
Abstract
Although hyperglycemia is common in patients with acute myocardial infarction (MI), the underlying mechanisms are largely unknown. Insulin signaling plays a key role in the regulation of glucose homeostasis. In this study, we test the hypothesis that rapid alteration of insulin signaling pathways could be a potential contributor to acute hyperglycemia after MI. Male rats were used to produce MI by ligation of the left anterior descending coronary artery. Plasma glucose and insulin levels were significantly higher in MI rats than those in controls. Insulin-stimulated tyrosine phosphorylation of insulin receptor substrate 1 (IRS1) was reduced significantly in the liver tissue of MI rats compared with controls, followed by decreased attachment of phosphatidylinositol 3-kinase (PI3K) p85 subunit with IRS1 and Akt phosphorylation. However, insulin-stimulated signaling was not altered significantly in skeletal muscle after MI. The relative mRNA levels of phosphoenolpyruvate carboxykinase (PEPCK) and G6Pase were slightly higher in the liver tissue of MI rats than those in controls. Rosiglitazone (ROSI) markedly restored hepatic insulin signaling, inhibited gluconeogenesis and reduced plasma glucose levels in MI rats. Insulin resistance develops rapidly in liver but not skeletal muscle after MI, which contributes to acute hyperglycemia. Therapy aimed at potentiating hepatic insulin signaling may be beneficial for MI-induced hyperglycemia.
Similar content being viewed by others

Introduction
Hyperglycemia is a common pathological feature in patients with acute myocardial infarction (MI), even in the absence of established diabetes mellitus (1,2). Acute elevation of plasma glucose is related causally to adverse outcomes after MI in both the thrombolytic and percutaneous coronary intervention (PCI) eras. Although acute hyperglycemia usually has been treated with intensive insulin therapy or glucose-insulin-potassium infusion, the benefits of these strategies are not always observed in clinical studies (2–4). In addition, the optimal glucose control level is still inconclusive to date. To resolve these problems in glucose management, it is wise to understand the molecular mechanisms by which acute hyperglycemia develops after MI.
Insulin acts as a key regulator of plasma glucose homeostasis, which increases glucose uptake in peripheral tissues such as skeletal muscle and inhibits hepatic glucose production through activating insulin signaling pathways (5,6). Insulin binding to its specific insulin receptor (IR) leads to its rapid tyrosine phosphorylation, which permits recruitment of adaptor proteins such as insulin receptor substrates (IRSs), thereby activating the downstream signaling pathways (7,8). The main pathways stimulated by insulin are the IR/IRSs/phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways. If impaired, they would result in insulin resistance.
There has been evidence indicating that insulin resistance developed in insulin-responsive tissues in chronic diseases such as obesity and type 2 diabetes, including downregulated expression of insulin signaling proteins and decreased tyrosine phosphorylation of these proteins (7,9,10). However, it is unknown what happens to the insulin signaling proteins in the setting of MI and whether rapid alteration of insulin signaling is the potential contributor to acute hyperglycemia after MI.
In this study, we investigated insulin signaling pathways in insulin responsive tissues with the aim to clarify the potential mechanisms of hyperglycemia after MI. Our results demonstrated that impaired insulin signaling rapidly developed in liver tissue but not skeletal muscle, which might lead to hyperglycemia through increased hepatic gluconeogenesis after MI.
Materials and Methods
Experimental Animals
Eight-week-old male Sprague Dawley rats weighing 200 ± 20 g were purchased from the experimental animal center of Shandong University (Jinan, China). The rats were housed at constant temperature with a 12-h light:dark cycle and allowed free access to standard diet plus water. All animal experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (11) and were approved by the Institutional Animal Care and Use Committee of Shandong University.
Animals Model of MI
Rats were anesthetized with intraperitoneal (IP) administration of 30 mg/kg pentobarbital sodium before surgery. Left thoracotomy was performed in the fourth intercostal space. MI was produced by ligation of the left anterior descending (LAD) coronary artery. Sham control rats underwent the same surgical procedure without ligation. Myocardial infarct area was determined by 2,3,5-triphenyltetrazolium (TTC) staining as described previously (12).
