
Abstract
Adult-onset Still’s disease (AoSD) is a rare but clinically well-known, polygenic, systemic autoinflammatory disease. It is typically characterized by four main (cardinal) symptoms: spiking fever ≥39 °C, arthralgia or arthritis, skin rash, and hyperleukocytosis (≥10,000 cells/mm3). However, many other clinical features are possible, and it can appear in all age groups with potentially severe inflammatory onset accompanied by a broad spectrum of disease manifestation and complications. Hence, it remains a diagnostic challenge, and the clinician should first rule out infectious, tumoral, or inflammatory differential diagnoses. Determination of the total and glycosylated ferritin levels, although not pathognomonic, can help in diagnosis. New biomarkers have recently been described, but they need to be validated. The disease evolution of AoSD can be monocyclic, polycyclic, or chronic. In chronic disease, a joint involvement is often predominant, and erosions are noted in one-third of patients. Many progresses have been made in the understanding of the pathogenesis over the last decades. This chapter provides a comprehensive insight into the complex and heterogeneous nature of AoSD describing the identified cytokine signaling pathways and biomarkers. It also discusses the current evidence for the usage of biologics in AoSD to provide guidance for treatment decisions, taking into account both the efficacy and the safety of the different therapeutic options.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Adult-onset Still’s disease
- Autoinflammatory disorder
- Inflammasome
- Interleukin 1
- Pathogenesis
- Diagnosis
- Prognosis
- Treatment
6.1 Introduction
Adult-onset Still’s disease (AoSD was first described in the early 1970s, by Bywaters, as an inflammatory condition occurring in young adults. The disease was very similar to childhood-onset Still’s disease, today called systemic-onset juvenile idiopathic arthritis (SoJIA), described a century ago by Sir Still [1]. Although the exact pathogenic mechanisms of the disease remain unknown, substantial advances have been made first to confirm the homology between AoSD and SoJIA, and then the disease became the archetype of nonfamilial, or sporadic, systemic autoinflammatory disorders (SAID) [2,3,4].
By definition, AoSD affects people older than 16, either de novo or those with a history of systemic JIA [3, 5, 6]. In the latter, a disease-free interval between the childhood episode and the adulthood recurrence differentiates AoSD from persistent SoJIA. In most patients, AoSD is characterized by four cardinal symptoms: spiking fever, an evanescent salmon-pink maculopapular rash, arthritis, and a white blood cell count (WBC) ≥10,000/mm3, mainly neutrophilic polymorphonuclear cells (PMNs). Other features are sore throat or pharyngitis, myalgia, lymph node or spleen enlargement, pleuritis or pericarditis, multivisceral involvement, elevated levels of liver enzymes, and other hematologic abnormalities [3, 7, 8]. None of these symptoms is disease specific, so diagnosis of AoSD is difficult, and clinicians must first rule out neoplastic, infectious, or inflammatory conditions.
6.2 Epidemiology
To date, epidemiologic data about AoSD have been scarce and imprecise, because of the heterogeneous clinical presentation of the disease and the complex assessment of diagnosis. The disease occurs worldwide, and no specific familial aggregation has been reported. AoSD satisfies the definition of an orphan disease because historically its prevalence was estimated between one case per million in Europe [9] and ten cases per million in Japan [10]. In the Japanese study, the incidence was estimated at about one case per million in the mid-1990s. However, because of the substantial advances in AoSD diagnosis as well as differential diagnoses during the last two decades, these prevalence and incidence estimates lack robustness, and new studies are clearly needed to update these figures.
Initially, the disease was characterized as affecting exclusively young adults (i.e., 16–35 years of age [1]); however, more recent series identified cases in adults older than 35 and even elderly people [5, 11,12,13,14]. The sex ratio is almost balanced, with only a slight female predominance [5, 6, 11, 13,14,15].
6.3 Clinical Expression
6.3.1 AoSD Clinical Symptoms
AoSD is traditionally characterized by four “cardinal” symptoms (three are clinical, one is biological), with many other manifestations possibly associated [3, 7, 16].
6.3.1.1 Cardinal Symptom 1: Fever
Fever is constant when the disease is active, except in patients with established disease, for whom residual inflammation could remain without it. Typically, fever starts suddenly, and temperature rapidly reaches 39 °C or more, associated with shivers. It evolves with daily evening spikes for more than a week (Fig. 6.1). Patients usually experience rapid deterioration of general health as well as significant weight loss. Of importance, fever may be the only clinical symptom of AoSD as a potential diagnosis in patients with fever of unknown origin [17].

Typical temperature curve during an AoSD flare
6.3.1.2 Cardinal Symptom 2: Arthralgia or Arthritis
Joint pain or arthritis is the second most common symptom, with synovitis, usually with concomitant fever spikes, occurring in more than two-thirds of patients. All joints can be involved, including sacroiliac and distal interphalangeal joints. In some patients, the presentation is that of a bilateral symmetrical rheumatoid arthritis (RA)-like polyarthritis [3, 7]. Synovial fluid analysis displays high cellularity, >2000 cells/mm3, which confirms joint inflammation. When performed, synovium biopsy reveals only nonspecific synovitis. During the evolution, arthritis becomes erosive in one-third of patients; in these patients, isolated bilateral carpal ankylosis (i.e., without structural damage of metacarpophalangeal or proximal interphalangeal joints, in contrast to RA) is very suggestive of AoSD (Fig. 6.2) [1, 3, 7].

Ankylosing carpal arthritis in adult-onset Still’s disease
6.3.1.3 Cardinal Symptom 3: Skin Rashes
Skin rash is typically salmon pink-colored macular or maculopapular (Figs. 6.3 and 6.4). The rash is transient, mainly visible during fever spikes on the proximal limbs or trunk, and rarely involves the face, palms, or soles of the feet [3, 7, 18]. No specific histological feature has been described. Misdiagnosis with drug allergy is frequent and usually attributed to nonsteroidal anti-inflammatory drugs (NSAIDs) prescribed for joint symptoms or fever. Complete regression without scars is the rule.

Typical rash of the arm in adult-onset Still’s disease

Transient erythema of the elbow in adult-onset Still’s disease
Atypical patterns also reported are urticarial or pruritic with dermographism [3, 7, 18]. Presence of purpuric lesions should lead to urgent coagulation workup because these lesions are suggestive of the AoSD hematologic complications of hemophagocytic syndrome or reactive hemophagocytic lymphoproliferation (RHL), disseminated intravascular coagulation (DIC), or thrombotic microangiopathy (TMA), also called thrombocytic thrombocytopenic purpura or Moschcowitz syndrome [3, 7].
6.3.1.4 Other Clinical Symptoms
Sore throat (odynophagia) and sometimes pharyngitis are classical symptoms, concomitant with fever (and not just a few weeks previous, e.g., in post-streptococcal arthritis) [3]. All microbiological test results are negative. The presence of this symptom in such a systemic context seems to exclude lymphoma or another neoplastic disease [19].
Diffuse and symmetrical lymphadenopathy is possible, eventually associated with splenomegaly or even hepatomegaly. However, large and nonsymmetrical lymphadenopathy should lead to the exclusion of malignant lymphoma by biopsy: reactive polyclonal lymphoid hyperplasia is the usual pattern in AoSD [3, 7]. Kikuchi necrotizing lymphadenitis has been described in a few cases [20].
Myalgia is frequent, but myositis or polymyositis is exceptional [3, 7, 21, 22].
Finally, as in many other inflammatory disorders, nonspecific manifestations have been reported: abdominal pain related to deep lymphadenitis, aseptic peritonitis or rarely acute pancreatitis, pleural effusion or pericarditis, or interstitial lung infiltrates [3, 7].
6.3.2 AoSD Biological Abnormalities
6.3.2.1 Cardinal Symptom 4: Increased Leukocyte and Neutrophil Counts
A major increase in leukocyte and neutrophil counts is observed (i.e., leukocyte count >10,000–15,000/mm3, with neutrophils >80%). Greatly increased counts, up to 50,000/mm3, can be observed, associated with myelemia. Leucopenia has also been described, in isolation or with anemia and/or thrombopenia; it often reveals RHL or TMA [3, 7].
6.3.2.2 Acute-Phase Reactants
Besides increased leukocyte and neutrophil counts, a substantial increase in acute-phase reactants—ESR and levels of CRP, fibrinogen, and serum immunoglobulins—is always present during AoSD flares.
6.3.2.3 Liver Function Tests
Elevated serum liver enzyme levels, rarely fulminant hepatitis, are often present, related to systemic inflammation or the use of drugs, such as antibiotics or NSAIDs, as symptomatic treatment before diagnosis.
6.3.2.4 Serum Ferritin and Glycosylated Ferritin
High serum ferritin level, an indicator of macrophage activation, has been frequently reported [5, 12, 23,24,25,26]. Although hyperferritinemia was first considered suggestive of AoSD, several studies showed that hyperferritinemia, whatever the threshold used, has poor positive predictive value in isolation without a suggestive context [27, 28].
Besides total ferritin level, the diagnostic performance of glycosylated ferritin (GF) level has been investigated [29]. The GF level normally represents more than half of the total ferritin level. In inflammatory conditions, the concentration of GF decreases and usually ranges from 20% to 50%; this decrease has been related to the saturation of glycosylation mechanisms due to hyperferritinemia. However, in AoSD, GF level is typically markedly decreased (below 20%), which suggests a more specific phenomenon. More extensive data revealed a sensitivity and specificity of GF ≤20% for AoSD diagnosis of 78% and 64%, respectively [27]. The combination of both hyperferritinemia and GF level ≤20% yielded a sensitivity and specificity of 67% and 84%, respectively. Of note, although serum ferritin level fluctuates according to systemic inflammation, GF level remains low several weeks to several months after disease remission [30]. However, as mentioned previously, low GF level is not completely specific to AoSD and is observed in other inflammatory processes, such as severe systemic infections [27]. Moreover, the GF level is usually low, ≤20%, in hemophagocytic syndromes, regardless of the cause, such as infection, neoplasm, or inflammation [31,32,33].
