
Abstract
There is an expanding role for interleukin (IL)-1 in diseases from gout to cancer. More than any other cytokine family, the IL-1 family is closely linked to innate inflammatory and immune responses. This linkage is because the cytoplasmic segment of all members of the IL-1 family of receptors contains a domain, which is highly homologous to the cytoplasmic domains of all toll-like receptors (TLRs). This domain, termed “toll IL-1 receptor (TIR) domain,” signals as does the IL-1 receptors; therefore, inflammation due to the TLR and the IL-1 families is nearly the same. Fundamental responses such as the induction of cyclo-oxygenase type 2, increased surface expression of cellular adhesion molecules and increased gene expression of a broad number of inflammatory molecules characterizes IL-1 signal transduction as it does for TLR agonists. IL-1β is the most studied member of the IL-1 family because of its role in mediating autoinflammatory disease. However, a role for IL-1α in disease is being validated because of the availability of a neutralizing monoclonal antibody to human IL-1α. There are presently three approved therapies for blocking IL-1 activity. Anakinra is a recombinant form of the naturally occurring IL-1 receptor antagonist, which binds to the IL-1 receptor and prevents the binding of IL-1β as well as IL-1α. Rilonacept is a soluble decoy receptor that neutralizes primarily IL-1β but also IL-1α. Canakinumab is a human monoclonal antibody that neutralizes only IL-1β. Thus, a causal or significant contributing role can be established for IL-1β and IL-1α in human disease.
Similar content being viewed by others

Are IL-1 and Cachetin the Same Molecule?
With this question, I first met Anthony (Tony) Cerami. This first interaction resulted in an enduring scientific and personal friendship for over 30 years. We first met at a meeting at the National Institutes of Health. We had both been studying soluble mediators of inflammation released into the supernatants of mononuclear phagocytes. We had both worked to purify our respective molecules. But neither Tony nor my group with Shelly Wolff had N-terminal amino acid sequences to compare. This was the early 1980s and cDNAs were in their infancy. Researchers studying small (<20,000 Da) biologically active proteins were limited by the fact that these biologically very active proteins were present in small amounts of the supernatants, making purification difficult. The name of the molecule that I was studying was called leukocytic pyrogen, the endogenous fever-producing protein, and Tony named the molecule that he was studying “cachectin.” Were these the same molecule?
IL-1: More than the “Fever Molecule”
During the late 1970s, what is now called IL-1 had been studied under different names such as leukocytic pyrogen, leukocyte endogenous mediator, mononuclear cell factor, catabolin and others. Each was characterized on the basis of a biological assay but a highly relevant bioassay. For example, the bioassay for leukocytic pyrogen was fever, and the bioassay for leukocyte endogenous mediator was a fall in serum zinc levels. The bioassay for the mononuclear cell factor was increased prostaglandin E2 (PGE2) and for catabolin was the degradation of cartilage. In each case, however, the source of the activity was found in the supernatants of mononuclear cells that had been stimulated in vitro. Others working with mononuclear supernatants described an activity of augmentation of T-cell proliferation to antigen challenge. They called the activity “lymphocyte activating factor.” In 1979, it was decided to group all the activities from macrophage supernatants and called these IL-1, whereas activities from lymphocyte supernatants were called IL-2. At the time, biochemical characterization of these activities from cell supernatants was limited—mostly gel filtration with similar molecular weights between 15,000 and 20,000 Da. In the case of leukocytic pyrogen, considerable progress had been made on the purification of supernatants from human monocytes (1) and rabbit peritoneal granulocytes (2).
Tumor Necrosis Factor as a Mediator of Inflammation
Although many were studying the tumor-killing activities of macrophage supernatants, starting in the early 1980s, a new activity in these supernatants was reported from Tony’s laboratory. The activity was the suppression lipoprotein lipase (LPL) (3). The discovery of LPL suppression was related to an observation that during infection, as well as during noninfectious inflammatory diseases, loss of lean body mass (LBM) takes place. This and other astute clinical observations are outlined in a review on the discovery of tumor necrosis factor (TNF) as an inflammatory mediator. The increase in serum lipids associated with infection and inflammatory diseases was known, but Tony’s papers linked suppression of LPL to an activity that was present in the supernatants of LPS-stimulated cells. Tony had clearly made a link between chronic inflammatory diseases, the loss of LBM and the LPL-suppressive activities of endotoxin-stimulated macrophages. In addition, he made a further link between the anemia that accompanies chronic diseases such as rheumatoid arthritis and LPL suppression. This loss in LBM takes place in cancer and is called “cancer cachexia.” Tony had named the LPL-suppressive activity in stimulated macrophage supernatants “cachectin” (3).
