
Abstract
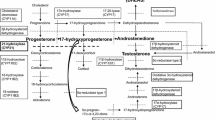
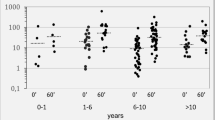
Congenital adrenal hyperplasia, both in its classic (CCAH) and non-classic form (NCAH), is a morbid condition sustained by the absent or reduced function of one of the enzymes involved in cortisol biosynthesis — mainly 21 hydroxylase — associated with different levels of clinical androgenization. In a wide group of relatives of patients affected by CCAH and NCAH (no.=222) and healthy volunteers (no.=30), a clinical, hormonal and genetic evaluation was performed in order to differentiate between the condition of heterozygous mutation carrier and non-carrier of any among 21-hydroxylase gene (CYP21) mutations. This study shows that clinical presentation and basal 17α-hydroxyprogesterone (17α-OHP) are not able to differentiate between heterozygous carriers and non-carriers, whereas 17α-OHP value after ACTH bolus is significantly different between heterozygous carriers and non-carriers: p<0.001 with a cut-off value of 3 ng/ml (90% sensitivity and 74,3% specificity). Moreover, our data indicate that 17α-OHP response to ACTH may be a useful tool to select subjects for genetic analysis.
Similar content being viewed by others
References
Stuckey MS, Boyne P, Macdonald WB, Christiansen FT, Houliston JB, Dawkins RL. HLA typing and ACTH stimulation in the detection of carriers for 21-hydroxylase deficiency. Aust N Z J Med 1980, 10: 552–4.
Mauseth RS, Hansen JA, Smith EK, Giblett ER, Kelley VC. Detection of heterozygotes for congenital adrenal hyperplasia: 21-hydroxylase deficiency- a comparison of HLA typing and 17-OH progesterone response to ACTH infusion. J Pediatr 1980, 97: 749–53.
Couillin P, Boué J, Bétuel H, Hors J, Gebuhrer L, Boué A Particular interest of HLA typing for genetic counselling in families with congenital adrenal hyperplsia (21-OH deficiency). Haematologia (Budap) 1987, 20: 25–30.
Oh BH, Park JK, Choi YM, Yang IM, Kim YS, Choi YK. Prenatal diagnosis of heterozygote of salt wasting congenital adrenal hyperplasia due to 21-hydroxylase deficiency by genetic linkage analysis. J Korean Med Sci 1988, 3: 73–7.
Escobar-Morreale HF, San Millán JL, Smith RR, Sancho J, Witchel SF. The presence of the 21-hydroxylase deficiency carrier status in hirsute woman: phenotipe-genotype correlation. Fertil Steril 1999, 72: 629–38.
Witchel SF, Lee PA. Identification of heterozygotic of 21-hydroxylase deficiency: sensitivity of ACTH stimulation test. Am J Med Genet 1998, 76: 337–42.
Ezquieta B, Cueva E, Varela J, Oliver A, Fernández J, Jariego C. Non-classical 21-hydroxylase deficiency in children: association of adrenocorticotropic hormone-stimulated 17-hydroxyprogesterone with the risk of compound heterozygosity with severe mutations. Acta Paediatr 2002, 91: 892–8.
Höppner W. Clinical impact of molecular diagnostics in endocrinology. Polymorphisms, mutations and DNA technologies. Horm Res 2002, 58(Suppl 3): 7–15.
Admoni O, Israel S, Lavi I, Gur M, Tenenbaum-Rakover Y. Hyperandrogenism in carriers of CYP21 mutation: the role of genotype. Clin Endocrinol (Oxf) 2006, 64: 645–51.
Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 1961, 21: 1440–7.
Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab 2000, 85: 1059–65.
Levine LS, Pang S. Prenatal diagnosis and treatment of congenital adrenal hyperplasia. J Pediatr Endocrinol 1994, 7: 193–200.
Dumić M. Congenital adrenal hyperplasia due to 21-hydroxylase enzyme deficiency. Lijec Vjesn 1996, 118(Suppl 1): 13–6.
Carlson AD, Obeid JS, Kanellopoulou N, Wilson RC, New MI. Congenital adrenal hyperplasia: update on prenatal diagnosis and treatment. J Steroid Biochem Mol Biol 1999, 69: 19–29.
New MI. Antenatal diagnosis and treatment of congenital adrenal hyperplasia. Curr Urol Rep 2001, 2: 11–8.
Nimkarn S, New MI. Prenatal diagnosis and treatment of congenital adrenal hyperplasia. Pediatr Endocrinol Rev 2006, 4: 99–105.
New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 2006, 91: 4205–14.
Ghizzoni L, Cesari S, Cremonini G, Melandri L. Prenatal and early postnatal treatment of congenital adrenal hyperplasia. Endocr Dev 2007, 11: 58–69.
Couillin P, Boue J, Nicolas H, Cheruy C, Boue A. Prenatal diagnosis of congenital adrenal hyperplasia (21-OH deficiency type) by HLA typing. Prenat Diagn 1981, 1: 25–33.
Potau N, Riqué S, Eduardo I, Marcos V, Ibañez L. Molecular defects of the CYP21 gene in Spanish girls with isolated precocius pubarche. Eur J Endocrinol 2002, 147: 485–8.
Ostlere LS, Rumsby G, Holownia P, Jacobs HS, Rustin MH, Honour JW. Carrier status for steroid 21-hydroxylase deficiency is only one factor in the variable phenotype of acne. Clin Endocrinol (Oxf) 1998, 48: 209–15.
Vottero A, Capelletti M, Giuliodori S, at al. Decreased androgen receptor gene methylation in premature pubarche: a novel pathogenetic mechanism? J Clin Endocrinol Metab 2006, 91: 968–72.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Napolitano, E., Manieri, C., Restivo, F. et al. Correlation between genotype and hormonal levels in heterozygous mutation carriers and non-carriers of 21-hydroxylase deficiency. J Endocrinol Invest 34, 498–501 (2011). https://doi.org/10.3275/7225
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3275/7225