Unless otherwise indicated, MI and control rats were assigned randomly to receive an intraperitoneal injection of either insulin (1 U/kg) or saline at the end points (30 min after ligation or sham surgery). Five minutes after the injection, liver, left quadriceps femoris and ischemic left and right cardiac ventricles were removed rapidly, snap-frozen in liquid nitrogen and stored at −80°C until analysis.
For rosiglitazone (ROSI) treatment, rats were administered with i.p. injection of ROSI (4 mg/kg) 10 min before ligation and all of them received an insulin injection before being killed.
Blood Analysis
During the experimental procedure, blood was collected from the jugular vein of the rats at the indicated time points. Blood was placed at room temperature, allowed to coagulate and then centrifuged at 5,000g for 15 min. The resulting plasma was stored at −80°C until analysis.
Plasma glucose levels were determined by glucose meter (LifeScan, Milpitas, CA, USA). Plasma creatine phosphokinase (CPK) levels and insulin levels were determined by enzymelinked immunosorbent assay (ELISA) (Millipore, Billerica, MA, USA). Tumor necrosis factor-α (TNF-α) levels also were measured by ELISA (R&D Systems, Minneapolis, MN, USA). Plasma cholesterol and triglyceride levels were measured by colorimetric assay (Roche Diagnostics, Mannheim, Germany). Plasma free fatty acid (FFA) levels were measured by an enzymatic colorimetric method (Wako Chemicals, Neuss, Germany). All the measurements were done in triplicate.
Immunoprecipitation and Western Blot Analysis
The frozen tissues were homogenized with a Polytron homogenizer in lysis buffer containing 20 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 0.1% SDS and 1% protease inhibitor cocktail. Protein concentrations were determined using the BCA method (Beyotime, Haimen, China).
For immunoprecipitation, 1 mg protein was incubated with anti-IRS1 overnight. The resulting immune complexes were absorbed to Protein A/G-agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for another 4 h and washed three times with lysis buffer. Immunoprecipitated proteins were immunoblotted with anti-IRS1, anti-phospho-tyrosine (P-Tyr-100) and anti-PI3K p85 subunit, respectively (Cell Signaling Technology, Beverly, MA, USA).
For Western blot, 30 µg protein was separated by SDS-PAGE and then transferred to PVDF membrane as described previously (13). The transfers were incubated with anti-IR β-subunit, anti-tyrosine-phospho-IR β-subunit and anti-Akt and anti-phospho-Akt (ser 473) respectively (Cell Signaling Technology), followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Protein bands were visualized by enhanced chemiluminescence (ECL) and quantified by densitometry using Image-J software.
Quantitative Real-time PCR
Total RNA was extracted by homogenizing frozen liver tissues in TRIzol reagent (Invitrogen [Thermo Fisher Scientific Inc., Waltham, MA, USA]) according to the manufacturer’s instruction. First-strand cDNA was synthesized from total RNA using the high capacity reverse transcription kit (Invitrogen [Thermo Fisher Scientific]). Quantitative RT-PCR assay was performed using cDNA (2 µl) and SYBR green (Life Technologies [Thermo Fisher Scientific]). Every reaction was performed in duplicate and the data were analyzed with the 2ΔΔCT method.
Primers for PEPCK, G6Pase, PGC1α and β-actin were as follows: PEPCK, forward 5′-GACTCGCCCTATGTGGTG-3′ and reverse 5′-TGGAGGCACTTGATAAACTC-3′; G6Pase, forward 5′-CTCCGTGCCTCTGATAAA-3′ and reverse 5′-CCACGAAAGATAGCGAGA-3′; PGC1α, forward 5′-GGGGCACATCTGTTCTTC-3′ and reverse 5′-CTCGGATTTCCTGGTCTT-3′; and β-actin, forward 5′-AAGAGAGGCATCCTCACCCT-3′ and reverse 5′-TACATGGCTGGGGTGTTGAA-3′.
Statistical Analysis
Data are presented as means ± SEM. Statistical significance was determined with one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls post hoc analysis. P < 0.05 was considered statistically significant. Data were analyzed with use of GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA).
