
Summary
Synopsis
Tinzaparin, a low molecular weight (LMW) heparin with an average molecular weight of 4.5 ± 1.5kD, has greater bioavailability and a longer duration of action than unfractionated heparin, allowing it to be administered once daily by subcutaneous injection for both prophylaxis and treatment of deep venous thrombosis (DVT). Like other members of its class, tinzaparin has a greater ratio of anti-factor Xa/anti- f actor IIa activity than unfractionated heparin, providing the theoretical advantage of similar antithrombotic efficacy with a diminished risk of haemorrhagic complications.
In a small number of clinical trials, tinzaparin was found to be more effective than intravenous dextran or oral warfarin sodium as prophylaxis against DVT in high-risk patients undergoing orthopaedic surgery, and more effective than subcutaneous heparin in both general surgical patients and medical patients with an immobilising illness. When used for the treatment of established DVT, tinzaparin was more effective in preventing DVT recurrence than intravenous heparin, both initially and during a 3- month follow-up period when patients received warfarin sodium. Tinzaparin was also used successfully in one small study to maintain the patency of haemodialysis circuits over a 6- month period, with favourable effects on the lipid profile of such patients.
Tinzaparin is well tolerated, the most frequent complication being injection site haematoma. In comparative trials, tinzaparin was associated with fewer major haemorrhagic complications than intravenous heparin (when used for treatment of venous thrombosis), but more than warfarin sodium. Other adverse events which have been reported in tinzaparin-treated patients include elevated liver enzyme levels and thrombocytopenia.
Thus, although clinical experience is limited at present, available data suggest that, in common with other LMW heparins, tinzaparin is likely to prove an effective alternative to unfractionated heparin for both the prevention and treatment of DVT, with the advantage of more convenient administration and decreased monitoring requirements.
Pharmacodynamic Properties
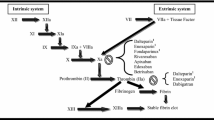
Tinzaparin is a low molecular weight (LMW) heparin formed by enzymatic depolymerisation of heparin. It has an average molecular weight of 4.5 ± 1.5kD. Its anti-factor Xa activity is 75 IU/mg and anti-factor IIa activity 50 IU/mg, with reference to the First International Standard for LMW heparins. LMW heparins vary in their content and structure and relative proportions of high and low molecular weight heparin molecules. These differences result in differing anti-factor Xa and anti-factor IIa activities and probably explain the differing antithrombotic, haemorrhagic and pharmacokinetic profiles observed in clinical practice. In common with other LMW heparins, tinzaparin has a greater anti-factor Xa/anti-factor IIa activity ratio than unfractionated heparin, and a less marked effect than heparin on thrombus formation in animal models. Tinzaparin demonstrated reduced activity relative to heparin in delaying production of thrombi ex vivo in blood samples from volunteers. LMW heparins may increase transport of lipoprotein lipase from extrahepatic sites to the liver, and tinzaparin was reported to have a beneficial effect on the lipid profile when substituted for heparin in patients undergoing haemodialysis. In common with other LMW heparins, tinzaparin has a lesser effect on platelet activation and is also less affected by platelet factor 4 than heparin. It also appears to act synergistically with tissue factor pathway inhibitor.
Unlike that of heparin, the anti-factor Xa activity of tinzaparin cannot be fully neutralised by a single dose of protamine sulfate, as observed with other LMW heparins.
Pharmacokinetic Properties
As with other LMW heparins, the pharmacokinetic properties of tinzaparin have been determined indirectly by measurement of anti-factor Xa and anti-factor IIa activities in plasma. Following subcutaneous injection, peak plasma anti-factor Xa and anti-factor IIa activities are reached after approximately 4 to 6 hours and are dose dependent within the range of tinzaparin doses in clinical use. The overall bioavailability, based on anti-factor Xa and anti-factor IIa activity, respectively, is 90% and 67%.
Peak plasma anti-factor Xa activity progressively increases when tinzaparin is administered for several days, but trough levels essentially return to baseline, indicating the absence of accumulation. The apparent volume of distribution is 3.9L for anti-factor Xa activity and 10.1L for anti-factor IIa activity. Tinzaparin does not cross the placenta during the second trimester of pregnancy, but otherwise the pattern and extent of tissue distribution in humans has not been reported.
