
Abstract
The genomes of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens, and Ophiostoma ips are presented in this genome announcement. Three of these genomes are from plant pathogens and otherwise economically important fungal species. Fusarium pininemorale and H. decipiens are not known to cause significant disease but are closely related to species of economic importance. The genome sizes range from 25.99 Mb in the case of O. ips to 4.82 Mb for H. lignivorus. These genomes include the first reports of a genome from the genus Hawksworthiomyces. The availability of these genome data will allow the resolution of longstanding questions regarding the taxonomy of these species. In addition these genome sequences through comparative studies with closely related organisms will increase our understanding of how these species or close relatives cause disease.
Similar content being viewed by others


IMA Genome-F 8A
Draft genome of an African isolate of the maize fungal pathogen, Cercospora zeina
Maize (Zea mays) is one of the most important staple food crops, especially in Sub-Saharan Africa. In the last three decades, Gray Leaf Spot (GLS) has become a widespread foliar disease of maize globally (Latterell & Rossi 1983, Ward et al. 1999, Meisel et al. 2009, Berger et al. 2014). Since GLS leads to substantial yield losses, it poses a threat to food security, especially in Africa (Meisel et al. 2009). Two causal agents of GLS have been identified, with Cercospora zeae-maydis the predominant pathogen in the USA (Wang et al. 1998), and to date, only Cercospora zeina reported from Africa (Crous et al. 2006, Meisel et al. 2009). These species belong to the class Dothidiomycete, that contains several important plant pathogens (Ohm et al. 2012). However, little is known about pathogenicity mechanisms of C. zeina.
Sequenced Strain
Zambia: Central region (Mkushi): isol. ex Zea mays (maize), March 2007, F.J. Kloppers & B. Meisel (CMW25467, MUCL 51677, CBS142763, PREM 61898 — dried culture).
Nucleotide Sequence Accession Number
The draft genome has been deposited at DDBJ/ENA/GenBank and is available under the accession number MVDW00000000; Biosample SAMN06067857; Bioproject PRJNA355276. This paper describes the first version of the genome. RNA sequencing data have been deposited in the NCBI Gene Expression Omnibus (Accession number GSE90705).
Materials and Methods
Conidia of Cercospora zeina were grown on V8 agar (20 % (v/v) Campbells V8 juice, 2 % (w/v) Bacterial Agar, 0.349 % (w/v) CaCO3) at ambient room temperature in constant darkness to promote conidiation. Conidia were collected and 1×105 conidia/ml cultured in Potato Dextrose Broth (PDB) at 25 °C with gentle shaking. Prior to cultures reaching the melanized stage DNA was isolated as described previously (Ma et al. 2010).
For RNA isolations C. zeina was cultured under seven different in vitro growth conditions. For solid media, conidia were transferred from V8 agar onto sterile cellophane sheets overlain onto the particular media by subculturing. For liquid media, conidia were washed from the V8 agar using the relevant media and a sterile L-spreader, and transferred to a flask containing the growth media. Media used included Complete media (1 % (w/v) glucose, 0.1 % (w/v) yeast extract, 0.1 % (w/v) casein hydrolysate, 0.1 % (w/v) Ca(NO3)2.4H2O, 1 % (v/v) mineral solution [2 % (w/v) KH2PO4, 2.5 % (w/v) MgSO4.7H2O, 1.5 % (w/v) NaCl]); 0.2x PDA (0.3 % (w/v) Potato Dextrose Agar (PDA), 1.2 % (w/v) Bacterial Agar), 0.2x PDA supplemented with 10mM NH4H2PO4; Cornmeal agar (1.7 % (w/v)); PDA pH8 (3.9 % PDA, pH adjusted with 0.29 % (w/v) Na2CO3 and 0.76 % NaHCO3); PDB pH3 (2.4 % (w/v) PDB, pH adjusted with 1.67 % (w/v) citric acid and 0.58 % (w/v) Na2HPO4); and Yeast Extract Peptone Dextrose (YPD, 0.05 % (w/v) peptone, 0.05 % (w/v) yeast extract, 0.5 % (w/v) glucose, 1.8 % (w/v) NaCl). With the exception of V8 agar, the cultures were kept in constant light at 25 °C for 7 d.
RNA was isolated using the Qiagen QIAzol Lysis Reagent and on-column DNase treatment and RNA purification was performed with the Qiagen RNeasy Mini kit, all according to the manufacturer’s specifications. Illumina HiSeq 2000 100 bp read-length sequencing was performed on three DNA libraries (paired-end (PE), 3 kb and 8 kb mate-pair). Sequence reads were quality filtered and trimmed using Trimmomatic v. 0.30 (Bolger et al. 2014) using the default parameters. Due to nucleotide content inconsistencies in the PE and 3 kb libraries the 8 kb mate-pair library reads were assembled as single-end reads using Velvet (Zerbino & Birney 2008), with optimal k-mer size of 57 nt determined with VelvetOptimizer. Contigs were scaffolded using SSPACE v. 2.0 (Boetzer et al. 2011) and scaffold gaps filled with Gapfiller v. 1.11 (Boetzer & Pirovano 2012) using all the sequenced libraries and allowing a 25 % error in library insert sizes. Completeness of the assembly was evaluated using BUSCO (Simao et al. 2015).