All supplementary materials are available online at https://doi.org/www.molmed.org.
Results
Increased Plasma Glucose and Insulin Levels in Rats after MI
A rat model of experimentally induced MI was established by permanent ligation of the LAD coronary artery. The success of ligation was confirmed by enlarged myocardial infarct size and increased CPK release (Figures 1A,B). Then, the plasma levels of glucose and insulin were measured at the indicated time points. We found that plasma glucose levels were elevated significantly at 5 min of ligation in MI rats (P < 0.01) and increased to 2.2-fold of that in controls at 30 min of ligation (Figure 1C). Similarly, plasma insulin levels increased rapidly in MI rats, which were significantly higher 10 min after ligation compared with control rats (P < 0.05 respectively) (Figure 1D).

Changes in plasma levels of glucose and insulin in rats after MI. (A) Representative photographs of heart sections stained by 2,3,5-triphenyltetrazolium (TTC). Red staining indicates viable tissues. Grey staining indicates infarct tissues. (B) Plasma creatine phosphokinase (CPK) levels at the indicated time points. (C) Plasma glucose levels at the indicated time points. (D) Plasma insulin levels at the indicated time points. Values presented are means ± SEM of 8 to 10 samples in each group. *P < 0.05 versus control group; **P < 0.01 versus control group.
Plasma cholesterol and triglyceride levels remained comparable between MI and control rats (Supplementary Figures S1A, B). Plasma FFA levels decreased gradually in MI rats, reaching the lowest level at 30 min of ligation when the difference became significant between MI and control groups (Supplementary Figure S1C).
Impaired Insulin Signaling Pathways in Liver after MI
To assess the early events in the insulin signaling pathways in liver tissue, rats were killed at 35 min of MI. 1 U/kg of insulin was used before tissues were harvested as described in Materials and Methods. Upon insulin stimulation, basal expression levels of insulin signaling proteins were comparable among the rats, but phosphorylation levels of these proteins were different.
Insulin-stimulated tyrosine phosphorylation of IR β-subunit, which is correlated with full activation of the IR tyrosine kinase (14,15), did not change significantly in liver tissue between MI and control rats (Figure 2A). By contrast, insulinstimulated tyrosine phosphorylation of IRS1 decreased by almost 70% more in MI rats than in controls (Figure 2B). In addition, we analyzed the fold effect of insulin within MI and control groups, respectively, since basal tyrosine phosphorylation of IRS1 also was markedly observed. As shown in Figure 2B, insulin induced a 1.7-fold increase in tyrosine phosphorylation of IRS1 in MI groups and 2.7-fold increase in control groups, suggesting insulin-stimulated tyrosine phosphorylation of IRS1 decreased in MI rats compared with controls. Upon insulin stimulation, the levels of tyrosine phosphorylation of IRS2 were comparable between MI and control rats (data not shown).

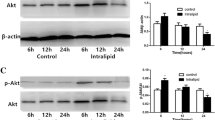
Alteration of insulin signaling cascades in liver tissue of rats after MI. (A) Tyrosine phosphorylation of IR β-subunit. Liver tissue lysates were separated by SDS-PAGE, and Western blot was performed using specific anti-tyrosine-phospho-IR β-subunit and anti-IR β-subunit. (B) Tyrosine phosphorylation of IRS1 and attachment of PI3K p85 subunit with IRS1. Liver tissue lysates were immunoprecipitated with specific anti-IRS1 and then subjected to Western blot using specific anti-IRS1, anti-phospho-tyrosine (P-Tyr-100) and anti-PI3K p85 subunit. (C) Serine phosphorylation of Akt (ser 473). Liver tissue lysates were separated by SDS-PAGE and Western blot was performed using specific anti-phospho-Akt (ser 473) and anti-Akt. Left panel, representative immunoblots are shown. Right panel, quantification of phosphorylation blots relative to their corresponding loading. (A) Tyrosine phosphorylation of IR β-subunit relative to IR β-subunit, (B) Tyrosine phosphorylation of IRS1 relative to IRS1 and IRS1-p85 subunit relative to IRS1, (C) Serine phosphorylation of Akt (ser 473) relative to Akt. IP immunoprecipitation; IB, immunoblotting; I, insulin (with insulin stimulation); B, basal (without insulin stimulation). Values presented are means ± SEM of 4 to 5 samples in each group. *P < 0.05 versus control group with insulin stimulation. #P < 0.05 versus control group without insulin stimulation.