Unlike that of heparin, the elimination of tinzaparin and other LMW heparins appears to occur primarily by an unsaturable renal mechanism. Data in humans are not available, but studies in the dog have shown that 80 to 90% of administered tinzaparin is recovered in the urine and 1 to 2% in the faeces. The elimination half-life of anti-factor Xa activity in volunteers after subcutaneous administration of tinzaparin was 82 minutes. The effects of renal or hepatic disease on the pharmacokinetics of tinzaparin are unknown.
Therapeutic Trials
In a small number of clinical trials, subcutaneous tinzaparin, administered once daily, was found to reduce the incidence of deep venous thrombosis (DVT) in both surgical and medical patients. In patients undergoing hip surgery, tinzaparin 50 IUaXa/kg/day was significantly more effective than placebo when used in conjunction with early mobilisation (4 days after surgery) and more effective than intravenous dextran (incidence of DVT 17.1 vs 28.7%). In a comparative study involving 1436 patients, tinzaparin 75 IUaXa/kg was of similar efficacy to oral warfarin in patients undergoing hip surgery, and more effective than warfarin in those undergoing knee surgery, although a high incidence of DVT was noted with both regimens in the latter setting (incidence of DVT 45 vs 54.9%). Tinzaparin was associated with more major haemorrhagic complications (2.8 vs 1.2% of patients) and wound haematomas (6.7 vs 3.6%) than warfarin.
In patients undergoing general surgery, tinzaparin 3500 IUaXa or 50 IUaXa/kg per day was reported to be effective in decreasing the incidence of DVT and tinzaparin 3500 IUaXa daily was shown to be equivalent to standard treatment with subcutaneous unfractionated heparin (5000IU twice daily) in patients with specific risk factors for developing DVT (incidence of DVT 2.6 vs 3.5%).
In a single study in medical patients at risk of developing DVT because of immobilising spinal cord injury, tinzaparin (3500 IUaXa daily) was more effective than heparin (5000IU 3 times daily) and tended to produce less bleeding when administered over an 8-week period.
Tinzaparin 150 or 175 IUaXa/kg/day was reported to prevent progression or recurrence of established DVT in 2 studies and the higher dose was more effective than intravenous adjusted-dose heparin, both in the immediate treatment period and during a 3-month follow-up period when patients had been switched to oral warfarin (2.8 vs 6.9% of patients). Furthermore, tinzaparin was associated with less major bleeding than heparin during initial treatment.
When substituted for heparin to maintain the patency of haemodialysis lines, tinzaparin 40 IUaXa/kg was shown in one small trial to have possible advantages over heparin with respect to its effects on lipoproteins.
Tolerability
Haemorrhagic complications with tinzaparin have not been a major problem in clinical trials to date. Although both blood loss and transfusion requirements were higher relative to placebo in patients undergoing hip surgery, none of the findings were considered to be clinically important. In a small number of comparative trials, tinzaparin was associated with a similar incidence of haemorrhagic complications to subcutaneous heparin or intravenous dextran, but more than warfarin sodium. Larger doses of tinzaparin (175 IUaXa/kg daily) used for treatment of established DVT produced significantly fewer haemorrhagic complications than did intravenous heparin (0.5 vs 5.0% of patients) during initial treatment, although delayed bleeding during long term follow up (after patients were switched to oral warfarin) was more common in tinzaparin recipients.
The most common adverse event during tinzaparin treatment has been the development of haematomas at the injection site. These appeared to be dose related, occurring in 1.2% of patients receiving tinzaparin 2500 IUaXa daily, 4.9% of patients receiving 3500 IUaXa daily and up to 80% of patients receiving 150 IUaXa/kg daily. Haematomas occurring with the largest dose of tinzaparin rarely exceeded lcm in diameter and in no case did a local haematoma require discontinuation of treatment.
Thrombocytopenia was reported in approximately 2% of patients in 2 studies. Transient increases in aspartate aminotransferase and alkaline phosphatase activities were reported in 35% and 16.5% of patients, respectively, receiving 50 IUaXa/kg tinzaparin as prophylaxis in hip replacement surgery.
Dosage and Administration
For the prevention of DVT in high-risk surgical patients, the usual dose of tinzaparin is 50 IUaXa/kg body weight, administered subcutaneously 2 hours prior to surgery, then daily for 7 to 10 days. Patients at moderate risk of developing DVT may receive 3500 IUaXa. A daily dose of 175 IUaXa/kg tinzaparin, commenced as soon as possible after diagnosis and continued until stabilisation of warfarin therapy, has proved effective in the treatment of established DVT.