6.3.2.5 Immunological Findings
Results of immunological workups performed to exclude other connective tissue diseases or inflammatory joint diseases must be negative.
6.4 Pathogenesis
6.4.1 A Systemic Autoinflammatory Disorder as Opposed to a Systemic Autoimmune Disease
Approximately ten years ago, McGonagle and McDermott formulated the hypothesis of two main pathogenic mechanisms underlying immune-mediated inflammation against the self and proposed a new classification for immunological diseases contrasting autoimmunity and autoinflammation [2, 34]. Autoimmunity referred to adaptive immunity and was defined as aberrant dendritic, B- and T-cell responses in primary and secondary lymphoid organs leading to tolerance break and development of immune reactivity toward native antigens (with autoantibodies in most cases), whereas autoinflammation referred to innate immunity and was defined as dysregulated activation of macrophages and neutrophils in response to a danger signal leading to tissue damage. These categories represent a continuum, which allowed for a new classification of immune-mediated inflammatory disorders that was refined in the following years [2, 4].
Although autoimmune diseases were rather easily diagnosed based on autoantibody or autoantigen-specific T and B cells, no specific biomarker exists for SAID. The definition mainly relies on similarities with monogenic, hereditary periodic fever syndromes, which were at the origin of the concept. They include several key features: intense inflammation with periodic fever, tissue inflammation depending on the disease, increased leukocyte and neutrophil count, elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level, and, more recently, a pathogenic role of inflammasome and response to interleukin-1 (IL-1) blockade [35]. Besides monogenic familial syndromes, Crohn’s disease was the first nonfamilial polygenic AID, mainly tissue specific, with predominant gut involvement [36, 37]. A few years later, Still’s disease was described as another nonfamilial AID, becoming one of the most characteristic polygenic systemic SAID [3].
6.4.2 A Pro-inflammatory Cascade
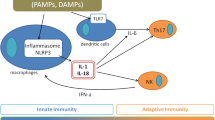
The mechanisms underlying AoSD pathogenesis are largely hypothetical, but, from the evident pathogenic pathway seen in SoJIA patients and the growing understanding of SAID, a pro-inflammatory cascade can be proposed (Fig. 6.5) [3, 7, 16, 38, 39].

Pathogenic pathways involved in AoSD (adapted from [16]). AGE advanced glycosylated end products, ATP adenosine triphosphate, ER endoplasmic reticulum, DAMP damage-associated molecular pattern, DC dendritic cells, HLA human leukocyte antigen, IL interleukin, MΦ macrophages, MIF macrophage inhibitory factor, NK natural killer, PAMP pathogen-associated molecular pattern, PMN polymorphonuclear neutrophil, RAMP resolution-associated molecular pattern, ROS reactive oxygen species, sRAGE soluble receptors of AGE products, TGF transforming growth factor, Th1 T-helper 1 cells, Treg T-regulatory cells
6.4.2.1 A Cytokine Storm
The starting point is likely specific danger signals, such as pathogen- or damage-associated molecular patterns (i.e., PAMPs or DAMPs). Danger signals are transmitted to macrophages and neutrophils via specific Toll-like receptors that activate specific inflammasomes, likely NOD-like receptor family pyrin domain containing 3 (NLRP3), leading to caspase activation and overproduction of active IL-1 [39,40,41,42]. This step seems to be central to the AoSD pathogenesis and leads to intense innate immune cell activation and overproduction of several pro-inflammatory cytokines including IL-6, IL-18, tumor necrosis factor (TNF), as well as IL-8 and IL-17. Several factors actively contribute to an amplified inflammatory response, often referred to as the cytokine “burst” or “storm” [3, 42]. In addition to IL-1 itself, conferring retrograde activation of macrophages and neutrophils; different alarmins, such as the S100 protein—S100A12 protein seeming to be more specific to Still’s disease in children—and advanced glycosylated end (AGE) products are involved in these processes [39, 42,43,44,45,46]. Besides amplification mechanisms, SAID pathogenesis has been suggested to involve deficiency or failure in regulatory or anti-inflammatory mechanisms: deficiency in T regulator or natural killer cells, insufficient IL-10 production, and deficiency in resolution lipid mediators, soluble receptors of AGEs (RAGEs), or other resolution-associated molecular patterns [47,48,49,50].
6.4.2.2 A Role for Infections as a Trigger
Bacteria or viruses are the usual suspects for the danger signals. Numerous case reports describe the occurrence of AoSD after viral infection (rubella; measles; mumps; Epstein-Barr virus; hepatitis A, B, or C virus; HIV; cytomegalovirus; parvovirus B19; adenovirus; echovirus; human herpes virus 6; influenza and parainfluenza viruses; Coxsackie virus) or bacterial infection (Yersinia enterocolitica, Campylobacter jejuni, Chlamydia trachomatis or pneumoniae, Mycoplasma pneumoniae, Borrelia burgdorferi) [3, 7, 39]. In addition, patients often experience odynophagia or pharyngitis just before the start of the disease or the relapse, which could correspond to the infectious danger signal that will trigger Toll-like receptors and start the intense inflammatory process.
6.4.2.3 A Role for a Genetic Background
In contrast to monogenic, hereditary, periodic fever syndromes, the underlying genetic background of AoSD is largely unknown. The disease is present in different geographic regions and different ethnic groups [3, 4, 7, 39, 51]. Association studies have suggested a potential predisposing genetic background. HLA-Bw35 was the first identified as a susceptibility antigen and associated with a mild self-limiting pattern of the disease [52]. HLA-DR4 was found more prevalent in AoSD cases versus healthy controls, and HLA-DRw6 was associated with the occurrence of proximal arthralgia [53]. However, no functional analysis has confirmed these hypotheses [3, 39, 53].
Recently, two major findings have been reported in SoJIA patients. Firstly, a homoallelic missense mutation in the enzyme laccase (multicopper oxidoreductase) domain-containing 1—LACC1—has been identified in 13 SoJIA patients from five Saudi consanguineous families [54]. Although familial SoJIA is not the rule, this study raises the potential role of this laccase in the pathogenesis of SoJIA. Secondly, a large association study performed in 982 SoJIA children and 8010 healthy controls identified a strong association between SoJIA and different HLA-DRB1∗11 haplotypes which all contain glutamate 58 [55]. This finding is more challenging since it would involve adaptive immunity in the AoSD pathogenesis which was not expected [56].
Finally, substantial advances have been made in exploring the human genome, which will facilitate the identification of potential mutations in genes involved in autoinflammatory pathways, including de novo germinal mutations or somatic mutations occurring during fetus development or after birth [57, 58].
6.5 Atypical Forms and Life-Threatening Complications
Several severe manifestations have been described in AoSD, which explains the potentially pejorative prognosis of the disease [3, 59]. The disease can be life-threatening because of the predominant involvement of one organ or of systemic complications with multiple organ failure from the outset, while the diagnosis isn’t clearly confirmed. The articular signs may often fall by the wayside because of the gravity of the complications of such clinical forms, which can contribute to misdirect the clinicians who are not familiar with AoSD.
These complications are given in Table 6.1, and the most frequent and relevant ones are discussed in more detail.
6.5.1 Reactive Hemophagocytic Lymphohistiocytosis (RHL)
This is a common complication of AoSD at the time of diagnosis, right after treatment introduction or during the course of the disease. In systemic-onset JIA, it is also called macrophage activation syndrome. This serious complication should be suspected in a patient with persisting fever (in contrast to evening spiking fever) and decrease in initially elevated leukocyte and neutrophil counts [3, 32, 59]. Several other manifestations can be associated.
The key issue with RHL is to determine whether its occurrence is related to AoSD intense inflammation or to concomitant infection, potentially favored by immunomodulators introduced because of AoSD. The list of possible infections is quite long, with viral reactivation at first place.
6.5.2 Coagulation Disorders
AoSD can be complicated by two serious coagulation disorders, mainly at the acute phase of the disease.
The first disorder, disseminated intravascular coagulation (DIC), is not rare and may occur at a frequency of 1–5% [8, 53, 59,60,61,62,63,64,65,66,67]. This diagnosis should be suspected with the combination of thrombotic events and cutaneous or mucosal bleeding and sometimes specific organ involvements, such as fulminant hepatitis, cardiac or respiratory failure, or stroke. Hemostasis workups reveal platelet and coagulation factor consumption, increased thromboplastin time, and high D-dimer levels.
The other rare but quite severe coagulation disorder is thrombotic microangiopathy (TMA). It must be suspected with unexplained multiorgan failure or stroke, related to multiple small thrombi leading to tissue ischemia and mechanical hemolytic anemia [60, 74, 75]. Acute vision impairment, such as blurred vision related to Purtscher-like retinopathy, is frequently the first symptom [74]. Key diagnostic tests display thrombocytopenia by platelet consumption, anemia, and schistocytes (fragmented red blood cells), which are specific to this diagnosis. Organ imaging may reveal multiple infarctions. In addition, ADAMTS13 enzymatic activity needs to be tested because acquired deficiency has been found to predispose to TMA [59, 96,97,98]. TMA has been mainly described during AoSD flare, related to intense inflammation or to concomitant infection with Shiga toxin-producing microorganisms [74].