The downstream key proteins in IRS1 signaling pathways, such as the attachment of PI3K p85 subunit with IRS1 and phosphorylation of Akt on ser473, were examined. Insulin-stimulated attachment of PI3K p85 subunit with IRS1 was reduced significantly in MI rats compared with controls. The fold effect of insulin on IRS1-PI3K p85 subunit association within MI and control groups was 1.6 and 12.7, respectively (see Figure 2B). Insulin-stimulated phosphorylation of Akt (ser473) was reduced significantly in MI rats compared with controls. However, the fold effect of insulin on Akt phosphorylation (ser473) was greater within MI rats than within controls (56.7-fold and 19.3-fold, respectively, Figure 2C).
Taken together, our data demonstrate that insulin signaling was not impaired at the level of the IR in liver tissue after MI, whereas a significant decrease of the downstream IRS1/PI3K/Akt cascades was detected.
Insulin Signaling Pathways in Skeletal Muscle and Heart Did Not Change Significantly after MI
The insulin signaling pathways in the skeletal muscle of rats were studied. As in the liver studies, 1 U/kg of insulin was used. Upon insulin stimulation, tyrosine phosphorylation of IR β-subunit was reduced slightly in MI rats, but the difference did not reach significance between MI and control rats. Besides this finding, we did not observe any changes for insulin signaling proteins in skeletal muscle of MI rats compared with controls (Figure 3).

Alteration of insulin signaling cascades in skeletal muscle of rats after MI. Tyrosine phosphorylation of IR β-subunit and IRS, and serine phosphorylation of Akt (ser 473). Skeletal muscle tissue lysates were subjected to immunoprecipitation and/or Western blot using specific antibodies. Left panel, representative immunoblots are shown. Right panel, quantification of phosphorylation blots relative to their corresponding loading. Tyrosine phosphorylation of IR β-subunit relative to IR β-subunit, tyrosine phosphorylation of IRS1 relative to IRS1, serine phosphorylation of Akt (ser 473) relative to Akt. IP immunoprecipitation. IB, immunoblotting. I, insulin (with insulin stimulation). B, basal (without insulin stimulation). Values presented are means ± SEM of 4 to 5 samples in each group. *P < 0.05 versus control group with insulin stimulation. #P < 0.05 versus control group without insulin stimulation.
Local insulin signaling in the heart, another insulin-sensitive organ, also was investigated. After 1 U/kg of insulin was injected, no measurable phosphorylation of IRS1 or Akt was detected. Next, we used different doses of insulin to evaluate its ability to induce Akt phosphorylation (ser473) in heart tissue. The response to insulin was graded, with slight phosphorylation of Akt at 0.5 U/kg of insulin injection and marked phosphorylation at 10 U/kg and 20 U/kg of insulin injection (Supplementary Figure S2). Thus, 10 U/kg of insulin was injected.
Insulin-stimulated tyrosine phosphorylation of IR β-subunit, tyrosine phosphorylation of IRS1, attachment of PI3K p85 subunit with IRS1 and phosphorylation of Akt (ser473) were comparable in heart tissue between MI and control rats. However, the fold effect of insulin on tyrosine phosphorylation of IR β-subunit, tyrosine phosphorylation of IRS1, IRS1-PI3K p85 subunit association and phosphorylation of Akt (ser473) within MI and control groups were different (tyrosine phosphorylation of IR β-subunit: 1.4-fold and 1.3-fold within MI and controls, respectively; IRS1-PI3K p85 subunit association: 0.9-fold and 2.5-fold within MI and controls, respectively; Akt phosphorylation: 3.3-fold and 17.2-fold within MI and controls, respectively). Interestingly, the basal Akt phosphorylation (ser473) was increased in heart tissue of MI rats compared with controls, indicating that ischemia induces Akt activation in the injured heart of MI rats independent of insulin stimulation (Figure 4).

