
Abstract
Vancomycin and teicoplanin are still the only glycopeptide antibiotics available for use in humans. Emergence of resistance in enterococci and staphylococci has led to restriction of their use to severe infections caused by Gram-positive bacteria for which no other alternative is acceptable (because of resistance or allergy). In parallel, considerable efforts have been made to produce semisynthetic glycopeptides with improved pharmacokinetic and pharmacodynamic properties, and with activity towards resistant strains. Several molecules have now been obtained, helping to better delineate structure-activity relationships. Two are being currently evaluated for skin and soft tissue infections and are in phases II/ III. The first, oritavancin (LY333328), is the 4′-chlorobiphenylmethyl derivative of chloroeremomycin, an analogue to vancomycin. It is characterised by: i) a spectrum covering vancomycin-resistant enterococci (VRE), methicillin-resistant Staphylococcus aureus (MRSA) and to some extent glycopeptide-intermediate S. aureus (GISA); ii) rapid bactericidal activity including against the intracellular forms of enterococci and staphylococci; and iii) a prolonged half-life, allowing for daily administration. The second molecule is dalbavancin (BI397), a derivative of the teicoplanin analogue A40926. Dalbavancin has a spectrum of activity similar to that of oritavancin against vancomycin-sensitive strains, but is not active against VRE. It can be administered once a week, based on its prolonged retention in the organism. Despite these remarkable properties, the use of these potent agents should be restricted to severe infections, as should the older glycopeptides, with an extension towards resistant or poorly sensitive bacteria, to limit the risk of potential selection of resistance.
Similar content being viewed by others


Glycopeptide antibiotics were introduced in clinical practice 50 years ago, with vancomycin as the only agent for almost 30 years. Teicoplanin was launched in Europe in the mid 1980s and these two molecules remain the only members in this class available for human use; however, related derivatives have been used widely as growth promotants for livestock. The emergence and spreading of resistance in enterococci and staphylococci towards vancomycin has stimulated active research for new glycopeptides over the last 10 years, leading to the production of a series of semisynthetic derivatives. Starting from vancomycin and moving through a short description of the various compounds obtained so far, this paper reviews the basis of the selection of oritavancin and dalbavancin as new glycopeptides for clinical development (the chemical structure of these products is presented in figure 1). We also discuss their advantages and potential role in the therapeutic armament.

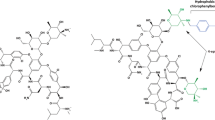
Chemical structure of glycopeptides. Upper panel: vancomycin and teicoplanin, which are the two glycopeptides currently available in clinical settings; lower panel: oritavancin and dalbavancin, two semisynthetic glycopeptides currently under clinical development. Middle panel: natural derivatives of vancomycin and teicoplanin used for the synthesis of oritavancin and dalbavancin, respectively. The part of the molecule that is common to all glycopeptides appears in bold. The arrows point to the differences between molecules.
1. Vancomycin and Teicoplanin
1.1 Mechanism of Action
Biochemical studies indicate that glycopeptides inhibit the late stages of peptidoglycan synthesis.[1] The biosynthetic pathway of this polymer involves three steps: (i) the synthesis of cytosolic precursors made of pentapeptides fixed on a disaccharide; (ii) the coupling of these precursors with a lipid carrier and the transfer of the resulting amphiphilic molecule to the outer surface of the membrane; and (iii) the reticulation between individual precursors by transpeptidation and transglycosylation reactions, accompanied by the release of the lipid carrier and its recycling to the inner face of the membrane. Bacteria incubated with vancomycin accumulate cytosolic precursors,[1] suggesting that glycopeptides interfere with the assembly of peptidoglycan and, in particular, with transglycosylation reactions (figure 2). At the molecular level, the primary target of vancomycin was shown to be the D-Ala-D-Ala terminus of the precursors. Molecular modelling and experimental studies[1–4] indicate that vancomycin forms a stoechiometric complex with the D-Ala-D-Ala dipeptide via the formation of five hydrogen bonds with the peptidic backbone of the glycopeptide. The formation of this complex prevents the transpeptidation reactions by steric hindrance. The tightness of the interaction between the glycopeptide and the D-Ala-D-Ala motif can be enhanced by two mechanisms.

Peptidoglycan synthesis in glycopeptide-susceptible bacteria (upper panel), in glycopeptide-resistant enterococci (middle panel) and in glycopeptide-intermediate staphylococci (lower panel).[2,5] In susceptible strains, peptidoglycan precursors are ending by D-Ala-D-Ala termini synthesised by D-Ala:D-Ala ligases (ddl). Glycopeptides interact with D-Ala-D-Ala termini of pentapeptides (through five hydrogen bonds) at the cell surface, preventing transglycosylation reactions and therefore peptidoglycan reticulation. The figure also illustrates how dimerisation of glycopeptide molecules or anchoring in the membrane by a hydrophobic tail may increase the tightness of this interaction. Hydrophobic derivatives may also inhibit the transglycosylation step without binding to peptidoglycan precursors. In glycopeptide-resistant enterococci, the presence of glycopeptides activates the signal-transducing system VanS (sensor) — VanR (regulator), which allows transcription of the resistance genes. These include enzymes involved in the hydrolysis of D-Ala-D-Ala termini (VanX and VanY) or in the synthesis of precursors ending by either D-Ala-D-Lac (VanH and VanA, VanB or Van D) or D-Ala-D-Ser (VanT and VanC, VanE or VanG), characterised by a lower affinity for glycopeptides. In glycopeptide-intermediate staphylococci (so-called VISA [vancomycin-intermediate Staphylococcus aureus] or GISA [glycopeptide-intermediate S. aureus] strains), more murein monomers are supplied and incorporated in the peptidoglycan, in which more free D-Ala-D-Ala residues remain present because of a decreased level of reticulation. More glycopeptide molecules are therefore trapped in the multiple layers of the thicker cell wall but fewer reach their target at the cell membrane. UDP = uridine diphosphate; UMP = uridine monophosphate; VRE = vancomycin-resistant enterococci.
-
Formation of homodimers between glycopeptide molecules, which has been demonstrated for glycopeptides of the vancomycin group, such as vancomycin itself,[6] eremomycin[7] or chloroeremomycin.[8] Organisation of molecules in dimers confers a structural rigidity that locks the binding pocket into the correct conformation and may allow for a cooperative binding to the ligand.[6,9–12] These dimers are formed by hydrogen bonds between the glycopeptide aglycone, which are maintained in the rigid conformational state favourable for ligand binding by the sugar residues on the glycopeptide molecule.[13]
-
Anchoring of the antibiotic in the membrane, as proposed for glycopeptides carrying a lipophilic tail, such as teicoplanin derivatives.[11] Lipid moieties are actually a common determinant in the structure of several antibiotics, such as ramoplanin, moenomycin and tunicamycin, which inhibit steps of peptidoglycan synthesis occurring close to the membrane. This suggests that such moieties help to maintain the drug close to their target. The importance of hydrophobic determinants has been further emphasised by the observation that vancomycin derivatives may inhibit transglycosylation without binding to D-Ala-D-Ala.[14,15]
1.2 Mechanisms and Importance of Resistance
The American National Committee for Clinical Laboratory Standards (NCCLS) has set up breakpoints for susceptibility and resistance, respectively, of ≤4 and ≥16 μg/mL for vancomycin and ≤8 and ≥32 μg/mL for teicoplanin (see table I for the main characteristics of the resistant strains). Over the years, two organisms of medical interest, namely the enterococci and Staphylococcus aureus, have developed a different, ingenuous resistance mechanism.