Transcriptome sequencing was performed at BGI (HK) using HiSeq 2000 100bp PE sequencing. RNAseq reads were mapped to the C. zeina reference genome using TopHat2 (Kim et al. 2013). Manual gene finding of 150 C. zeina genes using Genomeview (Abeel et al. 2012) was guided by BLAST (Altschul et al. 1990) alignments of C. zeae-maydis (JGI) genes to the C. zeina reference genome together with C. zeina transcriptome mapping information for gene and splice site identification. Automated gene prediction was performed using the MAKER (Cantarel et al. 2008) pipeline incorporating the SNAP (Korf 2004) and AUGUSTUS (Stanke et al. 2006) ab initio gene predictors. The SNAP training data were the 150 manually curated genes, while the AUGUSTUS training data comprised of high-confidence predicted genes from MAKER using only SNAP predictions (AED score <0.2).
To verify the species identity of the sequenced strain, the Translation Elongation Factor 1-alpha and ITS sequences for selected Cercospora species (Table 1) were concatenated and aligned with ClustalW (Thompson et al. 1994). The relevant sequences were extracted from the genome assembly using blastn from the C. zeina strain CMW 25467 data, and are listed under the genome assembly accession. The analysis workflow was similar to Meisel et al. (2009). The species relationships were inferred using the Maximum Parsimony method in MEGA7 (Kumar et al. 2016), with confidence at nodes gained using bootstrap analysis (Felsenstein 1985). Branches corresponding to partitions reproduced in less than 50 % bootstrap replicates were collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches (Felsenstein 1985). The MP tree was obtained using the Tree-Bisection-Regrafting algorithm (Nei & Kumar 2000) with search level 1 in which the initial trees were obtained by the random addition of sequences (10 replicates). All positions containing gaps and missing data were eliminated. Two strains of Mycosphaerella thailandica (CBS 116367 and CPC 10548) were used to root the cladogram (Fig. 1).

Cladogram to classify the sequenced Cercospora zeina genome with reference to related Cercospora species. Maximum parsimony analysis based on translation elongation factor 1-alpha and ITS sequences was performed, with percentage bootstrap (1000) values shown. The cladogram was rooted using Mycosphaerella thailandica.
Results and Discussion
Assembly of the sequenced reads yielded a draft genome 37 Mb in size in 10 027 scaffolds >200 bp with N50 of 161 kb. The average scaffold size was 4 059 bp with the largest contig of 938 kb. BUSCO evaluation using the Ascomycota dataset yielded a completeness report of C: 95.4 %: (95.4 % Complete and single-copy BUSCOs, D: 0.0 % duplicated BUSCOs, 2.1 % fragmented BUSCOs, M: 2.5 % missing BUSCOs, total 1315 genes evaluated).
Following manual gene curation, 10 193 protein-coding gene-models were predicted. The genome size and number of genes are similar to other genomes in the order Capnodiales (Stanke et al. 2006).
IMA Genome-F 8B
Draft genome sequence of Fusarium pininemorale
Introduction
The Fusarium fujikuroi species complex (FFSC) represents an assemblage of diverse fungal species. The so-called “American” clade of the FFSC contains known emerging pathogens of many cultivated crops and trees including pine (F. circinatum; Hepting & Roth 1946), maize (F. temperatum; Desjardins et al. 2000), and mango (F. parvisorum; Liew et al. 2016). Fusarium pininemorale, a recently recognized member of this clade, was isolated from diseased pine trees in plantations of Colombia (Herron et al. 2015). Even though this species shares numerous morphological and biological traits with other FFSC pine pathogens, F. pininemorale itself was not found to cause significant disease symptoms on pine as is the case for F. circinatum (Herron et al. 2015).
Overall, little is known regarding the biology and genetics of this species, even less so for the genetic and determinants of host range in the broader “American” clade. The exact genetic basis and genomic processes underlying pathogenicity also remains elusive. The aim of this study was to determine the full genome sequence for F. pininemorale, which will allow further studies to investigate genomic aspects of not only pathogenicity but also of its biology and evolution.
Sequenced Strain
Colombia: Angela Maria (Santa Rosa), Risaralda 75°360.2100′ W 4°490.1800′ N, Isolated from Pinus tecunumanii, 2012, (CBS 137240 = CMW 25243; FCC 5383 — cultures; Herron et al. 2015).
Nucleotide Accession Number
The Fusarium pininemorale whole genomic sequence data has been deposited at DDBJ/EMBL/GenBank under the accession NFZR00000000. The version described in this paper is version NFZR00000000.
Methods
The Fusarium pininemorale isolate was grown on ½ PDA for 7 d and DNA was extracted as described previously (Möller et al. 1992). A pair-end library (350 bp average insert size) and a mate-pair library (5 kb average insert size) were prepared and sequenced using Illumina HiSeq platforms at Macrogen (Seoul, Korea). All sequences had an average read length of 58 bp. Poor quality reads, terminal nucleotides, as well as duplicate reads were removed using CLC Genomics Workbench v. 8.0.1 (CLCBio, Aarhus). De novo genome assembly was performed using ABySS v. 1.3.7 (Simpson et al. 2009), further scaffolding was performed using SSPACE v. 2. 0 (Boetzer et al. 2011) and gapped genomic regions were closed using GapFiller v. 1.11 (Boetzer & Pirovano 2012). Genome completeness was assessed using the software BUSCO (Benchmarking Universal Single-Copy Orthologs) v2 with the Sordariomycete dataset (Simão et al. 2015). Chromosome-sized scaffolds were compared using LASTZ alignments (Harris 2007) against genomes of F. circinatum (Wingfield et al. 2012) and F. temperatum (Wingfield et al. 2015a). Lastly, gene prediction was performed using AUGUSTUS 2.5.5 (Hoff & Stanke 2013) based on gene models for F. graminearum (http://bioinf.uni-greifswald.de/augustus), together with mRNA sequence data from the F. circinatum genome (Wingfield et al. 2012).