TNF Induces IL-1
Of the many collaborations in my career, the studies on the ability of cachectin’s (TNFα’s) inducing IL-1 stands out (4). It brought me to work with Tony and to benefit from his unique insights into science. Our collaborative studies on TNFα’s inducing IL-1 has had far-reaching clinical impacts on cytokine-based therapeutics. However, exactly how this collaboration started is worth reviewing. Motivated by the ability of TNFα to killed tumor cells in vitro, Genentech scientists isolated the cDNA for TNFα and soon thereafter, Tony and his postdoctoral student, Bruce Beutler, published that cachetin and TNFα were the same molecule. In an instant, the biology of TNFα was transformed from a cytokine that caused tumor cell death to a major mediator of inflammation. Few saw or appreciated this transformation, but for Tony, this came as no surprise. Genentech certainly did not understand that TNFα was a highly active inflammatory molecule.
When Genentech injected recombinant TNFα into humans suffering with cancer, they observed high fevers and a fall in blood pressure. This result was unexpected, and using TNFα to treat human cancer patients would reveal the true nature of TNFα (that is, it is a highly inflammatory molecule).
In the early 1980s when the first recombinant proteins for human use were produced in E. coli, Genentech had injected healthy subjects with recombinant human growth hormone. Despite negative Limulus testing, the subjects experienced high fevers and shaking chills. The fevers were due to endotoxin contamination and the company failed to detect endotoxin in the preparations of recombinant human growth factor prepared for human use (5). But in the case of recombinant TNFα causing fever, it was an intrinsic property of TNFα to cause fever directly as well as to induce IL-1 (4).
How Much of the Benefit of Anti-TNFα Therapy Is due to Reducing IL-1?
In 1989, when Marc Feldmann and Fiona Brennan added a neutralizing monoclonal antibody to TNFα to cultured synovial tissue explants from rheumatoid arthritis patients, the readout was spontaneous IL-1 production (6). Indeed, anti-TNFα reduced the spontaneous production of IL-1, but it was a reduction in IL-1α. One year previously in 1988, Feldmann and Maini published their first paper on TNFα in rheumatoid arthritis; they measured mRNA of TNFα levels in synovial explants but also measured IL-1α, not IL-1β (7). In many ways, the Brennan-Feldmann study confirmed the observation that in a clinical setting TNFα induced IL-1 (4). Looking back, the Brennan-Feldmann study helped provide the rationale for testing anti-TNFα in patients with rheumatoid arthritis. But the concept that TNFα had a role in the pathogenesis of rheumatoid arthritis was already known well before 1988. Tony and Jean-Michel Dayer had provided a basis for using anti-TNFα in rheumatoid arthritis by showing that TNFα induced PGE2 and collagenase in synovial fibroblasts (8). Jeremy Saklatvala (9) also provided a rationale for a role of TNFα in rheumatoid arthritis. But in the early 1980s, Tony had already proposed that anti-cachetin monoclonal antibodies should be used to treat rheumatoid arthritis (10). Although blocking TNFα in rheumatoid arthritis is a highly effective treatment for rheumatoid arthritis, other “biologicals” are also effective. Antibodies to the IL-6 receptor, the CTLA4-IgG fusion protein, blocking the IL-1 receptor with the IL-1 receptor antagonist (IL-1Ra, anakinra), are approved for treating the signs and symptoms of rheumatoid arthritis, and antibodies to IL-1β (canakinumab) are also effective (11).