Alteration of insulin signaling cascades in heart tissue of rats after MI. Tyrosine phosphorylation of IR β-subunit and IRS1 and serine phosphorylation of Akt (ser 473). Heart muscle tissue lysates were subjected to immunoprecipitation and/or Western blot using specific antibodies. Left panel, representative immunoblots are shown, Right panel, quantification of phosphorylation blots relative to their corresponding loading. Tyrosine phosphorylation of IR β-subunit relative to IR β-subunit, tyrosine phosphorylation of IRS1 relative to IRS1, serine phosphorylation of Akt (ser 473) relative to Akt. IP immunoprecipitation; IB, immunoblotting; I, insulin (with insulin stimulation); B, basal (without insulin stimulation). Values presented are means ± SEM of 4 to 5 samples in each group. *P < 0.05 versus control group with insulin stimulation. #P < 0.05 versus control group without insulin stimulation.
Alteration of Gluconeogenesis in Liver Tissue after MI
Gluconeogenesis plays a key role in the regulation of hepatic glucose production, which is modulated by IRS1/PI3K/Akt signaling in hepatocytes (16,17). Thus, we further examined the mRNA expression of gluconeogenesis rate-limiting enzymes PEPCK, G6Pase and PGC1α in liver tissue by quantitative RT-PCR assay. We found that the relative mRNA levels of PEPCK and G6Pase were slightly higher in liver tissue of MI rats than those in controls, whereas the mRNA levels of PGC1α remained comparable between these two groups (Supplementary Figure 3). These results suggest that the gluconeogenesis is at least partially elevated in liver tissue of MI rats.
Increased Plasma TNF-α Levels in Rats after MI
TNF-α, an important proinflammatory cytokine, is associated with the development of insulin resistance in chronic diseases. However, it was unknown whether TNF-α would be increased enough to contribute to the insulin resistance rapidly developed after MI. Thus, we examined plasma TNF-α levels by ELISA. MI rats exhibited significantly increased plasma levels of TNF-α compared with controls (11.83 pg/mL and 4.06 pg/mL, P < 0.01; Figure 5).

Alteration of plasma TNF-α levels in rats after MI. Plasma TNF-α levels were measured by ELISA. Values presented are means ± SEM of 8 to 10 samples in each group. *P < 0.05 versus control group.
ROSI Ameliorated Hepatic Insulin Resistance after MI
To test whether impairment of insulin signaling pathways in liver tissue was responsible for elevated plasma glucose levels, rats were administrated with insulin sensitizer ROSI. As expected, ROSI significantly restored tyrosine phosphorylation of IRS1 and phosphorylation of Akt (ser473) in liver tissue of MI rats. Moreover, the restoration of Akt phosphorylation almost reached the levels shown in controls (Figure 6). Next, we determined the effect of ROSI on hepatic gluconeogenesis, and found that it significantly inhibited the mRNA expression of PEPCK and G6Pase (Supplementary Figure S4).

The effect of ROSI on insulin signaling cascades in liver tissue of rats after MI. (A) Tyrosine phosphorylation of IRS1. (B) Serine phosphorylation of Akt (ser 473). Rats were administered with ROSI (4 mg/kg) and all of them received insulin injection before being killed. Upper panel, representative immunoblots are shown. Lower panel, quantification of phosphorylation blots relative to their corresponding loading. Tyrosine phosphorylation of IRS1 relative to IRS1, serine phosphorylation of Akt (ser 473) relative to Akt. Values presented are means ± SEM of 4 to 5 samples in each group. *P < 0.05 versus control group. #P < 0.05 versus MI group.
ROSI Reduced Plasma Glucose and Insulin Levels after MI
Finally, we examined the effect of ROSI on plasma glucose and insulin levels. As shown in Figure 7, MI rats with ROSI administration exhibited decreased glucose levels, which were markedly lower 15 min after ligation than in rats without ROSI administration. Similar trends were observed for plasma insulin levels, with the difference becoming significant 20 min after ligation between MI rats with and without ROSI administration. Neither the plasma glucose or insulin levels in MI rats with ROSI administration were significantly different from controls. These results indicates that ROSI could ameliorate metabolic dysfunction after MI.