Results and Discussion
The draft nuclear genome of Fusarium pininemorale had an estimated genome size of 47 778 776 bp. The N50 value was 1 358 616 bp and the GC content was 46.0 %. The assembly consisted of 200 contigs which ranged between 200 bp to 6 144 005 bp in size. The average scaffold size was determined to be 310 252 bp in length of which 13 contigs were larger than 1 000 000 bp. The assembled genome was predicted to encode for 14 640 open reading frames (ORFs) with an average gene length of 1472 bp, and an overall gene density of 306 ORFs/MB was observed. BUSCO suggested that the assembly was 99 % complete (i.e., complete BUSCOs = 99 %; complete and single-copy BUSCOs = 98.2 %; complete and duplicated BUSCOs = 0.8 %; fragmented BUSCOs = 0.6 %; missing BUSCOs = 0.4 %; number of BUSCOs searched = 3725). The taxonomic identity of the genome was confirmed using a phylogenetic analysis using authenticated sequences (Fig. 2).

Maximum likelihood (ML) tree based on partial gene sequences of β-tubulin and translation elongation factor 1-α. Sequence alignments were assembled with MAFFT version 7 (Katoh & Standley 2013). The program jModelTest v 2.1.7 (Guindon & Gascuel 2003, Darribo et al. 2012) was used to determine the best-fit substitution model (GTR+I+G substitution model) with gamma correction (Tavaré 1986). The ML phylogenetic analysis was performed using PhyML v 3.1 (Guindon & Gascuel 2010). Values at branch nodes are the bootstrapping confidence values with those ≥ 85 % shown. Indicated in bold are sequences from the Fusarium genomes of F. pininemorale and those of F. nygamai, F. temperatum and F. circinatum strain GL1327 which have been previously published in IMA fungus (Wingfield et al. 2012, 2015a, 2015b).
The GC content and number of identified ORFs is comparable to that of other FFSC genomes (Ma et al. 2010, Wingfield et al. 2012, Wiemann et al. 2013, Wingfield et al. 2015a, 2015b, Niehaus et al. 2016). However, the genome assembly of F. pininemorale is notably larger than those of other species in the “American” clade for which genome sequences are available. In comparison, the genome sizes of F. circinatum and F. temperatum is 4 534 176 bp and 2 319 967 bp smaller than that of F. pininemorale, respectively. Nevertheless, sequence comparisons based on chromosome-sized scaffolds suggests that F. pininemorale harbours 12 chromosomes similar to that of species in the FFSC. The putative 12th chromosome of F. pininemorale is 968 722 bp size and spans two scaffolds in the current assembly. Also, the reciprocal chromosome translocation (involving parts of chromosomes 8 and 11) confirmed previously in F. circinatum and F. temperatum (De Vos et al. 2014, Wingfield et al. 2015a) is also present in F. pininemorale. Clearly, the addition of the F. pininemorale genome assembly would facilitate detailed studies of genome evolution, host adaption and pathogenicity in the “American” clade as well as the broader FFSC.
IMA Genome-F 8C
Draft genome sequence of Hawksworthiomyces lignivorus
Introduction
Hawksworthiomyces lignivorus (Ascomycota: Ophiostomatales) was first described from decaying Eucalyptus utility poles in South Africa (De Meyer et al. 2008). It was initially described in the genus Sporothrix which, under the dual nomenclature system, accommodated mycelial asexual morphs of Ophiostomatales (De Beer & Wingfield 2013, De Hoog 1993). However, DNA sequences of multiple genes and more inclusive phylogenies showed that that this species was distinct from all other species residing in Sporothrix as redefined by De Beer et al. (2016a), and from other genera in Ophiostomatales (De Beer & Wingfield 2013). De Beer et al. (2016b) therefore described the new genus Hawksworthiomyces, to accommodate H. lignivorus together with four other newly described species, H. crousii, H. hibbettii, H. sequentia, and H. taylorii.
Species of Hawksworthiomyces differ from all other ophiostomatoid fungi in their biology and ecology. Most species residing in the numerous genera of Ophiostomatales are associates of bark or ambrosia beetles that inhabit the cambium and sapwood of trees (De Beer & Wingfield 2013). Many species of Sporothrix are exceptions to this norm and are found in soil and Protea infructescences (De Beer et al. 2016a, Roets et al. 2013). All species of Hawksworthiomyces have either been isolated from decaying wood or environments associated with degrading plant materials (De Beer et al. 2016b). An inoculation study conducted by De Beer et al. (2006) also suggested that H. lignivorus is capable of degrading lignocellulose components of wood. This is different to other species in Ophiostomatales that specifically degrade resinous compounds in wood (Farrell et al. 1993) and that do not display any marked lignocellulose degradation (Seifert 1993).
In this study, we generated the draft genome sequence for H. lignivorus, which will provide basal data required to explore its unique biology and ecology. As the type species of Hawksworthiomyces, the genome sequence of H. lignivorus will be useful for future phylogenomic studies aimed at a better understanding the evolutionary history of this genus and other genera in Ophiostomatales.