Main characteristics of glycopeptide-resistant bacteria
Resistance of enterococci to vancomycin was first reported in 1988.[27,28] The situation has evolved quite rapidly in the US, where resistance in blood isolates reached 13% in 1995 and 26% in 2000.[29] Colonisation by resistant strains is common in critically ill and immunosuppressed patients as well as in patients hospitalised in wards with high antimicrobial use. Hospitalisation is probably the main route for spread of these strains but, currently, outpatients are also often colonised and may, therefore, also represent another important source of contamination.[30] This alarming observation has stimulated drastic measures in terms of hygiene, restriction in antimicrobial use and surveillance to try to control or even reverse the situation.[29] In sharp contrast, resistance of enterococci in Europe is minimal (3%) and observed only in nosocomial infections;[31] outbreaks have been reported but remain sporadic. Carriage of resistant enterococci in animals, however, is frequent in Europe and is thought to be due to the massive use of avoparcin, a glycopeptide antibiotic used as growth promoter in animal feed,[32,33] but which has the same mode of action as vancomycin and, therefore, shares cross-resistance. The ban on avoparcin use instituted by EU countries has caused, in most countries, a fall in the prevalence of resistance in both animals and humans.[29,34–36]
The mechanism of resistance in enterococci relies on synthesis of peptidoglycan by an alternative pathway, which produces precursors ending in D-Ala-D-Lac or D-Ala-D-Ser instead of D-Ala-D-Ala and concomitantly eliminates precursors ending in D-Ala-D-Ala. The replacement of D-Ala by D-Lac suppresses one of the five hydrogen bonds between the glycopeptides and their target, which leads to a 1000-fold decrease in the binding affinity.[2,37] The substitution in D-Ser causes a conformational change, which also reduces vancomycin affinity, although not as markedly as with D-Lac. To be phenotypically detectable and significant, resistance requires the coordinated action of several enzymes (see figure 2). Thus, the bacteria need to synthesise D-Lac (VanH) or D-Ser (VanT) and D-Ala-D-Lac or D-Ala-D-Ser (VanA, B, D or C, E, G), and to degrade D-Ala-D-Ala (VanX) or to remove D-Ala from growing precursors (VanY) or both (VanXY). Moreover, a two-component regulatory system (VanS-VanR) resulting in induction by either vancomycin (VanB, C, E, G phenotype) or vancomycin and teicoplanin (VanA phenotype) plays a critical role.[2,16,22,23] The resistance proteins are encoded by genes physically grouped in operons that are located on plasmids or in the chromosome, and can be easily transferred, even between different species.[2,38,39] Six phenotypes of resistance have been described (see table I), which differ by the genetic support, the regulation of expression and the level of resistance conferred. An intriguing phenotype of glycopeptide dependence has also been described, in which synthesis of the D-Ala-D-Ala-ending peptidoglycan precursors is impaired through a mutation in the host ligase. Induction of production of the resistance proteins by glycopeptides restores peptidoglycan synthesis by allowing production of precursors ending in D-Ala-D-Lac.[40,41]
Resistance in S. aureus first emerged in the form of strains with elevated minimum inhibitory concentration (MIC) values towards vancomycin (or both vancomycin and teicoplanin), which have been named vancomycin-intermediate S. aureus (VISA) or glycopeptide-intermediate S. aureus (GISA). Originally described in Japan in 1996 in methicillin-resistant S. aureus (MRSA),[25] VISA and GISA strains have now been isolated in numerous countries, particularly from patients having received prolonged vancomycin therapy.[5,42] These bacteria are characterised by a thickened cell wall, production of an abundant extracellular material of still ill-characterised nature and an impaired ability to divide.[43] These phenotypic changes can be explained by the production of an altered peptidoglycan with an increased proportion of free D-Ala-D-Ala termini (less reticulation), which can trap vancomycin molecules and prevent their access to the target at the cytosolic membrane (figure 2). The thickened cell wall of a VISA strain may contain up to two to four times more D-Ala-D-Ala residues than that of a susceptible strain and is able to bind up to three to six times more vancomycin molecules before peptidoglycan synthesis becomes impaired.[5] This mechanism also implies a reduced cross-linkage of peptidoglycan. The latter has been suggested to result from decreased activity of penicillin-binding proteins (PBPs)[26] or from an alteration of murein precursors.[44] Yet multiple, additive, but not clearly identified mutations, are probably necessary to obtain resistance. VISA and GISA also need to import a larger amount of precursors than normal strains, which compromises their fitness in an antibiotic-free environment. This explains why VISA and GISA tend to lose their resistance when relieved from vancomycin pressure, giving rise to the so-called hetero-VISA phenotype.[45] Moreover, a glycopeptide-tolerant phenotype has been observed in clinical isolates of MRSA, in which MICs of glycopeptides are not affected but minimum bactericidal concentrations (MBCs) are considerably increased.[46] In a still more frightening fashion, two MRSA strains with high levels of resistance to vancomycin and teicoplanin have now been reported in two different hospital institutions in the US.[17–20] Both of these strains harbour the vanA gene cluster, indicating that they had acquired the corresponding set of genes from enterococci. Should such strains spread in hospitals, treatment options would rapidly become very limited.[47]
1.3 Pharmacological Properties
The spectrum of activity of glycopeptides covers essentially the Gram-positive organisms and a few anaerobes, and their activity against Gram-negative organisms is most often marginal (table II). Vancomycin and teicoplanin have a similar intrinsic activity, except against streptococci, which are more susceptible to teicoplanin.[48]


In vitro activity of oritavancin and dalbavancin compared with those of vancomycin and teicoplanina
The key pharmacokinetic properties of glycopeptides are summarised in table III. The most striking difference between vancomycin and teicoplanin is their capacity to bind serum proteins, which is much higher for teicoplanin than for vancomycin. This explains the prolonged half-life of teicoplanin in the organism. However, high protein binding also needs to be taken into account for activity predictions (table IV), since only the free serum fraction is directly active. Teicoplanin, perhaps because of its lipophilic character, is characterised by a higher volume of distribution than vancomycin, allowing it to reach therapeutic concentrations in fat, muscles (including pericardium and myocardium) and, to some extent, bone and cartilage, but not in the CNS.[63]

Pharmacokinetic properties of glycopeptides as observed after administration of conventional clinical dosesa

Pharmacodynamic parameters predictive of glycopeptide efficiency calculated for conventional dosages (see table III)
In vitro pharmacodynamic models show that vancomycin exhibits time-dependent bactericidal activity against most Gram-positive organisms. However, vancomycin is essentially bacteriostatic against enterococci[71] but may become bactericidal when combined with an aminoglycoside.[72,73] Vancomycin activity is adversely affected by a large inoculum but not by acidic pH. The drug also demonstrates faster killing rates against actively growing organisms, which can be easily understood on the basis of its mechanism of action.[74] Finally, glycopeptides produce persistent effects (postantibiotic effect lasting 1–6 hours[71,75]). While the dosage recommendations for glycopeptides have insisted on low dosages because of the fear of toxicity, recent developments in our understanding of pharmacodynamics have allowed a more rational basis for recommendations. The first models suggested that the free serum concentration of glycopeptides needs to remain above the MIC for the infecting organism for a prolonged period and, therefore, concluded that activity was primarily driven by the so-called ‘time above MIC’ parameter.[76,77] However, more recent and thorough studies have indicated that the parameter which best predicts efficacy is the area under the plasma concentration-time curve (AUC)/MIC ratio,[56] as usually found for most antimicrobials exhibiting time-dependent killing and significant postantibiotic effect.[78] However, in non-neutropenic animals, the ratio of the free serum peak concentration to the MIC (Cmax-free/MIC) also plays a determinant role in efficacy.[79] This Cmax-free/MIC ratio needs to reach at least 5–6 for vancomycin and 2–3 for teicoplanin (note that the determination of the peak and trough levels for these drugs, which are determined in routine in hospital laboratories, can be used for calculation of both Cmax/MIC and AUC/MIC ratios). As a consequence, glycopeptides are probably best given by discrete administrations with a total daily dose sufficiently large to match with the MIC of the infecting organism. A recent study comparing vancomycin 30 mg/kg once daily versus two administrations for the treatment of patients with bacteraemia or arthritis did not show a significant difference between the two groups in terms of clinical response (>92% in both cases;[80] however, no patients with endocarditis were included in this study). On a pharmacodynamic basis, there is therefore little reason to administer vancomycin by continuous infusion[81,82] even though other parameters such as more sustained concentrations in given tissues, or cost containment and ease of administration, have been advocated.[83,84] (Note that the stability and compatibility of vancomycin with other drugs in relation to its use by continuous infusion are currently under investigation.[85])
1.4 Clinical Use: Pros and Cons
Originally introduced into clinical practice as an agent active against β-lactamase-producing S. aureus, vancomycin remained largely unused because of development of less toxic alternatives, namely the β-lactamase-resistant penicillins (methicillin, isoxazolyl penicillins) and cephalosporins, as well as the introduction of β-lactamase inhibitors such as clavulanic acid (mainly in Europe) or sulbactam, which have become highly popular when combined with ampicillin or amoxicillin (mostly against Gram-negative bacteria, however). Yet orally administered vancomycin became popular for treatment of Clostridium difficile-associated diarrhoea and colitis.[86] The pandemic of nosocomial MRSA infections which started in the mid 1970s, and the fact that these strains were resistant not only to all β-lactam agents but often also to aminoglycosides, macrolides, lincosamides and fluoroquinolones,[87] heralded the return of vancomycin for systemic use. Alternatives such as fusidic acid, rifampicin and cotrimoxazole have indeed been poorly studied and did not compete in the face of the extensive data and the everyday experience available with glycopeptides.[88] Moreover, easy selection of resistant mutants (especially for rifampicin and fusidic acid when used alone) or toxicity problems has further limited their use. As we have seen, however, resistance developed in enterococci almost in parallel with the increase in vancomycin use in the US.[89–91] This led health authorities and concerned clinicians to try to restrict glycopeptide usage in order to maintain the activity of these useful agents for situations where alternatives were not usable.[90,92–95] The introduction of quinupristin/dalfopristin and linezolid in the late 1990s has not really changed this situation, since both drugs are expensive, have rare but potentially worrying adverse effects (haematological toxicity for long-course linezolid treatments, thrombophlebitis, arthralgias and/or myalgias, and drug interactions for quinupristin/dalfopristin), and are already facing emergence of resistance.[96–101] In particular, linezolid-resistant strains are now increasingly reported, not only in enterococci but also in S. aureus, which could become a clinically significant problem in the future.
The recommendations presented in table V were issued in 1995 by the Hospital Infection Control Practices Advisory Committee in the US (HICPAC[102]) and have been adopted, with adaptations, in most countries (see Gordts et al.[103] for Belgium). These recommendations have been further extended to special populations (see Nourse et al.[104] for children). In summary, and outside the field of bacteriologically documented MRSA infections, empirical therapy with glycopeptides should be limited to severe infections in immunocompromised patients (e.g. burn patients, clinical sepsis in the intensive care unit) when local epidemiological data show a high percentage of MRSA, and/or to life-threatening infections associated with the presence of foreign bodies (often infected with methicillin-resistant coagulase-negative staphylococci; e.g. indwelling percutaneous catheters, prosthetic cardiac valves). Indeed, animal models and clinical studies have shown that vancomycin is less bactericidal than isoxazolyl penicillins against methicillin-sensitive staphylococci (causing prolonged periods of fever and persistence of positive blood cultures). This should preclude its use (and, despite fewer data, the use of teicoplanin) when such strains are involved, especially in life-threatening infections (e.g. endocarditis). Therefore, their indiscriminate use in non-documented infections (or infections caused by Gram-positive strains susceptible to other common agents) should be discouraged with the exception of true β-lactam allergy. Of these severe infections by multiresistant staphylococci, the main indications for vancomycin are, therefore, restricted to: (i) serious diphtheroid infections (when the strain is penicillin resistant or the patient has IgE-mediated allergy to β-lactam agents); (ii) penicillin-resistant Streptococcus pneumoniae infections of the CNS in combination with cefotaxime or ceftriaxone; and (iii) antibiotic-associated colitis that is life-threatening or fails to respond to metronidazole (only oral vancomycin; low faecal concentrations by intravenous route and few data with teicoplanin). Routine prophylaxis with glycopeptides should also be prohibited[102,103] and restricted to prevention of bacterial endocarditis in penicillin/ampicillin-allergic patients at risk, undergoing gastrointestinal or genitourinary procedures (plus gentamicin), or dental procedures (vancomycin alone). In selected surgical wards with a high incidence of methicillin-resistant staphylococci infections, a single dose before surgery may be used in prosthetic surgery, but one should always try to solve the underlying fundamental hygiene problem. Teicoplanin could be used in most of the same indications, with the exception of the CNS infection, because of the poor penetration of the drug in the infected compartment. Its main advantages are the possibility of administering it by the intramuscular route with a once-daily schedule (because of its longer half-life) and a lower incidence of adverse effects. However, several indications are less documented than for vancomycin, and there are no well conducted, large, comparative studies between the two drugs. In severe infections (as in S. aureus endocarditis), teicoplanin needs to be given at a dose of at least 12 mg/kg/day once daily, after a minimum of three to four loading doses (each given every 12 hours).

