oneal PA. Acute chest syndrome: sickle cell disease. Eur J Haematol. 2011;87(3):191–207.
Quinn CT, Ahmad N. Clinical correlates of steady-state oxyhaemoglobin desaturation in children who have sickle cell disease. Br J Haematol. 2005;131(1):129–34.
Rees DC, Williams TN, Gladwin MT. Sickle cell disease. Lancet. 2010;376:218–31.
Strauss T, Sin S, Marcus CL, Mason TB, McDonough JM, Allen JL, et al. Upper airway lymphoid tissue size in children with sickle cell disease. Chest. 2012;142(1):94–100.
Sylvester KP, Patey RA, Milligan P, Dick M, Rafferty GF, Rees D, et al. Pulmonary function abnormalities in children with sickle cell disease. Thorax. 2004;59:67–70.
Valdivia G, Caussade S, Navarro H, Cerda J, Pérez E, Aquevedo A, et al. Influencia del nivel socioeconómico (N SE) en el asma bronquial y cambios en su prevalencia en población escolar en un periodo de 6 años. Rev Méd Chile. 2009;137:215–25.
Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855–65.
Wedderburn CJ, Rees D, Height S, Dick M, Rafferty G, Lunt A, et al. Airways obstruction and pulmonary capillary blood volume in children with sickle cell disease. Pediatr Pulmonol. 2013; https://doi.org/10.1002/ppul.22845.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Álvarez, O., Wietstruck, M.A. (2020). Sickle Cell Disease. In: Bertrand, P., Sánchez, I. (eds) Pediatric Respiratory Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-26961-6_52
Download citation
DOI: https://doi.org/10.1007/978-3-030-26961-6_52
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26960-9
Online ISBN: 978-3-030-26961-6
eBook Packages: MedicineMedicine (R0)