Sequenced Strain
South Africa: Western Cape: Stellenbosch: isolated from Eucalyptus pole at soil level, Oct. 2003, E.M. de Meyer (CMW 18600 = CBS 119148 = MUCL 55926 — living culture, PREM 59284 — dried culture).
Nucleotide Sequence Accession Number
The genomic sequence of Hawsworthiomyces lignivorus (CMW 18600, CBS 119148) has been deposited at DDBJ/EMBL/GenBank under accession no. NTMA00000000. The version described in this paper is version NTMA01000000.
Materials and Methods
The ex-holotype culture of Hawsworthiomyces lignivorus (CMW 18600 = CBS 119148) was obtained from the culture collection of the Forestry and Agricultural Biotechnology Institute, University of Pretoria (CMW). Genomic DNA was extracted using the method described by Duong et al. (2013). Two pair-end libraries of approximately 350 bp and 530 bp were prepared and sequenced using the Illumina HiSeq 2000 platform with 100 bp read length. Reads obtained were subjected to quality and adapters trimming using Trimmomatic v. 0.36 (Bolger et al. 2014). De-novo genome assembly was performed using SPAdes v. 3.9.0 (Bankevich et al. 2012). The scaffolds obtained from SPAdes were subjected to further scaffolding with SSPACE-Standard v. 3.0 (Boetzer et al. 2011). Assembly gaps were filled with GapFiller v. 1.10 (Boetzer & Pirovano 2012). The quality and completeness of the assembly was validated with Benchmarking Universal Single Copy Orthologs (BUSCO v. 2.0.1) program using the Soradiomyceta odb9 dataset (Simão et al. 2015). The number of protein coding genes was determined using AUGUSTUS v3.2.2 (Stanke et al. 2006).
Results and Discussion
Sequencing of the Hawksworthiomyces lignivorus DNA libraries yielded 14 203733 paired-end reads with average read length of around 100 bp. Trimming recovered 12 704 558 pair-end reads and 1 362 623 single reads. De-novo genome assembly with SPAdes resulted in an assembly of 43.81 Mb in size, distributed in 280 scaffolds larger than 500 bp. The number of scaffolds was further reduced to 214 after scaffolding with SSPACE and filling gaps with GapFiller. The current genome assembly of H. lignivorus has a total sequence length of 43 822 585 bases, with an N50 value of 383 563 and an average GC content of 51.27 %. Hawsworthiomyces lignivorus had the largest genome size when compared to all species of Ophiostomatales for which whole genome data are available; the smallest genome reported was that of Ceratocystiopsis minuta (21.3 Mb) (Wingfield et al. 2016a), and the second largest to H. lignivorus was that of of Sporothrix pallida (37.8 Mb) (D’Alessandro et al. 2016).
The assembly had a BUSCO completeness score of 95.7 %. Out of the 3725 BUSCO groups searched, 3556 were complete single-copy BUSCOs, nine were complete duplicated BUSCOs, 64 were fragmented BUSCOs, and 96 were missing BUSCOs. AUGUSTUS predicted a total of 11 216 protein-coding genes encoded by H. lignivorus genome. The taxonomic identity of the genome was confirmed using a phylogenetic analysis using authenticated sequences (Fig. 3).

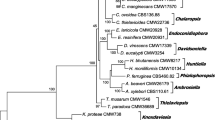
Identity verification of Ophiostomataceae isolates sequenced in this study and in all previous IMA Genome Announcements (IMA Genome-F: 3–7; Van der Nest et al. 2014a, Wingfield et al. 2015a, 2016a, 2015b, 2016b). Gene regions (LSU, βT) used for verification were extracted from assembled genomes. Other reference isolates and their corresponding sequences were obtained from published papers (De Beer et al. 2016, Linnakoski et al. 2012, Yin et al. 2015, Zipfel et al. 2006). The phylogeny was constructed using RAxML with the GTRGAMMA substitution model. Bootstrap values greater than 75 are indicated at the nodes.
IMA Genome-F 8D
Draft genome assembly for Huntiella decipiens
Introduction
The family Ceratocystidaceae as defined by De Beer et al. (2014) includes economically important plant pathogens, as well as agents of blue stain in timber, many of which result in substantial economic losses (Roux et al. 2000, Baker et al. 2003, Barnes et al. 2003, Van Wyk et al. 2007, Heath et al. 2009). De Beer et al. (2014), Fig. 4, revised this fungal family based on morphological, phylogenetic and ecological evidence and it now includes numerous clearly circumscribed genera such as Ceratocystis, Endoconidiophora, and Huntiella.

Identity verification of Ceratocystidaceae isolates sequenced in this study, and in all previous IMA Genome Announcements (IMA Genome-F: 1–7; Van der Nest et al. 2014a, 2014b, Wilken et al. 2013, Wingfield et al. 2015a, 2016a, 2015b, 2016b). Gene regions (60S, LSU, MCM7) used for verification were extracted from assembled genomes. Other reference isolates and their corresponding sequences were obtained from De Beer et al. (2014). The phylogeny was constructed using RAxML with the GTRGAMMA substitution model. Bootstrap values greater than 75 are indicated at the nodes.