Safety issues need also to be taken into account and may limit glycopeptide use in given populations. Earlier, vancomycin was notorious for toxicity related to impurities and to histamine release (causing the so-called ‘red man syndrome’). These adverse effects have been markedly reduced through better purification procedures and by giving the drug as a slow infusion over at least 1 hour.[108–110] With these precautions, vancomycin can be safely used, causing only relatively mild and self-limiting general toxicity.[111,112] However, the dose should be corrected in the case of renal insufficiency. The main adverse effects are phlebitis at the site of injection as well as nephrotoxicity and ototoxicity, the latter remaining the most problematic because it can be irreversible.[64] Both nephro- and ototoxicity are aggravated by the concomitant use of aminoglycosides or other drugs toxic for these organs.[112] Infrequent toxic manifestations include neutropenia, thrombocytopenia, fever, bullous dermatosis, necrotising cutaneous vasculitis and toxic epidermal necrolysis.[113] Teicoplanin has been claimed to be less toxic than vancomycin.[114] However, a difficulty relates to the fact that the recommended doses of teicoplanin have been increasing over time and that most comparative studies with vancomycin were done with teicoplanin doses (typically ≤6 mg/kg) that are now considered insufficient (table V). Therefore, further studies are needed to evaluate toxicity of teicoplanin in the current conditions of use.
2. New Developments in the Glycopeptide Area
Given the potential of glycopeptides in severe infections, considerable effort has been made to obtain new derivatives with improved pharmacological properties and activity against resistant strains. Because of a highly complex structure, which makes total synthesis of glycopeptides very challenging,[115] most of the new molecules obtained so far are semisynthetic derivatives of existing, natural glycopeptides. Structure-activity relationships (SARs) are presented in figure 3, based on our current understanding of the molecular mode of interaction between glycopeptides and their pharmacological target. Modifications can be subdivided into three main categories, each of them concerning distinct domains of the molecule and affecting the general mode of action of vancomycin in various ways. Other recent strategies have explored the formation of hybrid or dimerised molecules, or the synthesis of inhibitors of the resistance mechanisms to be administered in combination with unmodified glycopeptides.

Structure-activity relationship (SAR) of glycopeptides derived from vancomycin and teicoplanin. Upper panel: current view of the parts of the molecules involved in the various modes of action proposed for glycopeptides. Lower panel: most salient SARs that can be drawn for vancomycin (left) or teicoplanin (right) derivatives. ↑ indicates increased; ↓ indicates decreased.
2.1 Semisynthetic Modification of Natural Glycopeptides
2.1.1 Modification of the Binding Pocket
Binding studies have shown that five sites on the peptidic backbone of glycopeptides are involved in the interaction with the D-Ala-D-Ala termini (figure 3). Efforts have been made to change these amino acid residues, with the aim to generate compounds with increased affinity for both dipeptide and depsipeptide termini. These have included trimming or enlarging the heptapeptide backbone, epimerisation of the C3 chiral centre (in teicoplanin only), or substitution of amino acids 1 and 3.[116] Unfortunately, most of these modifications have resulted in a reduction of antibacterial activity. Nevertheless, the possibility of obtaining active compounds by this type of approach remains open, based on the observation that substitution of the Asn at position 3 by a hydrophobic amino acid enhances the affinity of vancomycin for D-Ala-D-Lac precursors.[117]
2.1.2 Modification or Addition of Functional Groups
A few functional groups can be modified out of the binding pocket, essentially causing changes in physicochemical properties. These studies have generated useful knowledge in SAR and have allowed changes in the natural compounds to be proposed.
Modification of Vancomycin
As stated earlier, the affinity of vancomycin for its target is increased by its capacity to dimerise. This process is facilitated by the addition of an amino sugar to residue 6 and by the presence of a chlorine atom in meta on the aromatic substituent of residue 2.[10,13,116] Other modifications at the extremities of the peptidic backbone have also brought significant pieces of information. Thus, replacement of the free carboxylate terminus by a carboxamide increases the activity on Staphylococcus epidermidis, whereas D-amino acids at the N-terminus appear more active than their corresponding L-isomers.[116]
Modification of Teicoplanin
As for vancomycin, structure-activity studies of teicoplanin have examined the influence of substituting the extremities of the peptidic backbone. At the C-terminus, substitution by a basic, positively charged amide considerably increases activity on staphylococci and also confers moderate activity on Gram-negative bacteria, because of improved ability to cross their outer membrane.[118] Esterification with hydrophobic alcohols also leads to marginal activity on Gram-negative bacteria. Similarly, basic amides of the A40,926 derivative of teicoplanin (which lacks the N-acetylglucosamine sugar; figure 1) show increased activity against staphylococci (particularly coagulase-negative) and streptococci. The most active compounds also show moderate activity against VanA enterococci.[119,120] At the N-terminus, acylation or alkylation causes either a slight increase or no modification in activity. Another important modification in the teicoplanin backbone is the introduction of chlorine atoms, which increase both target affinity and antibacterial activity.[116]
2.1.3 Alteration of Sugars
Despite their location apparently far from the binding pocket, sugars play a determinant role in glycopeptide activity, since aglycones of both vancomycin and teicoplanin are systematically less active than their parent compounds.[116,121] Substituting these sugars can actually have a considerable impact on glycopeptide pharmacological properties.
Modification of Vancomycin
Besides the beneficial addition of a 4-epi-vancosamine on the benzylic function of residue 6,[122] substitution of the vancosamine sugar by hydrophobic substituents proved mostly useful, probably by somehow mimicking the lipophilic side chain of teicoplanin derivatives. N-alkyl derivatives were found to be more active than N-acyl products, particularly against enterococci, streptococci and staphylococci.[116] When combined with the additional 4-epi-vancosamine (as in LY264826; see figure 1), an alkyl side chain confers to the molecule an unexpectedly rapid, concentration-dependent bactericidal activity, including against VanA- and VanB-type resistant enterococci.[123–125] The activity of these hydrophobic derivatives on resistant strains has been proposed to result from the combination of a facilitation of the binding to the peptidoglycan precursors due to their anchoring in the membrane[9,11] and of a direct inhibition of transglycosylases.[15,126] Recent studies also suggest that some of these derivatives may act as inhibitors not only of cell wall synthesis but also of phospholipid synthesis.[127] Acidic and hydrophilic substituents have also been added on the resorcinol in alpha of the free carboxylate of lipidated analogues of vancomycin, with favourable consequences on distribution and safety profiles, without negative effect on activity against MRSA or vancomycin-resistant enterococci (VRE).[128]
Modification of Teicoplanin
The impact of sugars on the activity is emphasised by the fact that the removal of N-acylglucosamine reduces activity, particularly against streptococci and enterococci, whereas removal of N-acetylglucosamine decreases the activity against staphylococci but may confer activity against Gram-negative as well as VanA-type enterococci. This is especially true for derivatives that also have a basic amide substitution of the C-terminus.[116]
Substituents on sugars also play important roles. As mentioned in the previous section, lipophilic substituents are of major interest for vancomycin activity. Teicoplanin contains naturally a lipophilic side chain that is responsible for its prolonged half-life. This side chain is also important for activity, since deacylated derivatives are only weak antibiotics.[116] The acyl chain has been proposed not only to favour anchoring in the membrane, and therefore the interaction with the target,[11] but also to circumvent VanB induction,[129] explaining teicoplanin activity against resistant strains of the VanB-type. On the other hand, the replacement of the N-acetylglucosamine by an N-acylglucuronic acid (as in A40926, see figure 1) has a detrimental effect on activity against VanA enterococci but improves anti-Gram-negative activity.[116]
2.2 Hybrid and Multivalent Glycopeptides
On the basis of current knowledge on the mode of action of glycopeptides, more innovative strategies have been explored to optimise activity against both susceptible and resistant strains. First, considering the potential cooperative binding occurring in multivalent drugs, as well as the enhanced activity of glycopeptides capable of self-association, dimers of vancomycin have been prepared using different linkers and various linkage positions.[130,131] Improved activity has been observed against Gram-positive organisms, including GISA and VRE strains, opening promising lines of investigation in this area. Secondly, taking into account the possibility of a dual target for glycopeptides, namely the dipeptide termini of peptidoglycan precursors, and the enzymes involved in the transglycosylation step, vancomycin derivatives have been prepared where the aglycone is separated from the sugars by linkers, with the aim to correctly position each part of the molecule.[132] This approach could now be extended to other glycopeptides in which both the aglycone and the carbohydrate moieties would be individually optimised for interaction with their own target, so as to generate very active compounds.
2.3 Inhibitors of Resistance Mechanisms
In the same way as clavulanic acid or sulbactam have been designed to restore the activity of β-lactam agents against β-lactamase-producing organisms, inhibitors of glycopeptide resistance mechanisms could be considered to specifically circumvent the mechanisms of resistance developed by enterococci (which, as described in section 1.2, may also apply to the recently described highly resistant S. aureus strains). This goal has been achieved by designing small molecules such as ε-aminopentanoylated prolinol,[133] which are able to selectively cleave the ester linkage of the D-Ala-D-Lac depsipeptide of resistant bacteria. Concomitant administration of this compound and vancomycin increases the activity of the glycopeptide in vitro against VanA-type enterococci, without affecting its activity against susceptible strains. However, activity remains globally low, probably because VanX hydrolyses the D-Ala-D-Ala dipeptide, so that there is no pentapeptide left for binding glycopeptides. These inhibitors will, therefore, need to be combined with inhibitors of the VanX D-D-peptidase.[134,135] They have been obtained with inhibitory potencies in the micromolar range, but microbiological data are still lacking to demonstrate the interest of combining these inhibitors with vancomycin.
3. Properties of the Ideal Glycopeptide
Based on an examination of the limitations of the current molecules and also on the promising properties of some of the semisynthetic derivatives described in section 2, we delineate in table VI the main desirable characteristics expected from new glycopeptides for future clinical development. Two molecules, oritavancin and dalbavancin, fulfil these conditions in various ways and are currently in phase II/III of clinical development.