Similar content being viewed by others


References
Nielsen JI, Ostergaard P. Chemistry of heparin and low molecular weight heparin. Acta Chirurg Scand 1988; Suppl. 543: 52–6
Barrowcliffe TW, Curtis AD, Johnson EA, et al. An international standard for low molecular weight heparin. Thromb Haemost 1988; 60: 1–7
Furie B, Furie BC. Molecular and cellular biology of blood coagulation. N Engl J Med 1992; 326: 800–6
Verstraete M. Haemostasis and thrombosis. In: Julian DG, et al., editors. Diseases of the heart, 2nd ed. London: WB Saunders, in press
Colman RW, Scott CF, Pixley RA, et al. Effect of heparin on the inhibition of the contact system enzymes. Ann NY Acad Sci 1989; 556: 95–103
Hirsh J, Levine MN. Low molecular weight heparin. Blood 1992; 79: 1–17
Wolf H. Low-molecular-weight heparin. Med Clin N Am 1994; 78: 733–43
Hemker HC. The mode of action of heparin in plasma. In: Verstraete et al., editors. Thrombosis and haemostasis. Leuven: International Society on Thrombosis and Haemostasis and Leuven University Press, 1987
Linhardt RJ, Loganathan D, Al-Hakim A, et al. Oligosaccharide mapping of low molecular weight heparins: structure and activity differences. J Med Chem 1990; 33: 1639–45
Ostergaard PB, Nilsson B, Bergqvist D, et al. The effect of low molecular weight heparin on experimental thrombosis and haemostasis — the influence of production method. Thromb Res 1987; 45: 739–49
Fareed J, Walenga JM, Hoppensteadt D, et al. Biochemical and pharmacologic inequivalence of low molecular weight heparins. Ann NY Acad Sci 1989; 556: 333–53
Abildgaard U, Norrheim L, Larsen AE, et al. Monitoring therapy with LMW heparin: a comparison of three chromogenic substrate assays and the Heptest clotting assay. Haemostasis 1990; 20: 193–203
Ofosu FA, Smith LM, Anvari N, et al. An approach to assigning in vitro potency to unfractionated and low molecular weight heparins based on the inhibition of prothrombin activation and catalysis of thrombin inhibition. Thromb Haemost 1988; 60: 193–8
Fareed J, Walenga JM, Racanelli A, et al. Validity of the newly established low-molecular-weight heparin standard in cross-referencing low-molecular-weight heparins. Haemostasis 1988; 18 Suppl. 3: 33–47
Doutremepuich C, Guyot M, Laianne MC, et al. Comparative pharmacology of low molecular weight heparins: implications of manufacturing processes on biological effects. Thromb Res 1989; 55: 419–26
Mätzsch T, Bergqvist D, Hedner U, et al. Effects of enzymati-cally depolymerized heparin as compared with conventional heparin in healthy volunteers. Thromb Haemost 1987; 57: 97–101
Leizorovicz A, Bara L, Samama MM, et al. Factor Xa inhibition: correlation between the plasma levels of anti-Xa activity and occurrence of thrombosis and haemorrhage. Haemostasis 1993; 23 Suppl. 1: 89–98
Mätzsch T, Bergqvist D, Fredin H, et al. Influence of a low molecular weight heparin on the inhibition of factor Xa and thrombin in hip surgery. Thromb Res 1989; 56: 559–64
Dhillon H, Walenga J, Hoppensteadt D, et al. Studies of the mechanism of the antithrombotic actions of a low molecular weight heparin (Logiparin®) [abstract 2803]. Federation of American Societies for Experimental Biology, New Orleans, Mar 28–Apr 1, 1993: A483
Padilla A, Gray E, Pepper DS, et al. Inhibition of thrombin generation by heparin and low molecular weight (LMW) heparins in the absence and presence of platelet factor 4 (PF4). Br J Haematol 1992; 82: 406–13
Sorensen JV, Borris LC, Lassen MR, et al. Levels of thrombin-antithrombin-III complex and factor VIII activity in relation to post-operative deep vein thrombosis and influence of prophylaxis with a low-molecular-weight heparin. Blood Coagul Fibrinol 1990; 1: 389–92
Sorensen JV, Lassen MR, Borris LC, et al. Reduction of plasma levels of prothrombin fragments 1 and 2 during thrombo-prophylaxis with a low-molecular-weight heparin. Blood Coagul Fibrinol 1992; 3: 55–9