Huntiella species previously formed part of the Ceratocystis moniliformis complex (De Beer et al. 2014). Species of Huntiella are generally weak pathogens or saprobes, although some cause sapstain which reduces the value of timber (Van Wyk et al. 2006, Kirisits et al. 2013). These fungi are associated with insects, particularly sapfeeding beetles (Nitidulidae) that are thought to primarily aid in their spread and distribution (Kirisits 2004). An unusual example of a Huntiella species is H. bhutanensis that lives in association with the bark beetle Ips schmutzenhoferi (Scolytidae) (Van Wyk et al. 2004, Kirisitset et al. 2013). In general, however, little is known regarding the biology or ecology of Huntiella species. For example, H. decipiens, that forms the basis of the present study, is known only from one region in the Limpopo Province of South Africa, where it was isolated from wounds on plantation-grown Eucalyptus species and from a Staphilinid (Staphylinidae) beetle found on freshly cut E. saligna stumps (Nkuekam et al. 2012).
The aim of this study was to produce a good quality draft genome assembly for H. decipiens. The genomes of several members of Ceratocystidaceae are already available in the public domain (Belbahri 2015, Van der Nest et al. 2014a, 2014b, Wilken et al. 2013, Wingfield et al. 2015a, 2015b, 2016) and the genome sequence for H. decipiens will provide valuable opportunities for comparative genomic studies on this important group of fungi.
Sequenced Strain
South Africa: Soutpansberg, isol. Staphilinid spp. infesting Eucalyptis saligna, Dec. 2008, K. Nkuekam (CMW 30855, CBS 129736 — cultures; PREM 60560 — dried culture).
Nucleotide Sequence Accession Number
The Huntiella decipiens isolate CMW 30855 Whole Genome Shotgun project has been deposited in GenBank under accession no. NETU00000000.
Materials and Methods
Genomic DNA was isolated from H. decipiens isolate CMW 30855 (Barnes et al. 2001) and sequenced using the Genomics Analyzer IIx, Illumina platform from the UC Davis Genome Centre (University of California, Davis, A). For this purpose, paired-end libraries of 350- p and 600 bp insert sizes were prepared and sequenced following the Illumina protocol. The CLC Genomics Workbench v. 8.0.1 (CLCBio, Aarhus) was employed to quality trim reads and denovo assemble a draft genome sequence using the default parameters. Thereafter, the assemblies were scaffolded using SSPACE v. 2.0 (Boetzer et al. 2011). GapFiller v. 2.2.1 (Boetzer & Pirovano 2012) was used to fill the gaps created during the scaffolding. The number of putative open reading frames (ORFs) was predicted with the web-based de novo gene prediction software AUGUSTUS using the Fusarium graminearum gene models (Stanke et al. 2008). The “create detailed mapping report” command of the CLC Genomics Workbench was used to produce statistics for the draft sequence. The Benchmarking Universal Single-Copy Orthologs (BUSCO v. 1.22) tool was used to assess the genome completeness (Simão et al. 2015) using the fungal data set.
Results and Discussion
The estimated size of the assembled Huntiella decipiens draft genome was 26.7 Mb, with 638 scaffolds larger than 500 bases. AUGUSTUS analysis predicted 7254 ORFs, which corresponds to an average gene density of 271.7 ORFs/Mb. The assembly contained 1 403 Complete Single-Copy BUSCOs, 90 Complete Duplicated BUSCOs, 11 Fragmented BUSCOs and 24 missing BUSCOs. The taxonomic identity of the genome was confirmed using a phylogenetic analysis using authenticated sequences (Fig. 4).
Relative to other species in Ceratocystidaceae, H. decipiens has a similar size genome than those of H. bhutanensis (26.7 Mb, 7261 ORFs) (Wingfield et al. 2016) and H. moniliformis (25.4 Mb, 6832 ORFs) (Van der Nest et al. 2014b). Compared to other species in the family, however, these three Huntiella genomes appeared to be smaller, encoding fewer genes. For example, the H. decipiens genome is smaller than those of H. omanensis (31.5 Mb, 8395 ORFs), Ceratocystis manginecans (31.7 Mb, 7494 ORFs), C. fimbriata (29.4 Mb, 7 266 ORFs), E. laricicola (33.3 Mb, 6 897 ORFs), and D. virescens (33.7 Mb, 6953 ORFs) (Van der Nest et al. 2014a, 2014b, Wilken et al. 2013, Wingfield et al. 2015, 2016). Whether these differences in genome size are linked to the different lifestyles of these fungi requires further research, which will be the subject of future studies.
IMA Genome-F 8E
Draft genome sequence of Ophiostoma ips
Introduction
Ophiostoma ips is an ascomycete fungus of the O. ips species complex (Ophiostomatales, Ascomycota) (De Beer & Wingfield 2013). The fungus was first described as the causal agent of blue stain on Pinus lumber in the USA (Rumbold 1931). Ophiostoma ips is commonly associated with coniferinfesting bark beetles in the genera Ips, Orthotomicus, and Hylurgus, which are native to the Northern Hemisphere. Outside of its native range, O. ips has been reported in various countries of the Southern Hemisphere including Australia, Chile, New Zealand, and South Africa, where it has been accidentally introduced with its bark beetle vectors (Wingfield & Marasas 1980, Zhou et al. 2001, 2004). A population genetic study using microsatellite markers (Zhou et al. 2007) revealed a high level of admixture between O. ips populations, suggesting frequent movement of this species between countries and continents. Although there have been some studies suggesting weak pathogenicity on conifers (Lieutier et al. 1989, Zhou et al. 2002), like most other Ophiostoma spp. and their relatives (Six & Wingfield 2011), O. ips is generally considered not to be a primary pathogen and its relevance is usually as a consequence of the reduced value of the wood due to sap stain (Seifert 1993).