Properties of an ideal glycopeptide
4. Glycopeptides Undergoing Clinical Evaluation
4.1 Oritavancin
Oritavancin (LY333328) was discovered at Eli Lilly (Indianapolis, IN, USA) but is now developed by InterMune (Brisbane, CA, USA)[136] [see figure 1 for structure]. This molecule was obtained by reductive alkylation with 4′-chloro-biphenyl-carboxaldehyde of A82846B (chloroeremomycin; LY264826), a natural glycopeptide that differs from vancomycin by the addition of a 4-epi-vancosamine sugar and the replacement of the vancosamine of the disaccharide moiety by an epi-vancosamine.[122] The design of this drug was based on previous observations that chloroeremomycin was more potent than vancomycin because of its stronger tendency to self-associate,[3] and that substitution of the vancosaminyl moiety of vancomycin by a lipophilic side chain resulted in enhanced activity and in some cases restored activity towards vancomycin-resistant enterococci.[121,137]
4.1.1 Microbiological and Pharmacological Properties
Spectrum of Activity and Resistance
The activity of oritavancin (table II) is similar to that of vancomycin against staphylococci, with MICs ranging between 0.03 and 8 mg/L; no differences are seen between methicillin-susceptible and -resistant stains. Against enterococci, oritavancin is more active than vancomycin or teicoplanin, with MICs consistently lower than 1 mg/L. Remarkably, it is as active against glycopeptide-resistant strains as against glycopeptide-susceptible isolates. Oritavancin is also very potent against pneumococci (both penicillin-susceptible and -resistant). It shows low MICs against other Gram-positive species including other streptococci, Listeria spp., Clostridium spp. and corynebacteria,[50] but is not active against Gram-negative bacteria, including Haemophilus influenzae. Resistance of S. aureus to oritavancin has so far not been described in clinical isolates,[51,52,138] including VISA strains. In contrast, enterococci with reduced susceptibility to oritavancin (MIC 8–16 mg/L) have been obtained in vitro, using strains harbouring the vanA or the vanB gene clusters.[139] Three mechanisms seem to operate:[139] (i) the complete elimination of D-Ala-ending precursors by overexpression of the vanA gene cluster or by reduced expression of the host D-Ala:D-Ala ligase; (ii) mutations in the VanSB sensor of the vanB cluster (which can occur in clinical isolates[140]); or (iii) the expression of VanZ, a protein encoded by a gene present in the resistance transposon but the precise function of which is still unknown. A clinical strain of oritavancin-dependent Enterococcus faecalis has also been isolated but the underlying mechanism should be different from that conferring dependence to vancomycin and teicoplanin, since the growth of this strain is not restored in the presence of D-Ala-D-Ala precursors.[141]
Pharmacodynamic Properties in Relation to the Mode of Action
Oritavancin is bactericidal in vitro, with MBCs only 1- to 8-fold higher than the corresponding MICs against most of the covered microorganisms.[50] Killing curve experiments have revealed a striking difference between vancomycin and oritavancin concerning the rate of killing and its concentration-dependent character. Oritavancin indeed shows very rapid and highly concentration-dependent bactericidal activity (3-log reduction in bacterial counts after 1–8 hours) in conditions where vancomycin requires at least 8–24 hours to reach the same effect.[50,142] However, oritavancin acts more slowly against VRE.[143] These properties suggest that oritavancin could have another mode of action than conventional glycopeptides. The current view is that oritavancin combines the advantages of a high capacity to dimerise and to interact with the membrane, and therefore may cooperatively bind to peptidoglycan residues of both susceptible and resistant strains. It may also directly perturb the membrane properties and inhibit transglycosylation reactions.[125]
Oritavancin also displays a concentration-dependent postantibiotic effect, increasing from ~2 hours at 1 × MIC to 4 hours against VRE and 8 hours against MRSA.[143] As with vancomycin, the combination of oritavancin with gentamicin is synergistic.[143–145] The combination with ampicillin enhances the bactericidal activity of oritavancin, without being truly synergistic. It also prolongs its postantibiotic effect against VRE from 18 to 23 hours at 10 × MIC.[146]
Oritavancin activity is negatively affected by large inocula,[143] but not by acid pH or by the growth phase of the bacteria.[147] However, as pointed out by Mercier et al.,[147] activity might be slightly reduced against VRE in stationary growth phase, as observed in animal model infective endocarditis or in acidic foci of infection.
Pharmacokinetic Profile and Relationship with Pharmacodynamic Properties
The pharmacokinetic properties of oritavancin are presented in table III in comparison with those of the other glycopeptides. Like teicoplanin, oritavancin is characterised by high protein binding to albumin, which explains its prolonged retention in mammals, but also reduces the rate and extent of its bactericidal activity and shortens the postantibiotic effect.[146,148] This could be important when serum levels become close to the MIC for the infecting organism. Further studies should address pharmacokinetic-pharmacodynamic relationships for these drugs, but we may anticipate, on the basis of its concentration-dependent bactericidal activity in vitro, that high serum levels or AUC relative to MIC will correlate with optimal efficacy. Studies on cultured cells also show that oritavancin accumulates in eukaryotic cells to exceptional levels (the apparent cellular concentration reaching values as high as 400 times the extracellular ones), which may be an advantage for the eradication of intracellular infections.[149]
Models of Infection
The promising in vitro activity of oritavancin has prompted evaluation of its activity in models of difficult-to-treat infections. As anticipated, oritavancin given once daily is as effective as vancomycin given every 8 hours against MRSA rabbit endocarditis[150] and is bactericidal against S. pneumoniae rabbit meningitis,[151,152] even though the drug penetration in the CSF reaches only 5% of the serum concentration.[152] Oritavancin activity has also been studied in models of infections by glycopeptide-resistant enterococci. While oritavancin was effective in a model of rat central venous catheter infection,[153] its activity was more limited in a rabbit endocarditis model, causing a reduction in the number of bacteria in the vegetations but failing to sterilise them.[154] Increasing serum concentrations to values as high as 80 mg/L did not provide significant improvement and did not prevent the selection of resistant mutants, probably because of the heterogeneous distribution of the drug in the vegetations. Yet combination with gentamicin proved synergistic and bactericidal, and prevented the emergence of resistant mutants.[155] Intracellular bactericidal activity of oritavancin has been demonstrated in in vitro models of polymorphonuclear leucocytes infected by vancomycin-resistant enterococci[156] or by MRSA[157] as well as in macrophages infected by S. aureus.[158] Taken together, these data suggest a definite clinical interest for this drug, provided it can reach the infected compartment at sufficiently high concentrations. Efficacy against infections occurring in less accessible compartments as well as against intracellular organisms needs, therefore, to be examined in the light of the tissue distribution of the drug in vivo.
4.1.2 Clinical Studies
Oritavancin is currently in phase III development, with two studies completed in the treatment of patients with complicated skin and skin structure infection caused by Gram-positive pathogens including MRSA.[159] The intent-to-treat analysis of one of these, a double-blind, randomised study, has been presented as an abstract[160] and showed equivalent clinical success with oritavancin at 1.5 or 3 mg/kg for 3–7 days versus vancomycin (15 mg/kg twice daily for 3–7 days followed by oral cephalexin for up to 10–14 days in the vancomycin group).
4.1.3 Safety Profile
The only published data available so far are those of the phase I studies, in which oritavancin was well tolerated.[161] However, studies on cultured cells indicate that the drug could induce lipid storage, probably in relation to its high level of cellular accumulation,[162] which may suggest further evaluation in animal models.
4.2 Dalbavancin
Dalbavancin (BI397) is a semisynthetic derivative from the natural glycopeptide A40926 (see figure 1 for structure). It was discovered by Biosearch Italia and outlicensed for North America to Versicor. The two companies have now merged to create Vicuron Pharmaceuticals (King of Prussia, PA, USA)[163] and continue to develop the product. A40926 differs from teicoplanin by the absence of the acetylglucosamine in the benzylic position, replacement of the acylglucosamine in position 4 by an acylaminoglucuronic acid, the length of the fatty acid chain, the position of one chlorine atom and the terminal methylamino group.[164] In dalbavancin, a 3,3-dimethylaminopropylamide replaces the peptide carboxy group of A40926.[165] SAR studies of teicoplanin indicated an improvement in activity by derivatisation of its carboxy group, and improved activity against VanA-type enterococci is expected to occur when removing the acetylglucosamine.[48] The most active compound in the series was actually the 6β-decarboxy-6β-hydroxymethyl amide of A40926,[165] but development of this compound was halted because of its poor tolerability in animals (causing adverse effects typical of histamine release, which have been ascribed to its basic character[48]). Unfortunately, most data on dalbavancin have been published only as abstracts or as short quotations in review papers. This makes an in-depth comparison of dalbavancin with oritavancin difficult.
4.2.1 Microbiological and Pharmacological Properties
Spectrum of Activity and Resistance
In general (table II), dalbavancin is more potent than vancomycin, teicoplanin and, to some extent, oritavancin against staphylococci (with MICs ranging from <0.03 to 1 mg/L for susceptible and methicillin-resistant strains, respectively) and S. pyogenes (MIC in the <0.002–0.06 mg/L range).[49,53,57] It is also very active against S. pneumoniae, with MICs 2- to 4-fold higher than those of oritavancin. It is as active as oritavancin or teicoplanin against vancomycin-susceptible enterococci but does not offer the same advantage as oritavancin against glycopeptide-resistant strains. Thus, the MIC distribution range towards VanB- and VanA-type strains includes more susceptible strains but the MIC50 values remain only marginally (4-fold) lower than those of teicoplanin.[49] Therefore, this drug represents a significant improvement except against glycopeptide-resistant enterococci.[48]
Subpopulations of staphylococci that have acquired low levels of resistance (2- to 4-fold increase in MIC) have been selected upon serial passage at sub-MIC of dalbavancin.[166] In vivo selection of such mutants may, however, be more difficult, since trough concentrations of dalbavancin are above the current MIC of the targeted microorganisms.
Pharmacodynamic Properties in Relation to the Mode of Action
Like oritavancin, dalbavancin is bactericidal, with MBC/MIC ratios close to 1, even in the presence of 30% serum.[66] It is synergic with ampicillin, including against VanA-type enterococci.