6.5.3 Cardiac and Pulmonary Involvements
Although pleural effusion or pericarditis is frequent, other serious cardiac or pulmonary manifestations have been described.
Specific attention should be dedicated to a rare and very severe AoSD manifestation, pulmonary arterial hypertension (PAH), with several cases recently reported [77,78,79,80,81,82,83,84,85,86,87,88]. PAH, either idiopathic or occurring with AoSD or other connective tissue diseases, is thought to be related at least in part to immune-related alteration of the endothelium with overproduction of IL-1 , IL-6, IL-18, and TNF [79, 80, 82, 83, 99]. PAH may occur at AoSD onset or later and seems to predominate in women. The diagnosis must be suspected with unexplained and often rapidly progressing dyspnea [59, 79, 81, 100, 101]. Electrocardiography is rarely contributive, eventually disclosing signs of right atrium hypertrophy. Echocardiography is more useful for detection by measuring systolic pulmonary artery pressure, which is >35 mmHg, and the exclusion of left ventricle dysfunction. The formal PAH diagnosis remains the right heart catheterization [100, 101].
6.5.4 Hepatitis
While liver abnormalities are most often limited to mild to moderate increases in aminotransferase activity which is frequent (up to 60% of cases), fulminant and fatal hepatitis have been reported [3, 59]. The potential role of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), or methotrexate has been mentioned by some authors [102]. Thus, clinicians should closely monitor liver function from the onset of the disease and particularly after prescription of potentially hepatotoxic drugs. Self-medication should be avoided.
Liver biopsy, if performed, reveals nonspecific portal infiltrates of lymphocytes, plasma cells, and polymorphonuclears [3, 59, 102]. Hepatocytic lesions or massive necrosis has been described in fulminant hepatitis with rapidly progressive hepatic insufficiency. Liver transplantation was necessary in a few exceptional cases [3, 103].
6.5.5 Exceptional Symptoms
Amyloid A amyloidosis is becoming extremely rare owing to the better ability of AoSD treatments to control inflammation [89,90,91,92,93]. It could be observed in refractory patients or in patients who did not have access to adequate treatments for long periods of time. Multiple organs can be involved.
Many other symptoms attributed to AoSD have appeared in the literature. As these are almost exclusively in case reports, drawing conclusions is difficult:
-
Ophthalmologic involvement related to sicca syndrome, conjunctivitis, uveitis, or episcleritis [1, 5, 102].
-
Neurological manifestations, such as ischemic stroke, aseptic meningitis, or encephalitis [3]. Such symptoms might reveal hepatic failure or coagulation dysfunction, as in DIC, RHL, or TMA.
-
Renal involvement limited to isolated proteinuria or related to glomerular or interstitial nephritis [102, 103]. Acute renal failure is possible in the context of severe myositis, hepatic insufficiency, or hematological complications [21, 74, 102,103,104].
-
Secondary amyloid A amyloidosis (AA amyloidosis) is nowadays exceptional and is related to sustained uncontrolled inflammation [89,90,91,92,93].
6.6 Differential Diagnoses
AoSD is an exclusion diagnosis. Because of presentation heterogeneity and of the lack of specificity of the clinical and biological manifestations, many other diagnoses might be evoked and afterward ruled out, before reasonably considering AoSD(Table 6.2) [3, 5, 6, 102]. Before finally diagnosing AoSD, it is thus essential to first rule out neoplastic, infectious, or inflammatory conditions that can mimic the disease.
6.6.1 Infections
Infectious diseases represent the most complex issue in differential diagnoses because AoSD therapies can be highly deleterious. The most misleading diagnoses are probably deep occult infections (e.g., biliary tract abscess or bone marrow infestation) or subacute endocarditis [3]. Whole blood culture, urine testing, and synovial fluid culture seem the minimal workup, while other tests might be considered depending on the context.
6.6.2 Malignant Diseases
Among malignancies, malignant lymphomas—either Hodgkin’s disease or non-Hodgkin’s lymphomas—are probably the most misleading and the first to exclude if nonsymmetrical, fixed, and indurated lymph node enlargement is present [3, 105]. Less frequently, systemic inflammatory conditions have been described in the course of lymphoid leukemia or in paraneoplastic syndromes associated with solid cancers of either the lung or the breast [3, 105].
6.6.3 Autoimmune or Autoinflammatory Disorders
Autoimmune diseases that can mimic AoSD include vasculitis, especially polyarteritis nodosa, and polymyositis [6]. For patients with mainly chronic arthritis, the distinction between seronegative RA and AoSD can be difficult.
Besides autoimmune diseases, hereditary or nonfamilial autoinflammatory syndrome (AIS) can present with symptoms close to AoSD because of the combination of fever, skin lesions, and arthritis in addition to other symptoms that differ among the AIS [106,107,108]. Although the hereditary AIS often begins during childhood, the diagnosis tends to be made when the individual is an adult; examples are hyperimmunoglobulin D (IgD) syndrome or TNF receptor-associated periodic syndrome (TRAPS). Different mutations have been associated with different hereditary AIS, but, to date, none has been associated with AoSD. However, the similarities between these diseases and AoSD might suggest common pathogenic pathways and place AoSD as a nonfamilial, sporadic form of AIS.
Finally, in rare cases, the severe and systemic drug hypersensitivity reaction can mimic AoSD. The presence of urticarial skin lesions and increased eosinophilic PMN count are suggestive features, as is exposure to a potential agent within the previous days or weeks.
6.7 Classification Criteria and Biomarkers
After comprehensive investigations, no clinical sign or biological abnormality can be considered AoSD specific enough to ascertain the diagnosis. Thus, classification criteria may be helpful, although they have been developed more for clinical research than diagnosis.
6.7.1 Classification Criteria
Two sets of criteria have been validated (Table 6.3). The Yamaguchi criteria set, published in 1992, is the most widely used [109]; however, it contains exclusion criteria that are problematic in clinical practice. The Fautrel criteria set has the advantage of including ferritin and GF levels as diagnostic biomarkers and does not require exclusion criteria [13]. In a recent validation study, both sets showed high sensitivity and specificity [14].
6.7.2 AoSD Biomarkers
The most relevant serum biomarkers identified to date are listed in Table 6.4.
6.7.2.1 Biomarkers for Routine Clinical Setting
Ferritins are the major biomarkers for AoSD. Serum ferritin level is a key biomarker to assess disease activity, but probably less relevant for diagnosis since its specificity is limited and no clear threshold has been identified so far [27, 28]. The diagnostic value of glycosylated ferritin level, with the threshold of GF≤20%, is much more interesting [27, 30].
Procalcitonin, a marker of severe systemic infection, was also found elevated in patients with active AoSD and does not appear relevant to distinguish acute infection from AoSD flare [42, 113].
Serum level of calprotectin (i.e., S100A8/S100A9 protein) could be an additional disease activity biomarker because alarmins seem to play a key role in AoSD pathogenesis. However, it is not specific to AoSD and may be elevated in many other inflammatory conditions [42, 44, 111, 112]. Finally, serum amyloid A protein (SAA) is an inflammatory biomarker found predictive of amyloidosis [42].
6.7.2.2 Potential Biomarkers for Future Clinical Research
Several research works assessed serum cytokine levels. High serum levels of IL-1 , IL-6, and IL-18 have been found in systemic forms of AoSD and can be considered activity biomarkers. However, their additional value on top of CRP level and other inflammatory biomarkers is unclear, and these cytokines are clearly not specific to AoSD. Thus, they are not recommended in routine investigation. Of importance, IL-18 seems to play a key role in RHL.
AGE products and RAGEs, involved in AoSD pathogenesis, may be elevated in other inflammatory disorders. High serum levels have been associated with polycyclic or chronic/progressive evolution patterns [42]. Serum level of soluble CD163, a macrophage activation biomarker, is elevated in AoSD but is not specific to the disease. Finally, several chemokines (C-X-C motif chemokine ligands 10 and 13, macrophage inhibitory factor) have been found to be diagnostic biomarkers for AoSD, which needs to be confirmed in larger patient samples [42].
6.8 Disease Evolution and Prognosis
AoSD evolution can be quite diverse and currently is impossible to predict. Several patterns of evolution have been described, mainly based on case series and not robust epidemiological studies [5, 9, 102, 132, 133]:
6.8.1 Evolution Patterns
6.8.1.1 Self-Limited, Monocyclic Evolution
It combines systemic manifestations and joint involvement: the initial flare evolves over a few weeks; remission is achieved with steroids or other immunomodulatory agents after a few days or weeks; and treatments can be progressively tapered and then stopped without relapse after a few months. The pattern may represent 19–44% of cases.
6.8.1.2 Recurrent or Polycyclic Evolution
It is characterized by AoSD relapses after a few months or years, under immunomodulatory treatment or after its discontinuation. In this pattern, one classical presentation is a first flare during childhood-diagnosed SoJIA, followed by sustained drug-free remission for several years, and then a relapse in adulthood. In most of these cases, recurrences combine systemic and joint involvement. The pattern represents 10–41% of cases.
6.8.1.3 Chronic and Progressive Evolution
In this form, a continuous inflammation is responsible for chronic and frequently erosive joint involvement with regular systemic flares. This pattern was the most frequent, estimated at 35–67% of cases, in old series [1, 102, 134]. Although no new estimates are available, this pattern should be less likely with the most recent targeted therapies.
6.8.2 Prognosis
The prognosis of AoSD is dominated by potential life-threatening events as well as progressive functional impairment.