We generated the genome sequence for O. ips, which adds to a growing number of genomes available from species of Ophiostomatales. Together, these genomes will serve as a valuable resource, enabling future comparative genomics studies seeking to gain insight into the biology, ecology and evolution of species in Ophiostomatales.
Sequenced Strain
USA: Louisiana: isol. Pinus taeda, 2004, X. Zhou (CMW 19371 = CBS 138721 — culture).
Nucleotide Sequence Accession Number
The genomic sequence of Ophiostoma ips (CMW 19371 = CBS 138721) has been deposited at DDBJ/EMBL/GenBank under the accession NTMB00000000. The version described in this paper is version NTMB01000000.
Methods
An isolate of Ophiostoma ips (CMW 19371 = CBS 138721) was obtained from the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute, University of Pretoria. DNA was extracted from a single conidium culture following a method described previously (Duong et al. 2013). Two pair-end libraries (350 bp and 550 bp medium insert sizes) were prepared and sequenced using the Illumina Hiseq 2500 platform. The program Trimmomatic v. 0.36 (Bolger et al. 2014) was used for quality and adapters trimming of pair-end reads. The genome was assembled from trimmed reads using Spades v. 3.10 (Bankevich et al. 2012) and was further placed into scaffolds using SSPACE Standard v. 3.0 (Boetzer et al. 2011). Gaps were filled or reduced with GapFiller v. 1.10 (Boetzer & Pirovano 2012). Several runs of genome assembly were conducted using different parameters. Assemblies obtained from these runs were subjected to quality and completeness assessment using the program BUSCO v. 2.0 (Simão et al. 2015) using the dataset for Sordariomycetes. The best assembly based on the best BUSCO statistics in term of completeness, was selected and presented in this study. The program AUGUSTUS v. 3.2.2 (Stanke et al. 2006) was used to estimate the number of protein coding genes encoded by the genome using the species model for Neurospora crassa.
Results and Discussion
The genome of Ophiostoma ips was assembled into 351 scaffolds. The genome is 25.99 Mb and the assembly has an N50 of 140.6 Kb. The average coverage across the whole genome was 80 times. The assembled O. ips genome has an average GC content of 56 %. The genome of O. ips has smaller genome size and higher GC genome content when compared to that of other Ophiostoma species such as O. ulmi (31.5 Mb, GC = 50.02 %; Khoshraftar et al. 2013), O. novo-ulmi (31.8 Mb, GC = 50.01 %; Forgetta et al. 2013) and O. piceae (32.8 Mb, GC = 52.8 %; Haridas et al. 2013). The taxonomic identity of the genome was confirmed using a phylogenetic analysis using authenticated sequences (Fig. 3). Assessment of the assembly using BUSCO with the Sodariomycetes dataset resulted in a completeness of 96.8 % (C:3606 [S:3603, D:3], F:42, M:77, n:3725), indicating that the assembly should cover most of the organism’s gene space. AUGUSTUS prediction using species model for Neurospora crassa resulted in 7607 protein coding genes, which is slightly lower than that of O. ulmi (8639 genes; Khoshraftar et al. 2013), O. novo-ulmi (8640 genes; Comeau et al. 2015), and O. piceae (8884 genes; Haridas et al. 2013).
The draft genome sequence from O. ips presented in this study will be a valuable addition to a number of genomes already available for species in Ophiostomatales. These will enable further studies to better understand this interesting group of fungi.
References
Abeel T, Van Parys T, Saeys Y, Galagan J, Van de Peer Y (2012) GenomeView: a next-generation genome browser. Nucleic Acids Research 40: e12.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. Journal of Molecular Biology 215: 403–410.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19: 455–477.
Barnes I, Gaur A, Burgess T, Roux J, Wingfield BD, Wingfield MJ (2001) Microsatellite markers reflect intra-specific relationships between isolates of the vascular wilt pathogen Ceratocystis fimbriata. Molecular Plant Pathology 2: 319–325.
Belbahri L (2015) Genome sequence of Ceratocystis platani, a major pathogen of plane trees. http://www.ncbi.nlm.nih.gov/nuccore/814603118.
Berger DK, Carstens M, Korsman JN, Middleton F, Kloppers FJ, et al. (2014) Mapping QTL conferring resistance in maize to gray leaf spot disease caused by Cercospora zeina. BMC Genetics 15: 60.
Boetzer M, Pirovano W (2012) Toward almost closed genomes with GapFiller. Genome Biology 13: R56.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579.
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30: 2114–2120.
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, et al. (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 18: 188–196.
Comeau AM, Dufour J, Bouvet GF, Jacobi V, Nigg M, et al. (2015) Functional annotation of the Ophiostoma novo-ulmi genome: insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biology and Evolution 7: 410–430.
Crous PW, Groenewald JZ, Groenewald M, Caldwell P, Braun U, et al. (2006) Species of Cercospora associated with grey leaf spot of maize. Studies in Mycology 55: 189–197.
Darribo D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: More models, new heuristics and parallel computing. Nature Methods 9: 772.
De Beer ZW, Duong TA, Barnes I, Wingfield BD, Wingfield MJ (2014) Redefining Ceratocystis and allied genera. Studies in Mycology 79: 187–219.
De Beer ZW, Wingfield MJ (2013) Emerging lineages in the Ophiostomatales. In: The Ophiostomatoid Fungi: expanding frontiers (Seifert KA, De Beer ZW, Wingfield MJ, et al., eds): 21–46. [CBS Biodiversity Series no. 12.] Utrecht: CBS-KNAW Fungal Biodiversity Institute.