Pharmacokinetic Profile and Relationship with Pharmacodynamic Properties
The pharmacokinetic profile of dalbavancin is compared with that of the other glycopeptides in table III. Phase I studies have used higher dosages, providing higher AUC values. However, when corrected for the free fraction, the AUC/MIC and Cmax/MIC ratios of dalbavancin are in the same order of magnitude as those of teicoplanin. Plasma dalbavancin concentrations still exceed the MBC values for staphylococci 1 week after administration of a single dose of 1000mg but free serum concentrations are close to the MIC.[66,67]
Models of Infection
The activity of dalbavancin has been assessed in models of infections by glycopeptide-susceptible Gram-positive organisms and by GISA. Preliminary reports indicate a reduction in bacterial load after a single-dose administration in models of penicillin-resistant S. pneumoniae pneumonia or of MRSA rat pouch infection.[48,167] Its activity is superior to that of conventional glycopeptides in models of endocarditis due to MRSA or GISA, and of acute septicaemia caused by staphylococci, streptococci or enterococci.[66,168]
4.2.2 Clinical Studies
No clinical data have been published so far but phase II/III clinical trials are ongoing to evaluate the efficacy and safety of dalbavancin for the treatment of complicated skin and soft tissue infections caused by Gram-positive organisms or the use of dalbavancin in a once-a-week mode of administration for other Gram-positive hospital infections.[66] In a first report of a phase II study,[169] administration of two doses (1000mg on day 1 and 500mg on day 8) was found to be as effective as comparators (7–21 days with clindamycin, ceftriaxone, vancomycin or cefazolin) for the treatment of deep skin and soft tissue infections caused by methicillin-susceptible S. aureus or MRSA.
4.2.3 Safety Profile
Preclinical studies in rats and dogs show that dalbavancin is well tolerated after intravenous bolus administration at doses several times higher than those expected to be used in humans.[48] Phase I or phase II clinical trials have not reported major adverse effects in the range of concentrations where dalbavancin is effective.[66,169] No dosage adjustments are necessary in case of mild renal insufficiency.[170]
5. New Glycopeptides in the Clinics: for Which Indications?
The improved pharmacological properties of new glycopeptides make them very potent agents, with potentially large clinical indications. However, their use should be as restricted as that of conventional glycopeptides if one wishes to avoid the rapid emergence of resistance.
The advantages of oritavancin that should prove to be useful in clinical practice include: (i) its fast rate of bacterial killing, its spectrum of activity, which covers VRE, methicillin-resistant staphylococci and to some extent the GISA strains; (ii) its activity against intracellular forms of enterococci and staphylococci; and (iii) its prolonged half-life. For dalbavancin, the main improvement seems to be its pharmacokinetic profile, which should allow once-weekly administration. Microbiological properties of dalbavancin are less favourable than those of oritavancin, since VRE are not covered.
On the basis of these properties, and also in view of the results from animal models, oritavancin may become a drug of interest for infections caused by VRE, while both oritavancin and dalbavancin could be considered for severe infections by multiresistant organisms, such as methicillin-resistant staphylococci or S. pneumoniae. In the latter case, however, other alternatives such as fluoroquinolones (moxifloxacin or gatifloxacin) should be examined first. In some countries resistance to fluoroquinolones has already reached alarming levels (15% in Southeast Asia [Hong Kong, South Korea[171]]). There are also situations such as CNS infections in which fluoroquinolones are poorly efficient.[172,173] Oritavancin and dalbavancin could, therefore, prove very useful in these ‘niche’ but important indications. Finally, their activity against GISA strains appears limited (table II), which could be a deterrent to recommending them in these situations. However, synergy has been demonstrated for dalbavancin when combined with β-lactam agents.[57] Encouraging results have also been obtained in animal models,[168] which should stimulate further testing of this molecule against GISA infections.
Therefore, reasonable indications could include the following: (i) nosocomial septicaemia or infections of deep organs caused by MRSA or VRE (oritavancin only); (ii) severe and recurrent skin and soft tissue infections caused by MRSA (in relation with activity against both extra- and intracellular bacteria [demonstrated so far only for oritavancin]); and (iii) endocarditis due to resistant organisms (but probably in combination with an aminoglycoside for oritavancin against VRE, based on disappointing animal data[155]). The usefulness of oritavancin in nosocomial CNS infections caused by MRSA, resistant S. pneumoniae or VRE may be suggested on the basis of its activity in animal models. No corresponding data exist for dalbavancin, for which penetration in CSF is low. The potential use of oritavancin in humans in this indication needs to be assessed, as well as the influence of corticosteroids on the CNS penetration of these new glycopeptides (influence of corticosteroids was found to be minor for vancomycin).
Globally speaking, clinical studies or additional animal data are needed to confirm the interest of new glycopeptides in these indications, since the proposals made above rely almost exclusively on an analysis of the pharmacological properties of these drugs and on the results obtained in animal models of infection.
6. Conclusion
Oritavancin, and to a lesser extent dalbavancin, offer considerable possibilities for improvement over vancomycin and teicoplanin, essentially in terms of pharmacokinetic and pharmacodynamic properties, as well as of activity against resistant strains (for oritavancin). Whether these properties can translate into improved clinical efficacy awaits completion of clinical studies. Should these molecules soon become approved for clinical use, it is vital that they are used rationally and prudently to protect against the rapid emergence of resistance and extend their useful life.
References
Reynolds PE. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 1989 Nov; 8(11): 943–50
Arthur M, Reynolds P, Courvalin P. Glycopeptide resistance in enterococci. Trends Microbiol 1996 Oct; 4(10): 401–7
Williams DH, Waltho JP. Molecular basis of the activity of antibiotics of the vancomycin group. Biochem Pharmacol 1988 Jan 1; 37(1): 133–41
Loll PJ, Axelsen PH. The structural biology of molecular recognition by vancomycin. Annu Rev Biophys Biomol Struct 2000; 29: 265–89
Hiramatsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis 2001 Oct; 1(3): 147–55
Schafer M, Schneider TR, Sheldrick GM. Crystal structure of vancomycin. Structure 1996 Dec 15; 4(12): 1509–15
Groves P, Searle MS, Mackay JP, et al. The structure of an asymmetric dimer relevant to the mode of action of the glycopeptide antibiotics. Structure 1994 Aug 15; 2(8): 747–54
Shiozawa H, Chia BC, Davies NL, et al. Cooperative binding interactions of glycopeptide antibiotics. J Am Chem Soc 2002 Apr 17; 124(15): 3914–9
Mackay JP, Gerhard U, Beauregard DA, et al. Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signalling. J Am Chem Soc 1994; 116: 4581–90
Mackay JP, Gerhard U, Beauregard DA, et al. Dissection of the contributions towards dimerization of glycopeptide antibiotics. J Am Chem Soc 1994; 116: 4573–80
Beauregard DA, Williams DH, Gwynn MN, et al. Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob Agents Chemother 1995 Mar; 39(3): 781–5
Williams DH, Maguire AJ, Tsuzuki W, et al. An analysis of the origins of a cooperative binding energy of dimerization. Science 1998 May 1; 280(5364): 711–4
Kaplan J, Korty BD, Axelsen PH, et al. The role of sugar residues in molecular recognition by vancomycin. J Med Chem 2001 May 24; 44(11): 1837–40
Ge M, Chen Z, Onishi HR, et al. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science1999 Apr 16; 284(5413): 507–11
Printsevskaya SS, Pavlov AY, Olsufyeva EN, et al. Role of the glycopeptide framework in the antibacterial activity of hydrophobic derivatives of glycopeptide antibiotics. J Med Chem 2003 Mar 27; 46(7): 1204–9
Gholizadeh Y, Courvalin P. Acquired and intrinsic glycopeptide resistance in enterococci. Int J Antimicrob Agents 2000 Nov; 16 Suppl. 1: S11–7
Centers for Disease Control and Prevention. Staphylococcus aureus resistant to vancomycin: United States, 2002. MMWR Morb Mortal Wkly Rep 2002; 51: 565–7
Chang S, Sievert DM, Hageman JC, et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med 2003 Apr 3; 348(14): 1342–7
Bozdogan B, Esel D, Whitener C, et al. Antibacterial susceptibility of a vancomycin-resistant Staphylococcus aureus strain isolated at the Hershey Medical Center. J Antimicrob Chemother 2003 Nov; 52(5): 864–8
Tenover FC, Weigel LM, Appelbaum PC, et al. Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrob Agents Chemother 2004 Jan; 48(1): 275–80
Poyart C, Pierre C, Quesne G, et al. Emergence of vancomycin resistance in the genus Streptococcus: characterization of a vanB transferable determinant in Streptococcus bovis. Antimicrob Agents Chemother 1997 Jan; 41(1): 24–9
Perichon B, Reynolds P, Courvalin P. VanD-type glycopeptide-resistant Enterococcus faecium BM 4339. Antimicrob Agents Chemother 1997 Sep; 41(9): 2016–8