6.8.2.1 Life Prognosis
The life prognosis is dominated by the severity of the following visceral involvements, in isolation or combination: hematological complications, such as DIC, TMA, or severe RHL; fulminant hepatitis with rapid liver failure; and acute respiratory distress syndrome, extensive myocarditis, or multiorgan failure [3, 59]. Importantly, exacerbation of these serious symptoms has been reported within the first days of treatment initiation, especially of biological therapies. This necessitates a close and very careful monitoring of such patients at the start of treatment. In addition to organ involvements, the development of secondary amyloidosis of the AA type remains possible in long-standing refractory chronic articular AoSD [59].
6.8.2.2 Functional Prognosis
Functional prognosis depends mainly on erosion and joint destruction in chronic articular AoSD, which affects approximately one-third of patients [3, 7, 102]. No clear predictor of erosive joint disease has been identified, but, as joint involvement is often limited, functional prognosis in AoSD seems to be less severe than in RA or other inflammatory arthritides [135].
6.8.2.3 Iatrogenicity
Corticosteroid side effects are also common because steroids are usually prescribed in high doses and the long term, which might contribute to the overall prognosis in AoSD.
6.9 AoSD Treatment
AoSD is rare, and no randomized controlled trial has been performed. Thus, the only information on therapy is from observational studies and retrospective case series. Recently, the management of AoSD benefited from the proofs of the efficacy of targeted biotherapies.
6.9.1 Symptomatic Treatments
Although NSAIDs, including aspirin, have been effective in systemic JIA, they are rarely effective in AoSD; only 20% of patients have achieved control with this therapy [1, 102, 103].
Of the NSAIDs, indometacin 150–250 mg/day seems to be the most effective [3, 102]. Liver enzymes must be monitored at the initial stages of the disease because severe hepatitis has been suggested to be related to treatment with NSAIDs [3, 102, 103].
Acetaminophen may help to reduce fever, although it is often not enough to suspend it. Other analgesics might be necessary to relieve intense myalgia and joint paint.
6.9.2 Corticosteroids
Once the diagnosis is established, corticosteroids are usually required to induce symptom remission. Optimal dosing relies on medium to high doses (i.e., 0.5–1 mg/kg/day of prednisone equivalent) [3, 5, 6, 16, 102]. Patients with serious visceral involvement might achieve a quick response with intravenous infusion of high-dose methylprednisolone [3, 102, 103]. Response to corticosteroids is often dramatic—within a couple of hours or a few days [3, 102].
The consensus is lacking on a therapeutic tapering scheme once clinical remission is achieved. However, owing to the potentially serious side effects of cumulative corticosteroid therapy, many others currently recommend to achieve the dose of 0.1 mg/kg/day after 6 weeks of therapy and to stop completely after three months. If this is not possible, the response should be considered inadequate, and a disease-modifying treatment, especially a targeted therapy, should be started (Fig. 6.6) [16].

Therapeutic strategies to manage AoSD patients. CRP C-reactive protein, ESR erythrocyte sedimentation rate, IL interleukin, methylPDN methylprednisolone, NSAIDs nonsteroidal anti-inflammatory drugs, TNF tumor necrosis factor
6.9.3 Methotrexate
Methotrexate is used as an immunomodulatory agent in rheumatoid arthritis. It is efficient in controlling AoSD disease activity and allowing for steroid dose sparing [3, 136,137,138]. However, whether methotrexate can prevent or limit joint structural damage in the erosive form of AoSD, as has been shown for RA, is unclear. The presence of liver enzyme abnormalities does not contraindicate methotrexate prescription, but close biological monitoring is necessary [3, 102, 136, 137].
Of importance, methotrexate can be associated with anti-IL-1- or anti-IL-6-targeted therapies.
6.9.4 Targeted Therapies for AoSD
Different targeted therapies have been used to treat refractory AoSD (Table 6.5). The medical need is high, keeping in mind the unsatisfactory rates provided by classical therapies with NSAIDs or conventional disease-modifying antirheumatic drugs (DMARDs). The use of glucocorticoids is also limited by the known long-term safety issues, especially with higher doses. In fact, for at least 30–40% of patients, the disease cannot be controlled by glucocorticoids even when combined with conventional DMARDs, such as methotrexate.
6.9.4.1 IL-1 Inhibition
The use of IL-1 inhibitors for AoSD contributed to the revival of considering this mode of action in rheumatology and now represents the primary choice for treating autoinflammatory diseases in general. However, it took surprisingly long until clinical development moved forward from cohort studies to randomized, placebo-controlled trials. Currently, two IL-1 blocking compounds are the focus of phase 2/phase 3 studies with different designs.
Anakinra, a recombinant IL-1 receptor antagonist, was the first biologic showing beneficial effects in treating the systemic and articular features of AoSD in many case series, uncontrolled trials, and different national surveys. Although the evidence for its efficacy has not been proven by controlled trials, the overall number of more than 250 published cases provides convincing results. In fact, a clear and sustained improvement was described for systemic and also arthritic symptoms in most treated cases [68, 139,140,141,142,143]. Systemic features seem to show a more rapid response, and longer exposure seems usually required for improvement of arthritis. Of major importance for long-term safety were consistent reports of a reduction or even discontinuation of glucocorticoids use as well as NSAIDs. In this context, meta-analyses from eight case series and three national surveys meeting predefined quality standards and including more than 100 anakinra-treated AoSD patients revealed a remission rate of approximately 80% and a reduced use of steroids in approximately 35% of patients [69]. Recently, a large observational retrospective multicenter study from Italy added evidence for a strong impact on disease activity with the Pouchot score as well as clinical and serological features regardless of sex, age, disease pattern, or co-medication [46]. Since September 2017, a randomized, double-blind, placebo-controlled, multicenter, phase 3 study in the United States has recruited patients for investigating two different doses of anakinra (2 mg/kg/day [max 100 mg/day] or 4 mg/kg/day [max 200 mg/day]) to evaluate its efficacy and safety in patients newly diagnosed with SoJIA and AoSD. The primary end point, American College of Rheumatology 30 (ACR30) response, has already been evaluated at week 2; however, the overall treatment period is short, with only 12 weeks of exposure followed by a 4-week safety follow-up [167]. Of note, anakinra is the only IL-1 blocker for which we have substantial long-term results in terms of efficacy and safety in AoSD [144], and it has now been approved in this indication. In contrast to monogenic autoinflammatory diseases, in AoSD, remission can continue in some cases even after treatment is stopped. However, a relatively high withdrawal rate of 40% has been reported owing to loss of response over time and also to frequent injection site reactions to the required daily administrations.
The other approved biologic for AoSD is canakinumab, a fully human antibody against IL-1β [145]. Furthermore, canakinumab is also approved for treating other autoinflammatory diseases including cryopyrin-associated periodic syndromes, familial Mediterranean fever, TNF receptor-associated periodic syndrome, and hyper-IgD syndrome as well as SoJIA. Although the overall experience with canakinumab in AoSD is still limited, the reported response of systemic features and arthritis was rapid and sustained over many months to years in most patients, frequently allowing for tapering of steroids [145,146,147]. Of note, canakinumab was found highly effective for patients with AoSD who were difficult to treat, including those showing failure of NSAIDs, glucocorticoids, and other IL-1 inhibitors. With these promising results, a placebo-controlled, randomized, multicenter, phase 2 study of canakinumab in AoSD is ongoing [168]. The dosage of canakinumab with monthly injections of up to 4 mg/kg body weight (maximum dos of 150 mg) is based on the pharmacokinetic profile in adolescent patients with SoJIA. The primary aim is to investigate the efficacy of canakinumab in patients with AoSD and active joint involvement in terms of the proportion of patients with a clinically significant reduction in disease activity (Disease Activity Score in 28 joints [DAS28] > 1.2) following a treatment period of 12 weeks. The overall placebo-controlled period is 24 weeks, but nonresponders can be unblinded and show treatment rescue at week 12. The core study is followed by an optional long-term extension over 2 years to provide additional safety results in the adult population.
In summary, the published data for anti-IL-1 agents in refractory AoSD clearly show a consistently high rate of full or partial remission with the additional achievement of lowering or stopping glucocorticoid medication [169].
6.9.4.2 IL-6 Inhibition
Inhibition of IL-6 signaling is possible by two different approaches, the direct neutralization of the cytokine or the blockade of the respective receptor. None of these mechanisms has been investigated in AoSD in a controlled setting of clinical trials in detail. For the two different IL-6 receptor antagonists currently available in daily practice for treating rheumatic diseases, only the tocilizumab AoSD case series has been published and reported at conferences [155,156,157,158]. To summarize, the observed anti-inflammatory effects were strong, rapid, and sustained for most of the cases. Usually, the systemic features disappeared entirely, but also the arthritic manifestations improved, and the serologic activity decreased [159, 160]. However, because of limited data, the likelihood of response cannot be estimated as clearly as for IL-1 inhibitors and seems to be between 60% to 80% in terms of full remission. A recently published meta-analysis of ten original studies (147 individuals) on the efficacy of tocilizumab and AoSD revealed overall high partial and complete remission rates of 85–77%, respectively. Tocilizumab prevented new flares, was well tolerated, and could significantly reduce the need for corticosteroids [161, 162].
Whether other IL-6 inhibitors have similar effects remains unknown. As shown for RA treatment, with AoSD, differences could exist with respect to safety, especially in comparison to the direct cytokine inhibitors. Also, the advantages or disadvantages that distinguish IL-6 and IL-1 inhibition are unclear. Although clearly IL-6 as well as IL-1 inhibitors have high response rates and represent alternative approaches in AoSD, we have no way to predict the individual effects and define the ideal treatment algorithm.