The effect of ROSI on plasma levels of glucose and insulin in rats after MI. (A) Plasma glucose levels at the indicated time points, (B) Plasma insulin levels at the indicated time points. Values presented are means ± SEM of 8 to 10 samples in each group. *P < 0.05 versus control group. #P < 0.05 versus MI group.
Discussion
In the present study, we observed a rapid development of hyperglycemia in rats after MI, consistent with previous reports (2,18,19). Corticosteroids and catecholamines have been considered primary contributors to MI-induced hyperglycemia (2,20). However, studies aimed to establish correlation between plasma glucose levels and the change of corticosteroids and catecholamines in response to acute injury have produced inconsistent results, suggesting other possible mechanisms are involved (21,22).
We first investigated the early alteration of insulin signaling pathways in insulin responsive tissues after MI. Our results showed significantly impaired insulin signaling in liver tissue 30 min after MI, but no measurable impairments in skeletal muscle, suggesting that insulin signaling pathways are more sensitive to being affected in liver tissue. Moreover, it should be noted that this pattern of tissue-specific development of insulin resistance might be due to a time-dependent response to MI. Suppression of insulin signaling in liver tissue is achieved rapidly, whereas inhibition of signaling in skeletal muscle may be delayed. Studies on other acute conditions have demonstrated that severe impairment of insulin signaling occurred in liver tissue as soon as 15 min after trauma injury. By contrast, development of skeletal muscle insulin resistance was not detectable until 60 min after trauma (21,23–25). Thus, the undetected changes in insulin signaling pathways in skeletal muscle could not rule out the possibility that the pathways would be impaired as time continues; this needs be further confirmed.
This MI-induced hepatic insulin resistance might not result from an IR defect since there were no measurable changes in IR protein levels or insulin-stimulated tyrosine phosphorylation of the IR. By contrast, significantly compromised insulin-stimulated IRS1 tyrosine phosphorylation was detected after MI, followed by the impaired attachment of PI3K p85 subunit with IRS1. IRS1 has been reported to be a predominant target for the development of chronic insulin resistance, in which IRS1 protein levels, IRS1 tyrosine phosphorylation and/or IRS1-PI3K association have been reported to be downregulated (8,26,27). However, in our study, there were no changes in IRS1 protein levels after MI, but a marked decrease in insulin-stimulated IRS1 tyrosine phosphorylation. Thus, IRS1 tyrosine phosphorylation may be a sensitive site that is easily affected, characterized by rapid onset, developing in minutes in the setting of MI. There is a paradox regarding insulin-stimulated phosphorylation of Akt when analyzing the current data in different ways, which needs further confirmation. To sum up, we favor the idea that MI-induced hepatic insulin resistance is generated from modifications in IRS1 tyrosine phosphorylation, leading to its impaired function and, ultimately, to disruption of insulin signal transduction in the downstream.
We also assessed the transcriptional levels of the gluconeogenesis rate-limiting enzymes regulated by insulin signaling in liver tissue (16,17). The mRNA expression of PEPCK and G6Pase were elevated significantly, indicating that there is a rapid shift to increase hepatic glucose production in response to MI. Thus, hepatic insulin resistance would lead to an increase in hepatic gluconeogenesis and glucose production, which may be an important mechanism for increased plasma glucose levels after MI.
Hepatic insulin resistance acting as the potential contributor to hyperglycemia after MI was further confirmed by ROSI administration in our study. ROSI is a member of the thiazolidinedione class that serves as a potent peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist (28). It shows favorable effects on glycemic control by potentiating insulin signaling and ameliorating insulin resistance in the process of type 2 diabetes (29,30). In this study, we observed that ROSI markedly restored insulin signaling and inhibited gluconeogenesis in liver tissue of MI rats. In addition, plasma levels of glucose and insulin were reduced after ROSI administration. It appears that ROSI could be an appropriate candidate to improve MI-induced hepatic insulin resistance and ultimately reduce hyperglycemia. It should be noted that the dose of ROSI may be higher as compared to previous studies, in which ROSI was usually administered via oral gavage. Moreover, in a large scale meta-analysis, long-term use of ROSI has been proved to be associated with an increased risk in heart failure and myocardial infarction indicated in type 2 diabetic patients (31), whereas this association has not been supported by subsequent prospective clinical studies (32,33). Considering the controversy of ROSI in clinical practice, its beneficial effect on insulin signaling and hyperglycemia needs to be confirmed in further studies.