Fines M, Perichon B, Reynolds P, et al. VanE, a new type of acquired glycopeptide resistance in Enterococcus faecalis BM 4405. Antimicrob Agents Chemother 1999 Sep; 43(9): 2161–4
McKessar SJ, Berry AM, Bell JM, et al. Genetic characterization of vanG, a novel vancomycin resistance locus of Enterococcus faecalis. Antimicrob Agents Chemother 2000 Nov; 44(11): 3224–8
Hiramatsu K, Hanaki H, Ino T, et al. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 1997 Jul; 40(1): 135–6
Sieradzki K, Tomasz A. Gradual alterations in cell wall structure and metabolism in vancomycin-resistant mutants of Staphylococcus aureus. J Bacteriol 1999 Dec; 181(24): 7566–70
Leclercq R, Derlot E, Duval J, et al. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 1988 Jul 21; 319(3): 157–61
Uttley AH, Collins CH, Naidoo J, et al. Vancomycin-resistant enterococci. Lancet 1988 Jan 2; I(8575–6): 57–8
Bonten MJ, Willems R, Weinstein RA. Vancomycin-resistant enterococci: why are they here, and where do they come from? Lancet Infect Dis 2001 Dec; 1(5): 314–25
Kenner J, O’Connor T, Piantanida N, et al. Rates of carriage of methicillin-resistant and methicillin-susceptible Staphylococcus aureus in an outpatient population. Infect Control Hosp Epidemiol 2003 Jun; 24(6): 439–44
Schouten MA, Hoogkamp-Korstanje JA, Meis JF, et al. Prevalence of vancomycin-resistant enterococci in Europe. Eur J Clin Microbiol Infect Dis 2000 Nov; 19(11): 816–22
van den Bogaard AE, Mertens P, London NH, et al. High prevalence of colonization with vancomycin- and pristinamycin-resistant enterococci in healthy humans and pigs in The Netherlands: is the addition of antibiotics to animal feeds to blame? J Antimicrob Chemother 1997 Sep; 40(3): 454–6
van den Braak N, van Belkum A, van Keulen M, et al. Molecular characterization of vancomycin-resistant enterococci from hospitalized patients and poultry products in The Netherlands. J Clin Microbiol 1998 Jul; 36(7): 1927–32
Klare I, Badstubner D, Konstabel C, et al. Decreased incidence of VanA-type vancomycin-resistant enterococci isolated from poultry meat and from fecal samples of humans in the community after discontinuation of avoparcin usage in animal husbandry. Microb Drug Resist 1999; 5(1): 45–52
Aarestrup FM, Seyfarth AM, Emborg HD, et al. Effect of abolishment of the use of antimicrobial agents for growth promotion on occurrence of antimicrobial resistance in fecal enterococci from food animals in Denmark. Antimicrob Agents Chemother 2001 Jul; 45(7): 2054–9
van den Bogaard AE, Stobberingh EE. Epidemiology of resistance to antibiotics: links between animals and humans. Int J Antimicrob Agents 2000 May; 14(4): 327–35
Bugg TD, Wright GD, Dutka-Malen S, et al. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991 Oct 29; 30(43): 10408–15
Arthur M, Molinas C, Depardieu F, et al. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM 4147. J Bacteriol 1993 Jan; 175(1): 117–27
Quintiliani Jr R, Courvalin P. Conjugal transfer of the vancomycin resistance determinant vanB between enterococci involves the movement of large genetic elements from chromosome to chromosome. FEMS Microbiol Lett 1994 Jun 15; 119(3): 359–63
Baptista M, Depardieu F, Reynolds P, et al. Mutations leading to increased levels of resistance to glycopeptide antibiotics in VanB-type enterococci. Mol Microbiol 1997 Jul; 25(1): 93–105
Van Bambeke F, Chauvel M, Reynolds PE, et al. Vancomycin-dependent Enterococcus faecalis clinical isolates and revertant mutants. Antimicrob Agents Chemother 1999 Jan; 43(1): 41–7
Hamilton-Miller JM. Vancomycin-resistant Staphylococcus aureus: a real and present danger? Infection 2002 Jun; 30(3): 118–24
Hamilton-Miller JM. Glycopeptide-resistant staphylococci. Int J Antimicrob Agents 1999 Sep; 13(1): 63–5
Cui L, Murakami H, Kuwahara-Arai K, et al. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu 50. Antimicrob Agents Chemother 2000 Sep; 44(9): 2276–85
Hiramatsu K, Okuma K, Ma XX, et al. New trends in Staphylococcus aureus infections: glycopeptide resistance in hospital and methicillin resistance in the community. Curr Opin Infect Dis 2002 Aug; 15(4): 407–13
May J, Shannon K, King A, et al. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother 1998 Aug; 42(2): 189–97
Johnson AP, Woodford N. Glycopeptide-resistant Staphylococcus aureus. J Antimicrob Chemother 2002 Nov; 50(5): 621–3
Malabarba A, Ciabatti R. Glycopeptide derivatives. Curr Med Chem 2001 Dec; 8(14): 1759–73
Candiani GP, Abbondi M, Borgonovi M, et al. In-vitro and in-vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J Antimicrob Chemother 1999 Aug; 44(2): 179–92
Biavasco F, Vignaroli C, Lupidi R, et al. In vitro antibacterial activity of LY333328, a new semisynthetic glycopeptide. Antimicrob Agents Chemother 1997 Oct; 41(10): 2165–72
Zeckel ML, Preston DA, Allen BS. In vitro activities of LY333328 and comparative agents against nosocomial gram-positive pathogens collected in a 1997 global surveillance study. Antimicrob Agents Chemother 2000 May; 44(5): 1370–4
Garcia-Garrote F, Cercenado E, Alcala L, et al. In vitro activity of the new glycopeptide LY333328 against multiply resistant gram-positive clinical isolates. Antimicrob Agents Chemother 1998 Sep; 42(9): 2452–5
Jones RN, Biedenbach DJ, Johnson DM, et al. In vitro evaluation of BI 397, a novel glycopeptide antimicrobial agent. J Chemother 2001 Jun; 13(3): 244–54
Jones RN, Barrett MS, Erwin ME. In vitro activity and spectrum of LY333328, a novel glycopeptide derivative. Antimicrob Agents Chemother 1997 Feb; 41(2): 488–93
Noviello S, Ianniello F, Esposito S. In vitro activity of LY333328 (oritavancin) against gram-positive aerobic cocci and synergy with ciprofloxacin against enterococci. J Antimicrob Chemother 2001 Aug; 48(2): 283–6
Aeschlimann JR, Allen GP, Hershberger E, et al. Activities of LY333328 and vancomycin administered alone or in combination with gentamicin against three strains of vancomycin-intermediate Staphylococcus aureus in an in vitro pharmacodynamic infection model. Antimicrob Agents Chemother 2000 Nov; 44(11): 2991–8
Hackbarth CJ, Lopez S, Trias J, et al. In vitro activity of the glycopeptide BI 397 against Staphylococcus aureus and Staphylococcus epidermidis [abstract no. 1283]. 39th Interscience Conference on Antimicrobial Agents and Chemotherapy; 1999 Sep 26–28; San Francisco (CA)
Tenover FC, Lancaster MV, Hill BC, et al. Characterization of staphylococci with reduced susceptibilities to vancomycin and other glycopeptides. J Clin Microbiol 1998 Apr; 36(4): 1020–7
Schwalbe RS, McIntosh AC, Qaiyumi S, et al. In vitro activity of LY333328, an investigational glycopeptide antibiotic, against enterococci and staphylococci. Antimicrob Agents Chemother 1996 Oct; 40(10): 2416–9
Harland S, Tebbs SE, Elliott TS. Evaluation of the in-vitro activity of the glycopeptide antibiotic LY333328 in comparison with vancomycin and teicoplanin. J Antimicrob Chemother 1998 Feb; 41(2): 273–6
Sillerstrom E, Wahlund E, Nord CE. In vitro activity of LY 333328 against anaerobic gram-positive bacteria. J Chemother 1999 Apr; 11(2): 90–2
Goldstein EJ, Citron DM, Merriam CV, et al. In vitro activities of Dalbavancin and nine comparator agents against anaerobic gram-positive species and corynebacteria. Antimicrob Agents Chemother 2003 Jun; 47(6): 1968–71
Wilson AP. Clinical pharmacokinetics of teicoplanin. Clin Pharmacokinet 2000 Sep; 39(3): 167–83
Feketi R. Vancomycin, teicoplanin, and the streptogramins: quinupristin and dalfopristin. In: Mandell GE, Bennett JE, Dolin R, editors. Principles and practice of infectious diseases. Philadelphia (PA): Churchill Livingstone, 2000: 382–92
Rowe PA, Brown TJ. Protein binding of 14C-oritavancin [abstract no. A-2193]. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; 2001 Dec 16–18; Chicago (IL)
Steiert M, Schmitz FJ. Dalbavancin (Biosearch Italia/Versicor). Curr Opin Investig Drugs 2002 Feb; 3(2): 229–33
Dowell JA, Gottlieb AB, Van Sanders C, et al. The pharmacokinetics and renal excretion of dalbavancin in healthy subjects [abstract no. A-1386]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Cavaleri M, Cooper A, Nutley MA, et al. Protein binding of dalbavancin using isothermal titration microcalorimetry [abstract no. A-1385]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Owen J. Population pharmacokinetic model. Brisbane (CA): InterMune, Inc., 2003. (Data on file)
Thomasson HR. Study ARKK. Brisbane (CA): InterMune, Inc., 1997. (Data on file)
Lowdin E, Odenholt I, Cars O. In vitro studies of pharmacodynamic properties of vancomycin against Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob Agents Chemother 1998 Oct; 42(10): 2739–44
Houlihan HH, Stokes DP, Rybak MJ. Pharmacodynamics of vancomycin and ampicillin alone and in combination with gentamicin once daily or thrice daily against Enterococcus faecalis in an in vitro infection model. J Antimicrob Chemother 2000 Jul; 46(1): 79–86
Fantin B, Carbon C. Importance of the aminoglycoside dosing regimen in the penicillin-netilmicin combination for treatment of Enterococcus faecalis-induced experimental endocarditis. Antimicrob Agents Chemother 1990 Dec; 34(12): 2387–91
Lamp KC, Rybak MJ, Bailey EM, et al. In vitro pharmacodynamic effects of concentration, pH, and growth phase on serum bactericidal activities of daptomycin and vancomycin. Antimicrob Agents Chemother 1992 Dec; 36(12): 2709–14
Drabu YJ, Blakemore PH. The post-antibiotic effect of teicoplanin: monotherapy and combination studies. J Antimicrob Chemother 1991 Apr; 27 Suppl. B: 1–7
Chambers HF, Kennedy S. Effects of dosage, peak and trough concentrations in serum, protein binding, and bactericidal rate on efficacy of teicoplanin in a rabbit model of endocarditis. Antimicrob Agents Chemother 1990 Apr; 34(4): 510–4