6.9.4.3 IL-18 Inhibition
A novel compound is Tadekinig alfa, a recombinant human IL-18 binding protein [163]. This drug was tested in healthy volunteers and patients with psoriasis and RA in phase I studies and demonstrated a good safety and tolerability profile with only mild adverse events at the injection site. Because of the postulated role of IL-18 in the pathogenesis of AoSD, it was a logical step to investigate the effects of Tadekinig alfa in this condition. A first open-label, dose-finding phase 2 study involving multiple centers in Europe was designed to capture safety information as the primary outcome. Two doses (80 and 160 mg) were administered during 12 weeks, and patients were followed up for 4 more weeks. As a secondary outcome measurement, the efficacious dose at 3 weeks was defined as normalization of body temperature and decrease of CRP level by 50% or more of baseline values.
Although limited by the low patient number and short observation period in the study, the safety profile of Tadekinig alfa was overall acceptable, with injection site reactions and infections being the most frequent adverse events. In terms of efficacy, reduced level of CRP as well as other inflammatory markers (including IL-18, ferritin) was detected in some patients. Furthermore, an improvement in skin rash and a slight but significant reduction in the prednisolone doses were reported. In summary, inhibition of IL-18 by Tadekinig alfa could be a promising new approach in ASOD that needs to be investigated in a controlled setting.
6.9.5 Other Potential Agents with Less Validated Indication
Many other treatments have been tried in AoSD, with varying degrees of success.
6.9.5.1 TNF Inhibition
Stimulated by encouraging results from other chronic inflammatory joint diseases, especially RA, TNF inhibitors have been the first biologics used for AoSD [149,150,151,152,153,154]. However, driven by the deceptive impression that AoSD, with a predominant arthritic phenotype, is a subgroup of seronegative RA, the results from uncontrolled trials involving usually small cohorts of patients were inconsistent. Since favorable outcomes have been seen in only a few patients without any specific characteristics, TNF inhibitors can only be considered third-line drugs, preferentially for patients with chronic arthritis.
6.9.5.2 Cyclosporine A
Cyclosporine A has been proposed before the era of biotherapies, in patients with systemic features of AoSD or with RHL, with an interesting efficacy. However, the tolerance of this drug limits its use, but it certainly can be of interest in complex or refractory situations.
6.9.5.3 Intravenous Immunoglobulins
There is henceforth no place for intravenous immunoglobulins in AoSD treatment, owing to the efficacy of the other therapies available.
6.9.6 Treatment Strategies
The strategy we propose for AoSD patients is summarized in Fig. 6.6.
6.9.7 Treatment and Management of Life-Threatening Complications
6.9.7.1 Reactive Hemophagocytic Lymphohistiocytosis (RHL)
Management should include, firstly, symptomatic care in a medicine department or intensive care unit (ICU); secondly, active and rapid search for an infection; and, thirdly, introduction of high-dose steroids. Since RHL has described under IL-1 therapy, it appears prudent to introduce this treatment only after RHL control by a few days of high-dose steroids.
6.9.7.2 Coagulation Disorders
DIC is a medical emergency whose care combines:
-
1.
Supportive measures, which could include platelet transfusion, coagulation factors (or fresh frozen plasma), and fibrinogen (in cryoprecipitate) infusion to control severe bleeding or sometimes heparin if no active bleeding is present [70].
-
2.
Specific anti-inflammatory agents often decided in multidisciplinary rounds, with mainly high-dose corticosteroids, either intravenous (methylprednisolone 15 mg/kg/day) or oral (prednisone 1 mg/kg/day) [59]. To limit steroid exposure, the addition of other immunomodulatory agents, such as IL-1 or IL-6 blockers or cyclosporine, is recommended [68, 69, 71,72,73].
TMA treatment is complex and mandatorily includes [60]:
-
1.
Supportive care in an ICU, with plasma exchange being central [75]
-
2.
High-dose steroids intravenously and then orally
Other immunomodulatory agents proposed for refractory cases include anakinra [74], tocilizumab [76], intravenous immunoglobulins, cyclophosphamide, azathioprine, cyclosporine A, and rituximab [60, 74]. Despite this treatment, mortality associated with TMA remains high, up to 20% [60].
6.9.7.3 Cardiac and Pulmonary Involvements
Their treatment combines supportive care, high-dose steroids, and frequently IL-1 or IL-6 inhibitors.
Regarding PAH, early diagnosis is central for the prognosis of this complication, with mortality remaining as high as 40% [60]. The diagnosis includes [80, 82, 83, 85, 86, 100, 101]:
-
1.
Hospitalization in an ICU and rapid referral to a specific reference center for multidisciplinary round discussions.
-
2.
Vasodilating therapies frequently combining endothelin receptor antagonists (bosentan, ambrisentan), phosphodiesterase-5 inhibitors and other guanylate cyclase stimulators (sildenafil or others), and prostacyclin analogues.
-
3.
High-dose steroids, initially intravenously and then orally.
-
4.
Rapid introduction of biologic immunomodulatory agents targeting IL-1 or IL-6 or other immunosuppressive agents, such as methotrexate, azathioprine, cyclophosphamide, cyclosporine, or rituximab. However, these treatments must be carefully monitored because a few cases of PAH worsening have been described after their introduction.
PAH deserves to be well known by physicians caring for AoSD patients because it was not described in most of the initial AoSD case series for several reasons: now better diagnostic tools for PAH; differential efficacy of large-spectrum immunomodulatory agents, such as steroids, as compared with more targeted therapies, such as IL-1 or IL-6 blockers; and exposure to microorganisms as an additional target whose ecology may have evolved in recent years.
6.9.7.4 Amyloidosis
Even administered late, AoSD therapy, such as steroids and above all IL-1 and IL-6 blockers, may at least in part improve the clinical and biological symptoms.
6.10 Conclusion
In the last 15 years, substantial progress has been made in the diagnosis and prognosis of AoSD. The coming years will likely contribute to a better understanding of the disease and its complications, notably in disease pathogenesis, identification of diagnostic and prognostic biomarkers, and new targets for preventing both life-threatening manifestations and chronicity.
References
Bywaters EG. Still’s disease in the adult. Ann Rheum Dis. 1971;30(2):121–33.
McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3(8):e297.
Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22(5):773–92.
Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune-mediated disease and autoinflammation: innate immune-mediated disease and autoinflammation. J Pathol. 2017;241(2):123–39.
Ohta A, Yamaguchi M, Tsunematsu T, Kasukawa R, Mizushima H, Kashiwagi H, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990;17(8):1058–63.
Ohta A, Yamaguchi M, Kaneoka H, Nagayoshi T, Hiida M. Adult Still’s disease: review of 228 cases from the literature. J Rheumatol. 1987;14(6):1139–46.
Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13(7):708–22.
Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91–9.
Magadur-Joly G, Billaud E, Barrier JH, Pennec YL, Masson C, Renou P, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587–90.
Wakai K, Ohta A, Tamakoshi A, Ohno Y, Kawamura T, Aoki R, et al. Estimated prevalence and incidence of adult Still’s disease: findings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7(4):221–5.
van de Putte LB, Wouters JM. Adult-onset Still’s disease. Baillieres Clin Rheumatol. 1991;5(2):263–75.
Ota T, Higashi S, Suzuki H, Eto S. Increased serum ferritin levels in adult Still’s disease. Lancet. 1987;1(8532):562–3.
Fautrel B, Zing E, Golmard J-L, Le Moel G, Bissery A, Rioux C, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81(3):194–200.
Lebrun D, Mestrallet S, Dehoux M, Golmard JL, Granger B, Georgin-Lavialle S, et al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin Arthritis Rheum. 2018;47(4):578–85.
Masson C, Le Loët X, Lioté F, Renou P, Dubost JJ, Boissier MC, et al. Adult Still’s disease: Part I. Manifestations and complications in sixty-five cases in France. Rev Rhum Engl Ed. 1995;62(11):748–57.
Feist E, Mitrovic S, Fautrel B. Adult-onset Still’s disease: new mechanisms, biomarkers and targets. Nat Rev Rheumatol. 2018;14(10):603–18.
Crispín JC, Martínez-Baños D, Alcocer-Varela J. Adult-onset Still disease as the cause of fever of unknown origin. Medicine (Baltimore). 2005;84(6):331–7.
Zuelgaray E, Battistella M, Sallé de Chou C, Vignon-Pennamen M-D, Rybojad M, Petit A, et al. Increased severity and epidermal alterations in persistent versus evanescent skin lesions in adult onset Still’s disease. J Am Acad Dermatol. 2018;79(5):969–71.
Nguyen KH, Weisman MH. Severe sore throat as a presenting symptom of adult onset Still’s disease: a case series and review of the literature. J Rheumatol. 1997;24(3):592–7.
Lyberatos C. Two more cases of Still’s disease and Kikuchi’s. J Rheumatol. 1990;17(4):568–9.
Samuels AJ, Berney SN, Tourtellotte CD, Artymyshyn R. Coexistence of adult onset Still’s disease and polymyositis with rhabdomyolysis successfully treated with methotrexate and corticosteroids. J Rheumatol. 1989;16(5):685–7.
Moreno-Alvarez MJ, Citera G, Maldonado-Cocco JA, Taratuto AL. Adult Still’s disease and inflammatory myositis. Clin Exp Rheumatol. 1993;11(6):659–61.
Gonzalez-Hernandez T, Martin-Mola E, Fernandez-Zamorano A, Balsa-Criado A, de Miguel-Mendieta E. Serum ferritin can be useful for diagnosis in adult onset Still’s disease. J Rheumatol. 1989;16(3):412–3.