The fold effect of insulin on phosphorylation of Akt within MI rats was markedly lower than that within controls, though insulin-stimulated phosphorylation of Akt was comparable between MI and control rats. It can be deduced that the fold effect of insulin was largely due to increased basal phosphorylation of Akt in the injured heart tissue. As one of the most important protein kinases at the core of cell signaling downstream, Akt is activated by a number of factors, including insulin, insulin-like growth factor and cytokines (34). The activation of Akt raises the possibility that it plays an important role in injured cardiac tissue, which is independent of insulin stimulation. Studies have shown that acute hyperglycemia favored enhancing the concentration gradient-dependent facilitative glucose transport into injured heart tissue to maintain energy supply in the setting of MI (35). It merits further investigation into whether this process is via activating Akt signaling.
That the impairment of insulin signaling occurs in the remote organs following MI is certainly recognized but poorly understood. Since circulating and local inflammatory cytokines are always elevated after MI in humans and experimental models (36), they may serve as important mediators of tissue-to-tissue cross-talk, which may extend beyond the injured heart to other tissues. Indeed, we found plasma TNF-α increased in MI rats. Another example for tissue-to-tissue cross-talk is cardio-renal syndromes in which MI induces decreased function in kidney (37). Thus, there are various systemic interactions and network in the body.
Conclusion
In summary, we demonstrate that impairment of insulin signaling pathways rapidly develops in liver tissue but not skeletal muscle in a rat MI model. Hepatic insulin resistance leads to hyperglycemia through increased hepatic gluconeogenesis after MI. These findings may improve our understanding of the molecular mechanisms responsible for acute hyperglycemia development after MI and provide new insights for future therapeutic targets. Therapy aimed at potentiating hepatic insulin signaling may be beneficial for MI-induced hyperglycemia.
Disclosure
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Stranders I, et al. (2004) Admission blood glucose level as risk indicator of death after myocardial infarction in patients with and without diabetes mellitus. Arch. Intern. Med. 164:982–8.
Ishihara M. (2012) Acute hyperglycemia in patients with acute myocardial infarction. Circ. J. 76:563–71.
Diaz R, et al. (2007) Glucose-insulin-potassium therapy in patients with ST-segment elevation myocardial infarction. JAMA. 298:2399–405.
de Mulder M, et al. (2013) Intensive glucose regulation in hyperglycemic acute coronary syndrome: results of the randomized BIOMarker study to identify the acute risk of a coronary syndrome-2 (BIOMArCS-2) glucose trial. JAMA Intern. Med. 173:1896–904.
Saltiel AR, Kahn CR. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 414:799–806.
Carrera BCA, Martinez-Moreno JM. (2013) Pathophysiology of diabetes mellitus type 2: beyond the duo “insulin resistance-secretion deficit.” Nutr. Hosp. 28 Suppl 2:78–87.
Zick Y. (2001) Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell. Biol. 11:437–41.
Yi Z, et al. (2007) Global assessment of regulation of phosphorylation of insulin receptor substrate-1 by insulin in vivo in human muscle. Diabetes. 56:1508–16.
Truglia JA, Livingston JN, Lockwood DH. (1985) Insulin resistance: receptor and post-binding defects in human obesity and non-insulin-dependent diabetes mellitus. Am. J. Med. 79:13–22.
Liu YF, et al. (2001) Insulin stimulates PKCzeta — mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J. Biol. Chem. 276:14459–65.
Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. (2011) Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press.
Su H, et al. (2007) Acute hyperglycemia exacerbates myocardial ischemia/reperfusion injury and blunts cardioprotective effect of GIK. Am. J. Physiol. Endocrinol. Metab. 293:E629–35.