De Vos L, Steenkamp ET, Martin SH, Santana QC, Fourie G, et al. (2014) Genome-wide macrosynteny among Fusarium species in the Gibberella fujikuroi complex revealed by amplified fragment lengthp-olymorphisms. PloS One 9: e114682.
Desjardins A, Plattner R, Gordon T (2000) Gibberella fujikuroi mating population A and Fusarium subglutinans from teosinte species and maize from Mexico and Central America. Mycological Research 104: 865–872.
Duong TA, De Beer ZW, Wingfield BD, Wingfield MJ (2013) Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biology 117: 411–421.
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791.
Forgetta V, Leveque G, Dias J, Grove D, Lyons R, et al. (2013) Sequencing of the Dutch Elm Disease fungus genome using the Roche/454 GS-FLX Titanium system in a comparison of multiple genomics core facilities. Journal of Biomolecular Techniques 24: 39–49.
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52: 696–704.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology 59: 307–21.
Haridas S, Wang Y, Lim L, Alamouti SM, Jackman S, et al. (2013) The genome and transcriptome of the pine saprophyte Ophiostoma piceae, and a comparison with the bark beetle-associated pine pathogen Grosmannia clavigera. BMC Genomics 14: 373.
Harris RS (2007) Improved pairwise alignment of genomic DNA. PhD thesis, Pennsylvania State University.
Hepting GH, Roth ER (1946) Pitch canker, a new disease of some southern pines. Journal of Forestry 44: 742–744.
Herron DA, Wingfield MJ, Wingfield BD, Rodas C, Marincowitz S et al. (2015) Novel taxa in the Fusarium fujikuroi species complex from Pinus spp. Studies in Mycology 80: 131–150.
Hoff KJ, Stanke M (2013) WebAUGUSTUS — a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Research 41: W123–W128.
Khoshraftar S, Hung S, Khan S, Gong Y, Tyagi V, et al. (2013) Sequencing and annotation of the Ophiostoma ulmi genome. BMC Genomics. 14: 162.
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780.
Kile G (1993) Plant diseases caused by species of Ceratocystis sensu stricto and Chalara. St Paul, MN: American Phytopathological Society Press.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14: R36.
Kirisits T (2004) Fungal associates of European bark beetles with special emphasis on the ophiostomatoid fungi. In: Bark and Wood Boring Insects in Living Trees in Europe, a synthesis (Lieutier F, Day KR, Battisti A, Grégoire JC, Evans HF, eds): 181–123. Dordrecht: Kluwer Academic Publishers.
Kirisits T, Konrad H, Wingfield MJ, Chhetri DB (2013) Ophiostomatoid fungi associated with the Eastern Himalayan spruce bark beetle, Ips schmutzenhoferi, in Bhutan and their pathogenicity to Picea spinulosa and Pinus wallichiana. In: The Ophiostomatoid Fungi: expanding frontiers (Seifert KA, De Beer ZW, Wingfield MJ, eds): 99–112. [CBS Biodiversity Series no. 12.] Utrecht: CBS-KNAW Fungal Biodiversity Institute.
Korf I (2004) Gene finding in novel genomes. BMC Bioinformatics 5: 59.
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874.
Latterell FM, Rossi AE (1983) Gray leaf spot of corn: a disease on the move. Plant Disease 67: 842–847.
Lieutier F, Yart A, Garcia J, Ham M, Morelet M, et al. (1989) Champignons phytopathogènes associés à deux coléoptères scolytidae du pin sylvestre (Pinus sylvestris L.) et étude préliminaire de leur agressivité envers l’hôte. Annales des Sciences Forestières 46: 201–216.
Liew E, Laurence M, Pearce C, Shivas R, Johnson G, et al. (2016) Review of Fusarium species isolated in association with mango malformation in Australia. Australasian Plant Pathology: DOI 10.1007/s13313-016-0454-z1-13.
Ma LJ, Van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, et al. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464: 367–373.
Meisel B, Korsman J, Kloppers F, Berger D (2009) Cercospora zeina is the causal agent of grey leaf spot disease of maize in southern Africa. European Journal of Plant Pathology 124: 577–583.
Möller E, Bahnweg G, Sandermann H, Geiger H (1992) A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Research 20: 6115.
Nei M, Kumar S (2000) Molecular Evolution and Phylogenetics. New York: Oxford University Press.
Niehaus E-M, Münsterkötter M, Proctor RH, Brown DW, Sharon A, et al. (2016) Comparative “omics” of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biology and Evolution 8: 3574–3599.
Nkuekam GK, Wingfield MJ, Roux J (2012) Ceratocystis species, including two new taxa, from Eucalyptus trees in South Africa. Australasian Plant Pathology 42: 283–311.
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, et al. (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathogens 8: e1003037.
Rumbold CT (1931) Two blue-staining fungi associated with bark-beetle infestation of pines. Journal of Agricultural Research 43: 847–873.
Seifert K (1993) Sapstain of commercial lumber by species of Ophiostoma and Ceratocystis. In: Ceratocystis and Ophiostoma: taxonomy ecology and pathogenicity (Wingfield MJ, et al., eds): 141–151. St Paul, MN: American Phytopathological Society Press.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Research 19: 1117–1123.
Six DL, Wingfield MJ (2011) The role of phytopathogenicity in bark beetle-fungus symbioses: a challenge to the classic paradigm. Annual Review of Entomology. 56: 255–272.