Akritidis N, Giannakakis I, Giouglis T. Ferritin levels and response to treatment in patients with Adult Still’s disease. J Rheumatol. 1996;23(1):201–2.
Schiller D, Mittermayer H. Hyperferritinemia as a marker of Still’s disease. Clin Infect Dis. 1998;26(2):534–5.
Zandman-Goddard G, Shoenfeld Y. Ferritin in autoimmune diseases. Autoimmun Rev. 2007;6(7):457–63.
Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28(2):322–9.
Lee MH, Means RT. Extremely elevated serum ferritin levels in a university hospital: associated diseases and clinical significance. Am J Med. 1995;98(6):566–71.
Van Reeth C, Le Moel G, Lasne Y, Revenant MC, Agneray J, Kahn MF, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still’s disease. J Rheumatol. 1994;21(5):890–5.
Vignes S, Le Moël G, Fautrel B, Wechsler B, Godeau P, Piette JC. Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still’s disease. Ann Rheum Dis. 2000;59(5):347–50.
Lambotte O, Costedoat-Chalumeau N, Amoura Z, Piette JC, Cacoub P. Drug-induced hemophagocytosis. Am J Med. 2002;112(7):592–3.
Lambotte O, Cacoub P, Costedoat N, Le Moel G, Amoura Z, Piette JC. High ferritin and low glycosylated ferritin may also be a marker of excessive macrophage activation. J Rheumatol. 2003;30(5):1027–8.
Fardet L, Coppo P, Kettaneh A, Dehoux M, Cabane J, Lambotte O. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome. Arthritis Rheum. 2008;58(5):1521–7.
McGonagle D, Savic S, McDermott MF. The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Semin Immunopathol. 2007;29(3):303–13.
McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97(1):133–44.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–6.
Maria ATJ, Le Quellec A, Jorgensen C, Touitou I, Rivière S, Guilpain P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: dichotomous view for cytokine and clinical expressions. Autoimmun Rev. 2014;13(11):1149–59.
Jamilloux Y, Gerfaud-Valentin M, Martinon F, Belot A, Henry T, Sève P. Pathogenesis of adult-onset Still’s disease: new insights from the juvenile counterpart. Immunol Res. 2015;61(1–2):53–62.
Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4(1):34–42.
Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–26.
Mitrovic S, Fautrel B. New markers for adult-onset Still’s disease. Joint Bone Spine. 2018;85(3):285–93.
Wittkowski H, Frosch M, Wulffraat N, Goldbach-Mansky R, Kallinich T, Kuemmerle-Deschner J, et al. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008;58(12):3924–31.
Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 2004;50(12):3762–71.
Foell D, Wittkowski H, Hammerschmidt I, Wulffraat N, Schmeling H, Frosch M, et al. Monitoring neutrophil activation in juvenile rheumatoid arthritis by S100A12 serum concentrations. Arthritis Rheum. 2004;50(4):1286–95.
Colafrancesco S, Priori R, Valesini G, Argolini L, Baldissera E, Bartoloni E, et al. Response to interleukin-1 inhibitors in 140 Italian patients with adult-onset Still’s disease: a multicentre retrospective observational study. Front Pharmacol [Internet]. 2017 Jun 13 [cited 2018];8. Available from: http://journal.frontiersin.org/article/10.3389/fphar.2017.00369/full.
Shields AM, Thompson SJ, Panayi GS, Corrigall VM. Pro-resolution immunological networks: binding immunoglobulin protein and other resolution-associated molecular patterns. Rheumatology (Oxford). 2012;51(5):780–8.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101.
Hofmann SR, Kubasch AS, Ioannidis C, Rösen-Wolff A, Girschick HJ, Morbach H, et al. Altered expression of IL-10 family cytokines in monocytes from CRMO patients result in enhanced IL-1β expression and release. Clin Immunol. 2015;161(2):300–7.
Chen D-Y, Chen Y-M, Lin C-C, Hsieh C-W, Wu Y-C, Hung W-T, et al. The potential role of advanced glycation end products (AGEs) and soluble receptors for AGEs (sRAGE) in the pathogenesis of adult-onset still’s disease. BMC Musculoskelet Disord. 2015;16:111.
Grateau G, Hentgen V, Stojanovic KS, Jéru I, Amselem S, Steichen O. How should we approach classification of autoinflammatory diseases? Nat Rev Rheumatol. 2013;9(10):624–9.
Terkeltaub R, Esdaile JM, Décary F, Harth M, Lister J, Lapointe N. HLA-Bw35 and prognosis in adult Still’s disease. Arthritis Rheum. 1981;24(12):1469–72.
Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61(235):1055–65.
Wakil SM, Monies DM, Abouelhoda M, Al-Tassan N, Al-Dusery H, Naim EA, et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2015;67(1):288–95.
Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas J-P, et al. HLA-DRB1∗11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A. 2015;112(52):15970–5.
Aksentijevich I, McDermott MF. Lessons from characterization and treatment of the autoinflammatory syndromes. Curr Opin Rheumatol. 2017;29(2):187–94.
Touitou I. Inheritance of autoinflammatory diseases: shifting paradigms and nomenclature. J Med Genet. 2013;50(6):349–59.
Sarrabay G, Touitou I. Autoinflammation. Management of hereditary recurrent fevers—SHARE experience. Nat Rev Rheumatol. 2015;11(10):567–9.
Mitrovic S, Fautrel B. Complications of adult-onset Still’s disease and their management. Expert Rev Clin Immunol. 2018;14(5):351–65.
Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014;33(3):305–14.
Colina M, Zucchini W, Ciancio G, Orzincolo C, Trotta F, Govoni M. The evolution of adult-onset Still disease: an observational and comparative study in a cohort of 76 Italian patients. Semin Arthritis Rheum. 2011;41(2):279–85.
Mimura T, Shimodaira M, Kibata M, Tsukadaira A, Shirahata K. Adult-onset Still’s disease with disseminated intravascular coagulation and hemophagocytic syndrome: a case report. BMC Res Notes. 2014;7:940.
Shinohara T, Hidaka T, Matsuki Y, Suzuki K, Ohsuzu F. Calcinosis cutis and intestinal pseudoobstruction in a patient with adult onset Still’s disease associated with recurrent relapses of disordered coagulopathy. Intern Med. 1999;38(6):516–20.
Yokoyama M, Suwa A, Shinozawa T, Fujii T, Mimori T, Akizuki M, et al. A case of adult onset Still’s disease complicated with adult respiratory distress syndrome and disseminated intravascular coagulation. Nihon Rinsho Meneki Gakkai Kaishi. 1995;18(2):207–14.
Yamashita S, Furukawa NE, Matsunaga T, Hirakawa Y, Tago M, Yamashita S-I. Extremely high serum ferritin: an instrumental marker of masquerading adult-onset Still’s disease with hemophagocytic syndrome. Am J Case Rep. 2017;18:1296–301.
Colina M, Govoni M, Trotta F. Fatal myocarditis in adult-onset still disease with diffuse intravascular coagulation. Rheumatol Int. 2009;29(11):1355–7.
Namas R, Nannapaneni N, Venkatram M, Altinok G, Levine M, Dhar JP. An unusual case of adult-onset Still’s disease with hemophagocytic syndrome, necrotic leukoencephalopathy and disseminated intravascular coagulation. Case Rep Rheumatol. 2014;2014:128623.
Kötter I, Wacker A, Koch S, Henes J, Richter C, Engel A, et al. Anakinra in patients with treatment-resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007;37(3):189–97.
Hong D, Yang Z, Han S, Liang X, Ma K, Zhang X. Interleukin 1 inhibition with anakinra in adult-onset Still disease: a meta-analysis of its efficacy and safety. Drug Des Devel Ther. 2014;8:2345–57.
Sakata N, Shimizu S, Hirano F, Fushimi K. Epidemiological study of adult-onset Still’s disease using a Japanese administrative database. Rheumatol Int. 2016;36(10):1399–405.
Matsumoto K, Nagashima T, Takatori S, Kawahara Y, Yagi M, Iwamoto M, et al. Glucocorticoid and cyclosporine refractory adult onset Still’s disease successfully treated with tocilizumab. Clin Rheumatol. 2009;28(4):485–7.
Park J-H, Bae JH, Choi Y-S, Lee H-S, Jun J-B, Jung S, et al. Adult-onset Still’s disease with disseminated intravascular coagulation and multiple organ dysfunctions dramatically treated with cyclosporine A. J Korean Med Sci. 2004;19(1):137–41.
Kumar A, Kato H. Macrophage activation syndrome associated with adult-onset Still’s disease successfully treated with anakinra. Case Rep Rheumatol. 2016;2016:3717392.
El Karoui K, Karras A, Lebrun G, Charles P, Arlet JB, Jacquot C, et al. Thrombotic microangiopathy and purtscher-like retinopathy associated with adult-onset Still’s disease: a role for glomerular vascular endothelial growth factor? Arthritis Rheum. 2009;61(11):1609–13.
Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–9.
Masuyama A, Kobayashi H, Kobayashi Y, Yokoe I, Sugimura Y, Maniwa K, et al. A case of adult-onset Still’s disease complicated by thrombotic thrombocytopenic purpura with retinal microangiopathy and rapidly fatal cerebral edema. Mod Rheumatol. 2013;23(2):379–85.
Chen CH, Chen HA, Wang HP, Liao HT, Chou CT, Huang DF. Pulmonary arterial hypertension in autoimmune diseases: an analysis of 19 cases from a medical center in northern Taiwan. J Microbiol Immunol Infect Wei Mian Yu Gan Ran Za Zhi. 2006;39(2):162–8.