Lindahl AK, Sandset PM, Abildgaard U. The present status of tissue factor pathway inhibitor. Blood Coagul Fibrinol 1992; 3: 439–49
Rapaport SI. The extrinsic pathway inhibitor. A regulator of tissue factor-dependent blood coagulators. Thromb Haemost 1991; 66: 6–16
Abildgaard U. Heparin/low molecular weight heparin and tissue factor pathway inhibitor. Haemostasis 1993; 23 Suppl. 1: 103–6
Valentin S, Ostergaard P, Kristensen H, et al. Synergism between full length TFPI and heparin: evidence for TFPI as an important factor for the antithrombotic activity of heparin. Blood Coagul Fibrinol 1992; 3: 221–2
Kristensen HI, Ostergaard PB, Nordfang O, et al. Effect of tissue factor pathway inhibitor (TFPI) in the Heptest® assay and in an amidolytic anti factor Xa assay for LMW heparin. Thromb Haemost 1992; 68: 310–4
Verstraete M, Boogaerts MA. Haematological disorders. In: Speight TM, editor. Avery’s drug treatment, 3rd ed. Auckland: ADIS Press, 1987: 958–1022
Lijnen HR, Collen D. Interactions between heparin and fibrinolysis. In: Bounameaux (editor). Low-molecular-weight heparins in prophylaxis and therapy of thromboembolic disease. New York: Marcel Dekker, 1994: 143–67
Neerstrand H, Ostergaard P, Bergqvist D, et al. TPA inhibitor, TPA: Ag, plasminogen, and α2-antiplasmin after low molecular weight heparin or standard heparin. Fibrinolysis 1987; 1: 39–43
Johnston RV, Orr M, Rumley A, et al. A study of the anti-throm-botic potential of low molecular weight heparin LHN-1 (Novo) in normal volunteers [abstract]. XIth International Congress on Thrombosis and Haemostasis. Thromb Haemost 1987; 58: 119
Fareed J, Breddin K, Murphy R, et al. Profibrinolytic actions of low molecular weight heparins: possible contribution in the mediation of the antithrombotic actions in man [abstract 135]. 10th International Congress on Fibrinolysis, Indianapolis, Aug 4–8, 1990. Fibrinolysis 1990; 4 Suppl. 3: 52
Hauch O, Jorgensen LN, Kolle TR, et al. Plasma cross-linked fibrin degradation products fraction D in patients undergoing elective abdominal surgery. Thromb Res 1988; 51: 385–9
Lassen M, Weber S, Murphy R, et al. Molecular markers of fibrinolytic activation during low molecular weight heparin (Logiparin) prophylaxis of deep venous thrombosis after total hip replacement [abstract 137]. 10th International Congress on Fibrinolysis, Indianapolis, Aug 4–8, 1990. Fibrinolysis 1990; 4 Suppl. 3: 52
Sorensen JV, Borris LC, Lassen MR, et al. Association between plasma levels of tissue plasminogen activator and postoperative deep vein thrombosis — influence of prophylaxis with a low molecular weight heparin. Thromb Res 1990; 59: 131–8
Bergqvist D, Nilsson B, Hedner U, et al. The effect of heparin fragments of different molecular weights on experimental thrombosis and haemostasis. Thromb Res 1985; 38: 589–601
Diness V, Nielsen JI, Pedersen PC, et al. A comparison of the antithrombotic and haemorrhagic effects of a low molecular weight heparin (LHN-1) and conventional heparin. Thromb Haemost 1986; 55: 410–4
Holst J, Lindblad B, Bergqvist D, et al. Antithrombotic properties of a truncated recombinant tissue factor pathway inhibitor in an experimental venous thrombosis model. Haemostasis 1993; 23 Suppl. 1: 112–7
Carrie D, Caranobe C, Boneu B. A comparison of the antithrombotic effects of heparin and of low molecular weight heparins with increasing antifactor Xa/antifactor IIa ratio in the rabbit. BrJ Haematol 1993; 83: 622–6
Harenberg J, Gnasso A, de Vries JX, et al. Inhibition of low molecular weight heparin by protamine chloride in vivo. Thromb Res 1985; 38: 11–20
Holmer E, Söderström G. Neutralization of unfractionated heparin and a low molecular weight (LMW) heparin fragment by protamine [abstract]. Thromb Haemost 1983; 50: 103
Hubbard AR, Jennings CA. Neutralization of heparin sulphate and low molecular weight heparin by protamine. Thromb Haemost 1985; 53: 86–9