Stanke M, Diekhans M, Baertsch R, Haussler D (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644.
Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34: W435–W439.
Tavaré S (1986) Some probabilistic and statistical problems in the analysis of DNA sequences. In: Some Mathematical Questions in Biology: DNA sequence analysis (Muira RM, ed.): 57–86. Providence, RI: American Mathematical Society.
Van der Nest MA, Beirn LA, Crouch JA, Demers JE, De Beer ZW, et al. (2014a) Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus 5: 473–486.
Van der Nest MA, Bihon W, De Vos L, Naidoo K, Roodt D, et al. (2014b) Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans, and Ceratocystis moniliformis. IMA Fungus 5: 135–140.
Van Wyk M, Roux J, Barnes I, Wingfield BD, Wingfield MJ (2006) Molecular phylogeny of the Ceratocystis moniliformis complex and description of C. tribiliformis sp. nov. Fungal Diversity 21: 181–201.
Van Wyk M, Roux J, Barnes I, Wingfield BD, Chhetri DB, et al. (2004) Ceratocystis bhutanensis sp. nov, associated with the bark beetle Ips schmutzenhoferi on Picea spinulosa in Bhutan. Studies in Mycology 50: 365–379.
Wang J, Levy M, Dunkle LD (1998) Sibling species of Cercospora associated with gray leaf spot of maize. Phytopathology 88: 1269–1275.
Ward JMJ, Stromberg EL, Nowell DC, Nutter FW (1999) Gray leaf spot: a disease of global importance in maize production. Plant Disease 83: 884–895.
Wiemann P, Sieber CM, Von Bargen KW, Studt L, Niehaus E-M, et al. (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens 9: e1003475.
Wilken PM, Steenkamp ET, Wingfield MJ, De Beer ZW, Wingfield BD (2013) IMA Genome-F 1: Draft nuclear genome sequence for the plant pathogen, Ceratocystis fimbriata. IMA Fungus 4: 357–358.
Wingfield BD, Barnes I, De Beer ZW, De Vos L, Duong TA, et al. (2015a) IMA Genome-F 5: Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus 6: 493–506.
Wingfield BD, Ades P, Al-Naemi F, Beim L, Bihon W, et al. (2015b) IMA Genome-F 4: Draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagono-sporopsis tanaceti, and Thielaviopsis punctulata. IMA Fungus 6: 233–248.
Wingfield BD, Ambler JM, Coetzee MPA, De Beer ZW, Duong TA. et al. (2016a) IMA Genome-F5: Draft genome sequences of Armillaria fuscipes, Ceratocystiopsis minuta, Ceratocystis adiposa, Endoconidiophora laricicola, E. polonica and Penicillium freii DAOMC 242723. IMA Fungus 7: 217–227
Wingfield BD, Duong TA, Hammerbacher A, Van der Nest MA, Wilson A, et al. (2016b) IMA Genome-F 7: Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus 7: 317–323.
Wingfield BD, Steenkamp ET, Santana QC, Coetzee MPA, Bam S, et al. (2012) First fungal genome sequence from Africa: a preliminary analysis. South African Journal of Science 108: 01–09.
Wingfield M J, Marasas WFO (1980) Ceratocystis ips associated with Orthotomicus erosus (Coleoptera: Scolytidae) on Pinus spp. in the Cape province of South Africa. Phytophylactica. 12: 65–68.
Yuan Z-Q, Mohammed C (2002) Ceratocystis moniliformopsis sp. nov, an early coloniser of Eucalyptus obliqua logs in Tasmania, Australia. Australian Systematic Botany 15: 125–133.
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 18: 821–829.
Zhou XD, Burgess TI, De Beer ZW, Lieutier F, Yart A, et al. (2007) High intercontinental migration rates and population admixture in the sapstain fungus Ophiostoma ips. Molecular Ecology 16: 89–99.
Zhou XD, De Beer ZW, Wingfield BD, Wingfield MJ (2001) Ophiostomatoid fungi associated with three pine-infesting bark beetles in South Africa. Sydowia 53: 290–300.
Zhou XD, De Beer ZW, Wingfield BD, Wingfield MJ (2002) Infection sequence and pathogenicity of Ophiostoma ips, Leptographium serpens and L. lundbergii to pines in South Africa. Fungal Diversity 10: 229–240.
Zhou XD, De Beer ZW, Ahumada R, Wingfield BD, Wingfield MJ (2004) Ophiostoma and Ceratocystiopsis spp. associated with two pine-infesting bark beetles in Chile. Fungal Diversity 15: 253–266.
Acknowledgements
Genome sequencing of Cercospora zeina was done by the Phillip SanMiguel, Purdue Genomics Core Facility (USA), and was funded by National Research Foundation (NRF) CPRR, South Africa. The genome sequencing assembly and analyses of the Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens, and Ophiostoma ips genomes were co-funded by the Tree Protection Co-operative Programme (TPCP) and the DST-NRF Centre of Excellence in Tree Health Biotechnology (CTHB) at the Forestry and Agricultural Biotechnology Institute (FABI), and the DST-NRF South African Research Chairs Initiative (SARChI) in Fungal Genomics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wingfield, B.D., Berger, D.K., Steenkamp, E.T. et al. Draft genome of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens and Ophiostoma ips. IMA Fungus 8, 385–396 (2017). https://doi.org/10.5598/imafungus.2017.08.02.10
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.5598/imafungus.2017.08.02.10