Peetermans WE, Hoogeterp JJ, Hazekamp-van Dokkum AM, et al. Antistaphylococcal activities of teicoplanin and vancomycin in vitro and in an experimental infection. Antimicrob Agents Chemother 1990 Oct; 34(10): 1869–74
Craig WA. Does the dose matter? Clin Infect Dis 2001 Sep 15; 33 Suppl. 3: S233–7
Knudsen JD, Fuursted K, Raber S, et al. Pharmacodynamics of glycopeptides in the mouse peritonitis model of Streptococcus pneumoniae or Staphylococcus aureus infection. Antimicrob Agents Chemother 2000 May; 44(5): 1247–54
Cohen E, Dadashev A, Drucker M, et al. Once-daily versus twice-daily intravenous administration of vancomycin for infections in hospitalized patients. J Antimicrob Chemother 2002 Jan; 49(1): 155–60
Klepser ME, Patel KB, Nicolau DP, et al. Comparison of bactericidal activities of intermittent and continuous infusion dosing of vancomycin against methicillin-resistant Staphylococcus aureus and Enterococcus faecalis. Pharmacotherapy 1998 Sep; 18(5): 1069–74
James JK, Palmer SM, Levine DP, et al. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother 1996 Mar; 40(3): 696–700
Byl B, Jacobs F, Wallemacq P, et al. Vancomycin penetration of uninfected pleural fluid exudate after continuous or intermittent infusion. Antimicrob Agents Chemother 2003 Jun; 47(6): 2015–7
Wysocki M, Delatour F, Faurisson F, et al. Continuous versus intermittent infusion of vancomycin in severe staphylococcal infections: prospective multicenter randomized study. Antimicrob Agents Chemother 2001 Sep; 45(9): 2460–7
Basma V, Van Bambeke F, Mingeot-Leclercq MP, et al. Stability and compatibility of vancomycin for administration by continuous infusion in intensive care patients [abstract]. 14th European Congress of Clinical Microbiology and Infectious Diseases; 2004 May 1–4; Prague. In press
Cheung RP, DiPiro JT. Vancomycin: an update. Pharmacotherapy 1986 Jul; 6(4): 153–69
Livermore DM. Antibiotic resistance in staphylococci. Int J Antimicrob Agents 2000 Nov; 16 Suppl. 1: S3–10
Michel M, Gutmann L. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci: therapeutic realities and possibilities. Lancet 1997 Jun 28; 349(9069): 1901–6
Kirst HA, Thompson DG, Nicas TI. Historical yearly usage of vancomycin. Antimicrob Agents Chemother 1998 May; 42(5): 1303–4
Centers for Disease Control and Prevention. Nosocomial enterococci resistant to vancomycin: United States, 1989–1993. MMWR Morb Mortal Wkly Rep 1993 Aug 6; 42(30): 597–9
Fridkin SK, Edwards JR, Courval JM, et al. The effect of vancomycin and third-generation cephalosporins on prevalence of vancomycin-resistant enterococci in 126 US adult intensive care units. Ann Intern Med 2001 Aug 7; 135(3): 175–83
Kumana CR, Ching TY, Kong Y, et al. Curtailing unnecessary vancomycin usage in a hospital with high rates of methicillin resistant Staphylococcus aureus infections. Br J Clin Pharmacol 2001 Oct; 52(4): 427–32
Shojania KG, Yokoe D, Platt R, et al. Reducing vancomycin use utilizing a computer guideline: results of a randomized controlled trial. J Am Med Inform Assoc 1998 Nov; 5(6): 554–62
Goeckner BJ, Hendershot E, Scott K, et al. A vancomycin monitoring program at a community hospital. Jt Comm J Qual Improv 1998 Jul; 24(7): 379–85
Hamilton CD, Drew R, Janning SW, et al. Excessive use of vancomycin: a successful intervention strategy at an academic medical center. Infect Control Hosp Epidemiol 2000 Jan; 21(1): 42–5
Linden PK. Treatment options for vancomycin-resistant enterococcal infections. Drugs 2002; 62(3): 425–41
Eliopoulos GM. Quinupristin-dalfopristin and linezolid: evidence and opinion. Clin Infect Dis 2003 Feb 15; 36(4): 473–81
Olsen KM, Rebuck JA, Rupp ME. Arthralgias and myalgias related to quinupristin-dalfopristin administration. Clin Infect Dis 2001 Feb 15; 32(4): e83–6
Rubinstein E, Prokocimer P, Talbot GH. Safety and tolerability of quinupristin/dalfopristin: administration guidelines. J Antimicrob Chemother 1999 Sep; 44 Suppl. A: 37–46
Gerson SL, Kaplan SL, Brass JB, et al. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother 2002 Aug; 46(8): 2723–6
Nilius AM. Have the oxazolidinones lived up to their billing?: future perspectives for this antibacterial class. Curr Opin Investig Drugs 2003 Feb; 4(2): 149–55
Centers for Disease Control and Prevention. Recommendations for preventing the spread of vancomycin resistance: Hospital Infection Control Practices Advisory Committee (HICPAC). Infect Control Hosp Epidemiol 1995 Feb; 16(2): 105–13
Gordts B, Firre E, Jordens P, et al. National guidelines for the judicious use of glycopeptides in Belgium. Clin Microbiol Infect 2000 Nov; 6(11): 585–92
Nourse C, Byrne C, Leonard L, et al. Glycopeptide prescribing in a tertiary referral paediatric hospital and applicability of hospital infection control practices advisory committee (HICPAC) guidelines to children. Eur J Pediatr 2000 Mar; 159(3): 193–7
Gruneberg RN, Antunes F, Chambers HF, et al. The role of glycopeptide antibiotics in the treatment of infective endocarditis. Int J Antimicrob Agents 1999 Aug; 12(3): 191–8
Manley HJ, Bailie GR, Frye RF, et al. Intravenous vancomycin pharmacokinetics in automated peritoneal dialysis patients. Perit Dial Int 2001 Jul; 21(4): 378–85
Stamatiadis D, Papaioannou MG, Giamarellos-Bourboulis EJ, et al. Pharmacokinetics of teicoplanin in patients undergoing continuous ambulatory peritoneal dialysis. Perit Dial Int 2003 Mar; 23(2): 127–31
Farber BF, Moellering Jr RC. Retrospective study of the toxicity of preparations of vancomycin from 1974 to 1981. Antimicrob Agents Chemother 1983 Jan; 23(1): 138–41
Wang LS, Liu CY, Wang FD, et al. Chromatographically purified vancomycin: therapy of serious infections caused by Staphylococcus aureus and other gram-positive bacteria. Clin Ther 1988; 10(5): 574–84
Sivagnanam S, Deleu D. Red man syndrome. Crit Care 2003 Apr; 7(2): 119–20
Sorrell TC, Collignon PJ. A prospective study of adverse reactions associated with vancomycin therapy. J Antimicrob Chemother 1985 Aug; 16(2): 235–41
Elting LS, Rubenstein EB, Kurtin D, et al. Mississippi mud in the 1990s: risks and outcomes of vancomycin-associated toxicity in general oncology practice. Cancer 1998 Dec 15; 83(12): 2597–607
Rocha JL, Kondo W, Baptista MI, et al. Uncommon vancomycin-induced side effects. Braz J Infect Dis 2002 Aug; 6(4): 196–200
Wood MJ. The comparative efficacy and safety of teicoplanin and vancomycin. J Antimicrob Chemother 1996 Feb; 37(2): 209–22
Boger DL. Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med Res Rev 2001 Sep; 21(5): 356–81
Malabarba A, Nicas TI, Thompson RC. Structural modifications of glycopeptide antibiotics. Med Res Rev 1997 Jan; 17(1): 69–137
Axelsen PH, Li D. A rational strategy for enhancing the affinity of vancomycin towards depsipeptide ligands. Bioorg Med Chem 1998 Jul; 6(7): 877–81
Hancock RE, Farmer SW. Mechanism of uptake of deglucoteicoplanin amide derivatives across outer membranes of Escherichia coli and Pseudomonas aeruginosa. Antimicrob Agents Chemother 1993 Mar; 37(3): 453–6
Kenny MT, Brackman MA, Dulworth JK. In vitro activity of the semisynthetic glycopeptide amide MDL 63, 246. Antimicrob Agents Chemother 1995 Jul; 39(7): 1589–90
Goldstein BP, Candiani G, Arain TM, et al. Antimicrobial activity of MDL 63,246, a new semisynthetic glycopeptide antibiotic. Antimicrob Agents Chemother 1995 Jul; 39(7): 1580–8
Nagarajan R. Structure-activity relationships of vancomycintype glycopeptide antibiotics. J Antibiot (Tokyo) 1993 Aug; 46(8): 1181–95
Cooper RD, Snyder NJ, Zweifel MJ, et al. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J Antibiot (Tokyo) 1996 Jun; 49(6): 575–81
Rodriguez MJ, Snyder NJ, Zweifel MJ, et al. Novel glycopeptide antibiotics: N-alkylated derivatives active against vancomycin-resistant enterococci. J Antibiot (Tokyo) 1998 Jun; 51(6): 560–9
Nicas TI, Mullen DL, Flokowitsch JE, et al. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob Agents Chemother 1996 Sep; 40(9): 2194–9
Allen NE, Nicas TI. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev 2003 Jan; 26(5): 511–32
Printsevskaya SS, Pavlov AY, Olsufyeva EN, et al. Synthesis and mode of action of hydrophobic derivatives of the glycopeptide antibiotic eremomycin and des-(N-methyl-D-leucyl)eremomycin against glycopeptide-sensitive and -resistant bacteria. J Med Chem 2002 Mar 14; 45(6): 1340–7
Debabov D, Pace J, Kaniga K, et al. A novel bactericidal antibiotic inhibits bacterial lipid synthesis [abstract no. F-364]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Leadbetter M, Linsell M, Fatheree P, et al. Difunctionalized vancomycin derivatives [abstract no. F-367]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Dong SD, Oberthur M, Losey HC, et al. The structural basis for induction of VanB resistance. J Am Chem Soc 2002 Aug 7; 124(31): 9064–5
Nicolaou KC, Hughes R, Cho SY, et al. Synthesis and biological evaluation of vancomycin dimers with potent activity against vancomycin-resistant bacteria: target-accelerated combinatorial synthesis. Chemistry 2001 Sep 3; 7(17): 3824–43
Griffin J, Linsell M, Nodwell M, et al. Multivalent drug design: vancomycin dimers [abstract no. F-369]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Sun B, Chen Z, Eggert US, et al. Hybrid glycopeptide antibiotics. J Am Chem Soc 2001 Dec 19; 123(50): 12722–3
Chiosis G, Boneca IG. Selective cleavage of D-Ala-D-Lac by small molecules: re-sensitizing resistant bacteria to vancomycin. Science 2001 Aug 24; 293(5534): 1484–7