Diness V, Ostergaard PB. Neutralization of a low molecular weight heparin (LHN-1) and conventional heparin by protamine sulfate in rats. Thromb Haemost 1986; 56: 318–22
Holst J, Lindblad B, Bergqvist D, et al. Partial in vivo neutralization of LMWH (Logiparin®) given IV and SC in thromboprophylactic and therapeutic doses by protamine sulphate to healthy volunteers [abstract C103]. Thromb Res 1992; 65 Suppl. 1: S54
Sobel M, Adelman B. Characterization of platelet binding of heparins and other glycosaminoglycans. Thromb Res 1988; 50: 815–26
Norrheim L, Abildgaard U, Larsen ML, et al. Involvement of the extrinsic pathway in the activities of low molecular weight heparins. Thromb Res 1991; Suppl. 14: 19–27
Olivecrona T, Bengtsson-Olivecrona G, Ostergaard P, et al. New aspects on heparin and lipoprotein metabolism. Haemostasis 1993; 23 Suppl. 1: 150–60
Liu G, Bengtsson-Olivecrona G, Ostergaard P, et al. Low-Mr heparin is as potent as conventional heparin in releasing lipo-protein lipase, but is less effective in preventing hepatic clearance of the enzyme. Biochem J 1991; 273: 747–52
Liu G, Hultin M, Ostergaard P, et al. Interaction of size-fractionated heparins with lipoprotein lipase and hepatic lipase in the rat. Biochem J 1992; 285: 731–6
Akiba T, Tachibana K, Ozawa K, et al. Long-term use of low molecular weight heparin ameliorates hyperlipidemia in patients on hemodialysis. ASAIO Journal 1992; 38: M326–30
Mätzsch T, Bergqvist D, Hedner U, et al. Effects of low molecular weight heparin and unfragmented heparin on induction of osteoporosis in rats. Thromb Haemost 1990; 63: 505–9
Pedersen PC, Ostergaard PB, Hedner U, et al. Pharmacokinetics of a low molecular weight heparin, Logiparin, after intravenous and subcutaneous administration to healthy volunteers. Thromb Res 1991; 61: 477–87
Siegbahn A, Y-Hassan S, Boberg J, et al. Subcutaneous treatment of deep venous thrombosis with low molecular weight heparin: a dose finding study with LMWH-Novo. Thromb Res 1989; 55: 767–78
Brindley CJ, Taylor T, Diness V, et al. Relationship between pharmacokinetics and pharmacodynamics of tinzaparin (Logiparin), a low molecular weight heparin, in dogs. Xenobiotica 1993; 23: 575–88
Mätzsch T, Bergqvist D, Bergqvist A, et al. No transplacental passage of standard heparin or an enzymatically depoly-merized low molecular weight heparin. Blood Coagul Fibrinol 1991; 2: 273–8
Omri A, Delaloye JF, Andersen H, et al. Low molecular weight heparin Novo (LHN-1) does not cross the placenta during the second trimester of pregnancy. Thromb Haemost 1989; 61: 55–6
Boneu B, Caranobe C, Sié P. Pharmacokinetics of heparin and low molecular weight heparin. Bail Clin Haematol 1990; 3: 531–44
de Swart CAM, Nijmeyer B, Roelefs JMM, et al. Kinetics of intravenously administered heparin in humans. Blood 1982; 60: 1251–8
Kandrotas R. Heparin pharmacokinetics and pharmacodynamics. Clin Pharmacokinet 1992; 22: 359–74
Bouchelouche PN, Ostergaard PB, Mortensen SA, et al. Comparative pharmacokinetics of a low molecular weight heparin (LHN-1, Novo) and conventional heparin after intravenous injection [abstract]. Thromb Res 1986; Suppl. 6: 89
Barradell LB, Buckley MM. Nadroparin calcium: a review of its pharmacology and clinical applications in the prevention and treatment of thromboembolic disorders. Drugs 1992; 44: 858–88
Buckley MM, Sorkin EM. Enoxaparin: a review of its pharmacology and clinical applications in the prevention and treatment of thromboembolic disorders. Drugs 1992; 44: 465–97
Verstraete M. The diagnosis and treatment of deep-vein thrombosis. N Engl J Med 1993; 329: 1418–20
Hull R, Raskob G, Pineo G, et al. A comparison of subcutaneous low-molecular-weight heparin with warfarin sodium for prophylaxis against deep-vein thrombosis after hip or knee implantation. N Engl J Med 1993; 329: 1370–6
Lassen MR, Borris LC, Christiansen HM, et al. Prevention of thromboembolism in 190 hip arthroplasties: comparison of LMW heparin and placebo. Acta Orthopaed Scand 1991; 62: 33–8