Menezes de Siqueira ME, Rodrigues RP, Kormann APM. Adult-onset Still’s disease and pulmonary arterial hypertension. Respir Med CME. 2009;2(2):70–2.
Guilleminault L, Laurent S, Foucher A, Poubeau P, Paganin F. Pulmonary arterial hypertension in adult onset Still’s disease: a case report of a severe complication. BMC Pulm Med. 2016;16(1):72.
Kadavath S, Zapantis E, Zolty R, Efthimiou P. A novel therapeutic approach in pulmonary arterial hypertension as a complication of adult-onset Still’s disease: targeting IL-6. Int J Rheum Dis. 2014;17(3):336–40.
Lowther GH, Chertoff J, Cope J, Alnuaimat H, Ataya A. Pulmonary arterial hypertension and acute respiratory distress syndrome in a patient with adult-onset Still’s disease. Pulm Circ. 2017;7(4):797–802.
Mehta MV, Manson DK, Horn EM, Haythe J. An atypical presentation of adult-onset Still’s disease complicated by pulmonary hypertension and macrophage activation syndrome treated with immunosuppression: a case-based review of the literature. Pulm Circ. 2016;6(1):136–42.
Mubashir E, Ahmed MM, Hayat S, Heldmann M, Berney SM. Pulmonary hypertension in a patient with adult-onset Still’s disease. Clin Rheumatol. 2007;26(8):1359–61.
Padilla-Ibarra J, Sanchez-Ortiz A, Sandoval-Castro C, Ramos-Remus C. Rituximab treatment for pulmonary arterial hypertension in adult-onset Still’s disease. Clin Exp Rheumatol. 2013;31(4):657–8.
Thakare M, Habibi S, Agrawal S, Narsimulu G. Pulmonary arterial hypertension complicating adult-onset Still’s disease. Clin Rheumatol. 2013;32(Suppl 1):S1–2.
Weatherald J, Lategan J, Helmersen D. Pulmonary arterial hypertension secondary to adult-onset Still’s disease: response to cyclosporine and sildenafil over 15 years of follow-up. Respir Med Case Rep. 2016;19:27–30.
Zen A, Yamashita N, Ueda M, Asakawa Y, Yoshikawa Y, Funai T, et al. A case of adult Still’s disease with pulmonary hypertension. Ryumachi. 1990;30(1):45–52.
Campos M, Schiopu E. Pulmonary arterial hypertension in adult-onset Still’s disease: rapid response to anakinra. Case Rep Rheumatol. 2012;2012:537613.
Uppal SS, Al-Mutairi M, Hayat S, Abraham M, Malaviya A. Ten years of clinical experience with adult onset Still’s disease: is the outcome improving? Clin Rheumatol. 2007;26(7):1055–60.
Ben Ghorbel I, Khanfir M, Houman MH. Amyloidosis in adult onset Still’s disease. Rev Med Interne. 2004;25(9):675–7.
Ishii T, Sasaki T, Muryoi T, Murai C, Hatakeyama A, Oosaki H, et al. Systemic amyloidosis in a patient with adult onset Still’s disease. Intern Med. 1993;32(1):50–2.
Oh YB, Bae SC, Jung JH, Kim TH, Jun JB, Jung SS, et al. Secondary renal amyloidosis in adult onset Still’s disease: case report and review of the literature. Korean J Intern Med. 2000;15(2):131–4.
Rivera F, Gil CM, Gil MT, Batlle-Gualda E, Trigueros M, Olivares J. Vascular renal AA amyloidosis in adult Still’s disease. Nephrol Dial Transplant. 1997;12(8):1714–6.
Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–20.
Drepper M, Rubbia-Brandt L, Spahr L. Tocilizumab-induced acute liver injury in adult onset Still’s disease. Case Rep Hepatol. 2013;2013:964828.
Bianchi V, Robles R, Alberio L, Furlan M, Lämmle B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100(2):710–3.
Hirata S, Okamoto H, Ohta S, Kobashigawa T, Uesato M, Kawaguchi Y, et al. Deficient activity of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura in the setting of adult-onset Still’s disease. Rheumatology (Oxford). 2006;45(8):1046–7.
Gopal M, Cohn CD, McEntire MR, Alperin JB. Thrombotic thrombocytopenic purpura and adult onset Still’s disease. Am J Med Sci. 2009;337(5):373–6.
Huertas A, Perros F, Tu L, Cohen-Kaminsky S, Montani D, Dorfmüller P, et al. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: a complex interplay. Circulation. 2014;129(12):1332–40.
McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–94.
Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–75.
Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118–36.
Reginato AJ, Schumacher HR, Baker DG, O’Connor CR, Ferreiros J. Adult onset Still’s disease: experience in 23 patients and literature review with emphasis on organ failure. Semin Arthritis Rheum. 1987;17(1):39–57.
Boki KA, Tsirantonaki MJ, Markakis K, Moutsopoulos HM. Thrombotic thrombocytopenic purpura in adult Still’s disease. J Rheumatol. 1996;23(2):385–7.
Cabane J, Lebas J, Wattiaux MJ, Imbert JC. Pseudo-Still disease and neoplasm. 2 cases. Rev Med Interne. 1988;9(1):81–4.
Sanmarti R, Cañete JD, Salvador G. Palindromic rheumatism and other relapsing arthritis. Best Pract Res Clin Rheumatol. 2004;18(5):647–61.
Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Baltimore). 1994;73(3):133–44.
Masson C, Simon V, Hoppé E, Insalaco P, Cissé I, Audran M. Tumor necrosis factor receptor-associated periodic syndrome (TRAPS): definition, semiology, prognosis, pathogenesis, treatment, and place relative to other periodic joint diseases. Joint Bone Spine. 2004;71(4):284–90.
Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–30.
Ichida H, Kawaguchi Y, Sugiura T, Takagi K, Katsumata Y, Gono T, et al. Clinical manifestations of adult-onset Still’s disease presenting with erosive arthritis: association with low levels of ferritin and interleukin-18. Arthritis Care Res. 2014;66(4):642–6.
Kim H-A, An J-M, Nam J-Y, Jeon J-Y, Suh C-H. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol. 2012;39(7):1399–406.
Kim HA, Han JH, Kim WJ, Noh HJ, An JM, Yim H, et al. TLR4 endogenous ligand S100A8/A9 levels in adult-onset Still’s disease and their association with disease activity and clinical manifestations. Int J Mol Sci. 2016;17(8) https://doi.org/10.3390/ijms17081342.
Scirè CA, Cavagna L, Perotti C, Bruschi E, Caporali R, Montecucco C. Diagnostic value of procalcitonin measurement in febrile patients with systemic autoimmune diseases. Clin Exp Rheumatol. 2006;24(2):123–8.
Kawashima M, Yamamura M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001;44(3):550–60.
Chen D-Y, Lan J-L, Lin F-J, Hsieh T-Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J Rheumatol. 2004;31(11):2189–98.
Choi J-H, Suh C-H, Lee Y-M, Suh Y-J, Lee S-K, Kim S-S, et al. Serum cytokine profiles in patients with adult onset Still’s disease. J Rheumatol. 2003;30(11):2422–7.
Priori R, Colafrancesco S, Alessandri C, Minniti A, Perricone C, Iaiani G, et al. Interleukin 18: a biomarker for differential diagnosis between adult-onset Still’s disease and sepsis. J Rheumatol. 2014;41(6):1118–23.
Rooney T, Murphy E, Benito M, Roux-Lombard P, FitzGerald O, Dayer J-M, et al. Synovial tissue interleukin-18 expression and the response to treatment in patients with inflammatory arthritis. Ann Rheum Dis. 2004;63(11):1393–8.
Conigliaro P, Priori R, Bombardieri M, Alessandri C, Barone F, Pitzalis C, et al. Lymph node IL-18 expression in adult-onset Still’s disease. Ann Rheum Dis. 2009;68(3):442–3.
Priori R, Barone F, Alessandri C, Colafrancesco S, McInnes IB, Pitzalis C, et al. Markedly increased IL-18 liver expression in adult-onset Still’s disease-related hepatitis. Rheumatology (Oxford). 2011;50(4):776–80.
Kawaguchi Y, Terajima H, Harigai M, Hara M, Kamatani N. Interleukin-18 as a novel diagnostic marker and indicator of disease severity in adult-onset Still’s disease. Arthritis Rheum. 2001;44(7):1716–7.
Jung K-H, Kim J-J, Lee J-S, Park W, Kim T-H, Jun J-B, et al. Interleukin-18 as an efficient marker for remission and follow-up in patients with inactive adult-onset Still’s disease. Scand J Rheumatol. 2014;43(2):162–9.
Sugiura T, Kawaguchi Y, Harigai M, Terajima-Ichida H, Kitamura Y, Furuya T, et al. Association between adult-onset Still’s disease and interleukin-18 gene polymorphisms. Genes Immun. 2002;3(7):394–9.
Maruyama J, Inokuma S. Cytokine profiles of macrophage activation syndrome associated with rheumatic diseases. J Rheumatol. 2010;37(5):967–73.
Rau M, Schiller M, Krienke S, Heyder P, Lorenz H, Blank N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J Rheumatol. 2010;37(11):2369–76.
Bae C-B, Suh C-H, An J-M, Jung J-Y, Jeon J-Y, Nam J-Y, et al. Serum S100A12 may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol. 2014;41(12):2403–8.
Han JH, Suh C-H, Jung J-Y, Nam J-Y, Kwon JE, Yim H, et al. Association of CXCL10 and CXCL13 levels with disease activity and cutaneous manifestation in active adult-onset Still’s disease. Arthritis Res Ther. 2015;17:260.