Wu Z, Walsh CT. Phosphinate analogs of D-, D-dipeptides: slow-binding inhibition and proteolysis protection of VanX, a D-, D-dipeptidase required for vancomycin resistance in Enterococcus faecium. Proc Natl Acad Sci U S A 1995 Dec 5; 92(25): 11603–7
Yang KW, Brandt JJ, Chatwood LL, et al. Phosphonamidate and phosphothioate dipeptides as potential inhibitors of VanX. Bioorg Med Chem Lett 2000 May 15; 10(10): 1085–7
InterMune Inc. [online]. Available from URL: http://www.intermune.com/ [Accessed 2004 Jan 1]
Nagarajan R. Antibacterial activities and modes of action of vancomycin and related glycopeptides. Antimicrob Agents Chemother 1991 Apr; 35(4): 605–9
Sanchez-Silos RM, Perez-Giraldo C, Blanco MT, et al. Resistance to vancomycin, LY333328, ciprofloxacin and trovafloxacin of community-acquired and nosocomial strains of Enterococcus faecalis isolated in Badajoz (Spain) with and without high-level resistance to streptomycin and gentamicin. Chemotherapy 2001 Dec; 47(6): 415–20
Arthur M, Depardieu F, Reynolds P, et al. Moderate-level resistance to glycopeptide LY333328 mediated by genes of the vanA and vanB clusters in enterococci. Antimicrob Agents Chemother 1999 Aug; 43(8): 1875–80
Aslangul E, Baptista M, Fantin B, et al. Selection of glycopeptide-resistant mutants of VanB-type Enterococcus faecalis BM4281 in vitro and in experimental endocarditis. J Infect Dis 1997 Mar; 175(3): 598–605
Wilson P, Koshy C, Minassian M. An LY333328-dependent strain of Enterococcus faecalis isolated from a blood culture. J Antimicrob Chemother 1998 Sep; 42(3): 406–7
Coyle EA, Rybak MJ. Activity of oritavancin (LY333328), an investigational glycopeptide, compared to that of vancomycin against multidrug-resistant Streptococcus pneumoniae in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 2001 Mar; 45(3): 706–9
Mercier RC, Houlihan HH, Rybak MJ. Pharmacodynamic eval-uation of a new glycopeptide, LY333328, and in vitro activity against Staphylococcus aureus and Enterococcus faecium. Antimicrob Agents Chemother 1997 Jun; 41(6): 1307–12
Hershberger E, Aeschlimann JR, Moldovan T, et al. Evaluation of bactericidal activities of LY333328, vancomycin, teico-planin, ampicillin-sulbactam, trovafloxacin, and RP59500 alone or in combination with rifampin or gentamicin against different strains of vancomycin-intermediate Staphylococcus aureus by time-kill curve methods. Antimicrob Agents Chemother 1999 Mar; 43(3): 717–21
Zelenitsky SA, Booker B, Laing N, et al. Synergy of an investigational glycopeptide, LY333328, with once-daily gentamicin against vancomycin-resistant Enterococcus faecium in a multiple-dose, in vitro pharmacodynamic model. An-timicrob Agents Chemother 1999 Mar; 43(3): 592–7
Baltch AL, Smith RP, Ritz WJ, et al. Comparison of inhibitory and bactericidal activities and postantibiotic effects of LY333328 and ampicillin used singly and in combination against vancomycin-resistant Enterococcus faecium. An-timicrob Agents Chemother 1998 Oct; 42(10): 2564–8
Mercier RC, Stumpo C, Rybak MJ. Effect of growth phase and pH on the in vitro activity of a new glycopeptide, oritavancin (LY333328), against Staphylococcus aureus and Enterococcus faecium. J Antimicrob Chemother 2002 Jul; 50(1): 19–24
Zhanel GG, Kirkpatrick ID, Hoban DJ, et al. Influence of human serum on pharmacodynamic properties of an investiga-tional glycopeptide, LY28, and comparator agents against Staphylococcus aureus. Antimicrob Agents Chemother 1998 Sep; 42(9): 2427–30
Van Bambeke F, Snoeck AS, Chanteux H, et al. Is LY333328 glycopeptide a new cell-associated antibiotic?: comparative studies with vancomycin and azithromycin in a model of J774 mouse macrophages [abstract no. 1245]. 11th European Congress of Clinical Microbiology and Infectious Diseases; 2001 Apr 1–4; Istanbul, Turkey
Kaatz GW, Seo SM, Aeschlimann JR, et al. Efficacy of LY333328 against experimental methicillin-resistant Staphylococcus aureus endocarditis. Antimicrob Agents Chemother 1998 Apr; 42(4): 981–3
Cabellos C, Fernandez A, Maiques JM, et al. Experimental study of LY333328 (oritavancin), alone and in combination, in therapy of cephalosporin-resistant pneumococcal meningitis. Antimicrob Agents Chemother 2003 Jun; 47(6): 1907–11
Gerber J, Smirnov A, Wellmer A, et al. Activity of LY333328 in experimental meningitis caused by a Streptococcus pneumoniae strain susceptible to penicillin. Antimicrob Agents Chemother 2001 Jul; 45(7): 2169–72
Rupp ME, Fey PD, Longo GM. Effect of LY333328 against vancomycin-resistant Enterococcus faecium in a rat central venous catheter-associated infection model. J Antimicrob Chemother 2001 May; 47(5): 705–7
Saleh-Mghir A, Lefort A, Petegnief Y, et al. Activity and diffusion of LY333328 in experimental endocarditis due to vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 1999 Jan; 43(1): 115–20
Lefort A, Saleh-Mghir A, Garry L, et al. Activity of LY333328 combined with gentamicin in vitro and in rabbit experimental endocarditis due to vancomycin-susceptible or -resistant Enterococcus faecalis. Antimicrob Agents Chemother 2000 Nov; 44(11): 3017–21
Al Nawas B, Swantes J, Shah PM. In vitro activity of LY333328, a new glycopeptide, against extracellular and intracellular vancomycin-resistant enterococci. Infection 2000 Jul; 28(4): 214–8
Al Nawas B, Shah PM. Intracellular activity of vancomycin and LY333328, a new semisynthetic glycopeptide, against methicillin-resistant Staphylococcus aureus. Infection 1998 May; 26(3): 165–7
Seral C, Van Bambeke F, Tulkens PM. Quantitative analysis of the activity of antibiotics (gentamicin, azithromycin, telithromycin, ciprofloxacin, moxifloxacin, oritavancin [LY333328]) against intracellular Staphylococcus aureus in mouse J774 macrophages. Antimicrob Agents Chemother 2003; 47(7): 2283–92
Loutit JS. Mode of action and current status of the glycopeptide oritavancin [abstract no. 617]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Wasilewski MM, Disch DP, McGill JM, et al. Equivalence of shorter course therapy with oritavancin vs vancomycin/ cephalexin in complicated skin/skin structure infections [abstract no. UL-18]. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; 2001 Dec 16–18; Chicago (IL)
Barrett JF. Oritavancin: Eli Lilly & Co. Curr Opin Investig Drugs 2001 Aug; 2(8): 1039–44
Van Bambeke F, Saffian J, Mingeot-Leclercq MP, et al. Oritavancin glycopeptide causes a lipid storage disorder and mixed morphological alterations of the vacuolar system in cultured rat embryo fibroblasts (FB) and J774 mouse macrophages (M) [abstract no. 2180]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Vicuron Pharmaceuticals, Inc. [online]. Available from URL: http://www.versicor.com/ [Accessed 2004 Jan 1]
Malabarba A, Ciabatti R, Kettenring J, et al. Amides of deacetylglucosaminyl-deoxy teicoplanin active against highly glycopeptide-resistant enterococci: synthesis and antibacterial activity. J Antibiot (Tokyo) 1994 Dec; 47(12): 1493–506
Malabarba A, Ciabatti R, Scotti R, et al. New semisynthetic glycopeptides MDL 63,246 and MDL 63,042, and other amide derivatives of antibiotic A-40,926 active against highly glycopeptide-resistant VanA enterococci. J Antibiot (Tokyo) 1995 Aug; 48(8): 869–83
Lopez S, Hackbarth CJ, White R, et al. In vitro susceptibility and population analysis of staphylococci after serial passage at sub-MIC levels of dalbavancin and other glycopeptides [abstract no. P1539]. 13th European Congress of Clinical Microbiology and Infectious Diseases; 2003 May 10–13; Glasgow
Candiani GP, Romano G, Brunati C, et al. Efficacy of a single dalbavancin dose compared with multiple linezolid doses against penicillin-resistant pneumococci in a lobar pneumonia model in the immunocompetent rat [abstract no. 989]. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy; 2001 Dec 16–18; Chicago (IL)
Lefort A, Pavie J, Garry L, et al. Activity of dalbavancin (BI 397) in vitro and in experimental endocarditis due to methicillin-resistant Staphylococcus aureus (MRSA) susceptible or intermediate to glycopeptides (GISA) [abstract no. B-278]. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002 Sep 27–30; San Diego (CA)
Seltzer E, Dorr MB, Goldstein BP, et al., for the Dalbavancin Skin and Soft-Tissue Infection Study Group. Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis 2003 Nov 15; 37(10): 1298–303
Dowell JA, Seltzer E, Stogniew M, et al. Dalbavancin dosage adjustments not required for patients with mild renal impairment [abstract no. P1224]. 13th European Congress of Clinical Microbiology and Infectious Diseases; 2003 May 10–13; Glasgow, UK
Felmingham D, Reinert RR, Hirakata Y, et al. Increasing prevalence of antimicrobial resistance among isolates of Streptococcus pneumoniae from the PROTEKT surveillance study, and comparative in vitro activity of the ketolide, telithromycin. J Antimicrob Chemother 2002 Sep; 50 Suppl. S1: 25–37
Vandecasteele SJ, Verhaegen J, Colaert J, et al. Failure of cefotaxime and meropenem to eradicate meningitis caused by an intermediately susceptible Streptococcus pneumoniae strain. Eur J Clin Microbiol Infect Dis 2001 Oct; 20(10): 751–2
Buckingham SC, Davis Y, English BK. Pneumococcal susceptibility to meropenem in a mid-south children’s hospital. South Med J 2002 Nov; 95(11): 1293–6
Acknowledgements
F. Van Bambeke is Chercheur Qualifié of the Belgian Fonds National de la Recherche Scientifique. The authors have provided no information on sources of funding or on conflicts of interest directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van Bambeke, F., Van Laethem, Y., Courvalin, P. et al. Glycopeptide Antibiotics. Drugs 64, 913–936 (2004). https://doi.org/10.2165/00003495-200464090-00001
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003495-200464090-00001