Mätzsch T, Bergqvist D, Fredin H, et al. Low molecular weight heparin compared with dextran as prophylaxis against thrombosis after total hip replacement. Acta Chirurg Scand 1990; 156: 445–50
Mätzsch T, Bergqvist D, Fredin H, et al. Comparison of the thromboprophylactic effect of a low molecular weight heparin versus dextran in total hip replacement. Thromb Haemorrhagic Disord 1991; 3: 25–9
Hauch O, Jorgensen LN, Kolle TR, et al. Low molecular weight heparin (Logiparin®) as thromboprophylaxis in elective abdominal surgery: a dose finding study. Acta Chirurg Scand 1988; Suppl. 543: 90–5
Leizorovicz A, Picolet H, Peyrieux JC, et al. Prevention of peri-operative deep vein thrombosis in general surgery: a multi-centre double blind study comparing two doses of Logiparin and standard heparin. Br J Surg 1991; 78: 412–6
Borris L, Lassen M, Nehen A-M, et al. Impact of interobserver variation in the interpretation of phlebographic examinations in a clinical trial [abstract]. International Symposium on Low Molecular Weight Heparins and Related Polysaccharides, Munich, Germany, Jun 27–28, 1991
Anon. Consensus conference. Prevention of venous thrombosis and pulmonary embolism. JAMA 1986; 256: 744–9
Clagett GP, Anderson FA, Levine MN, et al. Prevention of venous thromboembolism. Chest 1992; 102: 391–407
Green D. Prevention of thromboembolism after spinal cord injury. Semin Thromb Hemost 1991; 17: 347–50
Green D, Lee MY, Lim AC, et al. Prevention of thromboembolism after spinal cord injury using low-molecular-weight heparin. Ann Intern Med 1990; 113: 571–4
Green D, Chen D, Chmiel JS, et al. Prevention of thromboembolism in spinal cord injury. Role of low molecular weight heparin. Arch Phys Med Rehabil 1994; 75: 290–2
Cziraky MJ, Spinler SA. Low-molecular-weight heparins for the treatment of deep-vein thrombosis. Clin Pharm 1993; 12: 892–9
Hull RD, Raskob GE, Pineo GF, et al. Subcutaneous low-molecular-weight heparin compared with continuous intravenous heparin in the treatment of proximal-vein thrombosis. N Engl J Med 1992; 326: 975–82
US Renal Data System. USRDS annual data report. National Institutes of Health, National Institutes of Diabetes and Digestive and Kidney Diseases, Bethesda, 1990
Christiansen HM, Lassen MR, Borris LC, et al. Biological tolerance of Logiparin, a low molecular weight heparin used in patients undergoing total hip replacement. Semin Thromb Hemost 1991; 17 Suppl. 2: 224–7
Christiansen HM, Lassen MR, Borris LC, et al. Biologic tolerance of two different low molecular weight heparins. Semin Thromb Hemost 1991; 17: 450–4
Handeland GF, Abildgaard U, Holm HA, et al. Dose adjusted heparin treatment of deep venous thrombosis: a comparison of unfractionated and low molecular weight heparin. Eur J Clin Pharmacol 1990; 39: 107–12
Author information
Authors and Affiliations
Additional information
Various sections of the manuscript reviewed by: T. Akiba, Department of Internal Medicine, Tokyo Medical and Dental University, Tokyo, Japan; J.F Cade, Intensive Care Department, Royal Melbourne Hospital, Melbourne, Victoria, Australia; D. Green, Northwestern University Atherosclerosis Program, Rehabilitation Institute of Chicago, Chicago, Illinois, USA; J. Hirsh, Hamilton Civic Hospitals Research Centre, Henderson Hospital, Hamilton, Ontario, Canada; R.D. Hull, Department of Medicine, University of Calgary, Calgary, Alberta, Canada; T. Mätzsch, Department of Surgery, University of Lund, Malmö General Hospital, Malmö, Sweden; E.A. Ofosu, Department of Pathology, McMaster University, Hamilton, Ontario, Canada; M. Verstraete, Center for Molecular and Vascular Biology, Catholic University of Leuven, Leuven, Belgium.
Rights and permissions
About this article
Cite this article
Friedel, H.A., Balfour, J.A. Tinzaparin. Drugs 48, 638–660 (1994). https://doi.org/10.2165/00003495-199448040-00010
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003495-199448040-00010