Wang J, et al. (2011) Inhibition of aldehyde dehydrogenase 2 by oxidative stress is associated with cardiac dysfunction in diabetic rats. Mol. Med. 17:172–9.
Morgan DO, Roth RA. (1987) Acute insulin action requires insulin receptor kinase activity: introduction of an inhibitory monoclonal antibody into mammalian cells blocks the rapid effects of insulin. Proc. Natl. Acad. Sci. U. S. A. 84:41–5.
Alghamdi F, et al. (2014) A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell Signal. 26:1355–68.
Liu Y, et al. (2008) A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 456:269–73.
Kim DH, et al. (2011) FoxO6 integrates insulin signaling with gluconeogenesis in the liver. Diabetes. 60:2763–74.
Ceriello A. (2005) Acute hyperglycaemia: a ‘new’ risk factor during myocardial infarction. Eur. Heart J. 26:328–31.
Opie LH. (2008) Metabolic management of acute myocardial infarction comes to the fore and extends beyond control of hyperglycemia. Circulation. 117:2172–7.
Smit JW, Romijn JA. (2006) Acute insulin resistance in myocardial ischemia: causes and consequences. Semin. Cardiothorac. Vasc. Anesth. 10:215–9.
Ma Y, et al. (2003) Hemorrhage induces the rapid development of hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol 284:G107–15.
Carvalho LS, et al. (2012) High plasma HDL-C attenuates stress hyperglycemia during acute phase of myocardial infarction. Atherosclerosis 220:231–6.
Thompson LH, et al. (2008) Acute, muscle-type specific insulin resistance following injury. Mol. Med. 14:715–23.
Li L, Thompson LH, Zhao L, Messina JL. (2009) Tissue-specific difference in the molecular mechanisms for the development of acute insulin resistance after injury. Endocrinology. 150:24–32.
Corathers SD, Falciglia M. (2011) The role of hyperglycemia in acute illness: supporting evidence and its limitations. Nutrition. 27:276–81.
Giraud J, et al. (2007) Phosphorylation of Irs1 at SER-522 inhibits insulin signaling. Mol. Endocrinol. 21:2294–302.
Hoehn KL, et al. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7:421–33.
Vasudevan AR, Balasubramanyam A. (2004) Thiazolidinediones: a review of their mechanisms of insulin sensitization, therapeutic potential, clinical efficacy, and tolerability. Diabetes Technol. Ther. 6:850–63.
Jiang G, et al. (2002) Potentiation of insulin signaling in tissues of Zucker obese rats after acute and long-term treatment with PPARgamma agonists. Diabetes. 51:2412–9.
Potenza MA, et al. (2006) Treatment of spontaneously hypertensive rats with rosiglitazone and/or enalapril restores balance between vasodilator and vasoconstrictor actions of insulin with simultaneous improvement in hypertension and insulin resistance. Diabetes. 55:3594–603.
Singh S, Loke YK, Furberg CD. (2007) Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 298:1189–95.
Home PD, et al. (2009) Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 373:2125–35.
Bach RG, et al. (2013) Rosiglitazone and outcomes for patients with diabetes mellitus and coronary artery disease in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation. 128:785–94.
Manning BD, Cantley LC. (2007) AKT/PKB signaling: navigating downstream. Cell. 129:1261–74.
Vilahur G, et al. (2011) Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 50:522–33.
Ruparelia N, et al. (2013) Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflamm. Res. 62:515–25.
Ronco C, et al. (2008) Cardiorenal syndrome. J. Am. Coll. Cardiol. 52:1527–39.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81170136,81100147,81300103,81300219), the Taishan Scholar Program of Shandong Province (ts20130911), the Specialized Research Fund for the Doctoral Program of Higher Education (20130131110048), a grant from Department of Science and Technology of Shandong Province (2011GSF11806) and the Shandong Provincial Outstanding Medical Academic Professional Program, 1020 Program from the Health Department of Shandong Province, China.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Wang, J., Liu, B., Han, H. et al. Acute Hepatic Insulin Resistance Contributes to Hyperglycemia in Rats Following Myocardial Infarction. Mol Med 21, 68–76 (2015). https://doi.org/10.2119/molmed.2014.00240
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00240