Bleesing J, Prada A, Siegel DM, Villanueva J, Olson J, Ilowite NT, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–71.
Colafrancesco S, Priori R, Alessandri C, Astorri E, Perricone C, Blank M, et al. sCD163 in AOSD: a biomarker for macrophage activation related to hyperferritinemia. Immunol Res. 2014;60(2–3):177–83.
Zou Y-Q, Lu L-J, Li S-J, Zeng T, Wang X-D, Bao C-D, et al. The levels of macrophage migration inhibitory factor as an indicator of disease activity and severity in adult-onset Still’s disease. Clin Biochem. 2008;41(7–8):519–24.
Chen D-Y, Lan J-L, Lin F-J, Hsieh T-Y. Association of intercellular adhesion molecule-1 with clinical manifestations and interleukin-18 in patients with active, untreated adult-onset Still’s disease. Arthritis Rheum. 2005;53(3):320–7.
Efthimiou P, Kontzias A, Ward CM, Ogden NS. Adult-onset Still’s disease: can recent advances in our understanding of its pathogenesis lead to targeted therapy? Nat Clin Pract Rheumatol. 2007;3(6):328–35.
Masson C, Le Loët X, Lioté F, Renou P, Dubost JJ, Boissier MC, et al. Adult Still’s disease. Part II. Management, outcome, and prognostic factors. Rev Rhum Engl Ed. 1995;62(11):758–65.
Medsger TA, Christy WC. Carpal arthritis with ankylosis in late onset Still’s disease. Arthritis Rheum. 1976;19(2):232–42.
Sampalis JS, Esdaile JM, Medsger TA, Partridge AJ, Yeadon C, Senécal JL, et al. A controlled study of the long-term prognosis of adult Still’s disease. Am J Med. 1995;98(4):384–8.
Fujii T, Akizuki M, Kameda H, Matsumura M, Hirakata M, Yoshida T, et al. Methotrexate treatment in patients with adult onset Still’s disease—retrospective study of 13 Japanese cases. Ann Rheum Dis. 1997;56(2):144–8.
Aydintug AO, D’Cruz D, Cervera R, Khamashta MA, Hughes GR. Low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1992;19(3):431–5.
Fautrel B, Borget C, Rozenberg S, Meyer O, Le Loët X, Masson C, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–8.
Fitzgerald AA, LeClercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52(6):1794–803.
Naumann L, Feist E, Natusch A, Langen S, Krause A, Buttgereit F, et al. IL1-receptor antagonist anakinra provides long-lasting efficacy in the treatment of refractory adult-onset Still’s disease. Ann Rheum Dis. 2010;69(2):466–7.
Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis S-NC. Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann Rheum Dis. 2007;66(6):842–3.
Lequerre T, Quartier P, Rosellini D, Alaoui F, De Bandt M, Mejjad O, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2007;67(3):302–8.
Giampietro C, Ridene M, Lequerre T, Costedoat Chalumeau N, Amoura Z, Sellam J, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 2013;65(5):822–6.
Ortiz-Sanjuán F, Blanco R, Riancho-Zarrabeitia L, Castañeda S, Olivé A, Riveros A, et al. Efficacy of anakinra in refractory adult-onset Stillʼs disease: multicenter study of 41 patients and literature review. Medicine (Baltimore). 2015;94(39):e1554.
Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor, in refractory adult-onset Still’s disease. Semin Arthritis Rheum. 2012;42(2):201–5.
Lo Gullo A, Caruso A, Pipitone N, Macchioni P, Pazzola G, Salvarani C. Canakinumab in a case of adult onset Still’s disease: efficacy only on systemic manifestations. Joint Bone Spine. 2014;81(4):376–7.
Barsotti S, Neri R, Iacopetti V, d’Ascanio A, Talarico R, Tripoli A, et al. Successful treatment of refractory adult-onset still disease with canakinumab: a case report. J Clin Rheumatol. 2014;20(2):121.
Petryna O, Cush JJ, Efthimiou P. IL-1 Trap rilonacept in refractory adult onset Still’s disease. Ann Rheum Dis. 2012;71(12):2056–8.
Husni ME, Maier AL, Mease PJ, Overman SS, Fraser P, Gravallese EM, et al. Etanercept in the treatment of adult patients with Still’s disease. Arthritis Rheum. 2002;46(5):1171–6.
Kiyonaga Y, Maeshima K, Imada C, Haranaka M, Ishii K, Shibata H. Steroid-sparing effects of etanercept in a patient with steroid-dependent adult-onset Still’s disease. Intern Med. 2014;53(11):1209–13.
Kaneko K, Kaburaki M, Muraoka S, Tanaka N, Yamamoto T, Kusunoki Y, et al. Exacerbation of adult-onset Still’s disease, possibly related to elevation of serum tumor necrosis factor-alpha after etanercept administration: exacerbation of AOSD after etanercept. Int J Rheum Dis. 2010;13(4):e67–9.
Kraetsch HG, Antoni C, Kalden JR, Manger B. Successful treatment of a small cohort of patients with adult onset of Still’s disease with infliximab: first experiences. Ann Rheum Dis. 2001;60(Suppl 3):iii55–7.
Cavagna L, Caporali R, Epis O, Bobbio-Pallavicini F, Montecucco C. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin Exp Rheumatol. 2001;19(3):329–32.
Fautrel B, Sibilia J, Mariette X, Combe B, Club Rhumatismes et Inflammation. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: an observational study of 20 cases. Ann Rheum Dis. 2005;64(2):262–6.
Puéchal X, DeBandt M, Berthelot J-M, Breban M, Dubost J-J, Fain O, et al. Tocilizumab in refractory adult Still’s disease: refractory ASD and tocilizumab. Arthritis Care Res. 2011;63(1):155–9.
Elkayam O, Jiries N, Dranitzki Z, Kivity S, Lidar M, Levy O, et al. Tocilizumab in adult-onset Still’s disease: the Israeli experience. J Rheumatol. 2014;41(2):244–7.
Cipriani P, Ruscitti P, Carubbi F, Pantano I, Liakouli V, Berardicurti O, et al. Tocilizumab for the treatment of adult-onset Still’s disease: results from a case series. Clin Rheumatol. 2014;33(1):49–55.
Thonhofer R, Hiller M, Just H, Trummer M, Siegel C, Dejaco C. Treatment of refractory adult-onset Still’s disease with tocilizumab: report of two cases and review of the literature. Rheumatol Int. 2011;31(12):1653–6.
Li T, Gu L, Wang X, Guo L, Shi H, Yang C, et al. A pilot study on tocilizumab for treating refractory adult-onset Still’s disease. Sci Rep [Internet]. 2017. [cited 2018 Feb 4];7(1). Available from: http://www.nature.com/articles/s41598-017-13639-y.
Song ST, Kim JJ, Lee S, Kim H-A, Lee EY, Shin KC, et al. Efficacy of tocilizumab therapy in Korean patients with adult-onset Still’s disease: a multicentre retrospective study of 22 cases. Clin Exp Rheumatol. 2016;34(6. Suppl 102):S64–71.
Ma Y, Wu M, Zhang X, Xia Q, Yang J, Xu S, et al. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: a meta-analysis. Mod Rheumatol. 2018;28(5):849–57.
Nishina N, Kaneko Y, Kameda H, Takeuchi T. The effect of tocilizumab on preventing relapses in adult-onset Still’s disease: a retrospective, single-center study. Mod Rheumatol. 2015;25(3):401–4.
Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77(6):840–7.
Belfeki N, Smiti Khanfir M, Said F, Hamzaoui A, Ben Salem T, Ben Ghorbel I, et al. Successful treatment of refractory adult onset Still’s disease with rituximab. Reumatismo. 2016;68(3):159–62.
Ostrowski RA, Tehrani R, Kadanoff R. Refractory adult-onset still disease successfully treated with abatacept. J Clin Rheumatol. 2011;17(6):315–7.
Quartuccio L, Maset M, De Vita S. Efficacy of abatacept in a refractory case of adult-onset Still’s disease. Clin Exp Rheumatol. 2010;28(2):265–7.
US National Library of Medicine. ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/study/NCT03265132 (2017). [Internet]. Available from: https://clinicaltrials.gov/show/NCT03265132.
US National Library of Medicine. ClinicalTrialsgov. 2017. https://clinicaltrials.gov/ct2/show/NCT02204293.
Junge G, Mason J, Feist E. Adult onset Still’s disease—the evidence that anti-interleukin-1 treatment is effective and well-tolerated (a comprehensive literature review). Semin Arthritis Rheum. 2017;47(2):295–302.
Acknowledgments
Conflicts of Interest:
S.M. has received consultant fees from BMS and Pfizer.
E.F. has received consultant fees from AbbVie, Biogen, BMS, Celgene, Janssen, Lilly, Medac, MSD, Nordic Pharma, Novartis, Pfizer, Roche, Sanofi-Aventis, Swedish Orphan BioVitrum, and UCB, as well as a research grant from BMS, Lilly, Novartis, Pfizer, and Roche.
B.F. has received consultant fees from AbbVie, Biogen, BMS, Celgene, Janssen, Lilly, Medac, MSD, Nordic Pharma, Novartis, Pfizer, Roche, Sanofi-Aventis, Swedish Orphan BIovitrum, and UCB, as well as research grants from AbbVie, Lilly, MSD, and Pfizer.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mitrovic, S., Feist, E., Fautrel, B. (2020). Adult-Onset Still’s Disease. In: Cimaz, R. (eds) Periodic and Non-Periodic Fevers. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-030-19055-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-19055-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-19054-5
Online ISBN: 978-3-030-19055-2
eBook Packages: MedicineMedicine (R0)