A Broadening Role for IL-1-Blocking Therapies
With the availability of specific IL-1-targeting therapies, a broadening list of diseases has revealed the pathologic role of IL-1-mediated inflammation. Monotherapy blocking IL-1 activity results in a rapid and sustained reduction in disease severity. In common conditions such as heart failure (12,13) and acute gouty arthritis (14–17), IL-1 blockade can be highly effective therapy compared to standards of therapy. Three IL-1 blockers have been approved: anakinra, rilonacept and canakinumab. The IL-1 receptor antagonist, anakinra, blocks the IL-1 receptor and therefore reduces the activity of IL-1α and IL-1β. Rilonacept is a soluble decoy receptor, and canakinumab is a neutralizing monoclonal anti-interleukin-1β antibody. A monoclonal antibody directed against the IL-1 receptor and a neutralizing anti-IL-1α are in clinical trials. The antibody to IL-1α has been administered to patients with cancer cachexia and resulted in a reduction in the wasting syndrome, decreased IL-6 levels and a nonsignificant prolongation of life (18). By specifically blocking IL-1, we have learned a great deal about the role of this cytokine in inflammation, but equally important, reducing IL-1 activity has lifted the burden of disease for many patients.
Autoimmune Versus Autoinflammatory Diseases
All autoimmune diseases have a significant inflammatory component that is due to the production of IL-1β as well as IL-1α from myeloid cells, particularly macrophages. Membrane IL-1α from mesenchymal cells may also contribute to the inflammatory component in autoimmune disorders. Therefore, antiinflammatory strategies that target IL-1 will show efficacy when used to treat classic autoimmune diseases. However, what distinguishes autoimmune diseases from autoinflammatory diseases? In autoimmune diseases, the immunological dysfunction is due to autoreactive T cells that produce IL-12, TNFα, IL-17 and other cytokines, but there are few examples that T cells, particularly CD4+ T cells from the peripheral blood IL-1β. B cells produce IL-1β and there is IL-1α from T cells. Biologicals that target T-cell cytokines are highly effective in treating autoimmune diseases. CTLA4-Ig is also effective for autoimmune diseases. However, during the past 10 years, a concept has emerged that several chronic inflammatory diseases are due to monocyte/macrophage dysregulated IL-1β and have been termed “autoinflammatory” diseases. These diseases are uniquely responsive to blocking IL-1β but far less response to blocking TNFα. In fact, in some diseases, such as heart failure, blocking TNFα may worsen heart failure. The primary sources of IL-1β are the myeloid cells, primarily monocytes, macrophages and neutrophils. Unlike autoimmune diseases, autoinflammatory diseases are not due to autoreactive T cells. Autoimmune diseases exhibit distinct major histocompatibility complex (MHC)-associated haplotype susceptibilities, are progressive rather than periodic and are not strongly associated with environmental stress triggers. Moreover, many autoinflammatory diseases include illnesses due to specific mutations in proteins that regulate IL-1β activity and not TNFα activity. As autoimmune diseases respond to therapies such as TNFα neutralization, CTLA4-Ig, anti-IL-6 receptor, depletion of CD20 B cells and anti-IL-12/IL-23 and IL-17 neutralizing antibodies, these agents show no benefit in patients with autoinflammatory diseases.
Mechanisms in the Pathogenesis of Autoinflammatory Diseases
In general, there is considerable evidence that the caspase-1 inflammasome and the release of IL-1β are major players in the pathogenesis of autoinflammatory diseases. In circulating human blood monocytes, caspase-1 is present in an active state (19); as soon as the monocyte is stimulated, cleavage of the precursor takes place and mature IL-1β is secreted. Caspase-1 is also constitutively active in highly metastatic human melanoma cells (20). However, the release of active IL-1β from blood monocytes is tightly controlled; <20% of the total synthetic IL-1β precursor is processed and released (21). In contrast, monocytes from patients with autoinflammatory syndromes release more processed IL-1β, but the increase is modest (that is, <10-fold greater than that of monocytes from healthy subjects) (22,23). Only fivefold more IL-1β is produced each day in patients with cryopyrin-associated periodic syndrome (CAPS) compared with healthy controls (24).
Although it is possible to demonstrate that blood monocytes from patients with autoinflammatory diseases process and release significantly more IL-1β than monocytes from healthy controls, the basis for the increase varies. Several autoinflammatory diseases have a mutation in NACHT, LRR and PYD domains-containing protein 3 (NALP3) resulting in an active inflammasome (25–30), but many do not have mutations in genes known to regulate caspase-1 or the secretion of IL-1β. For example, the secretion of IL-1β requires triggering of the P2X7 purinergic receptor by adenosine triphosphate (ATP) (31) and active phospholipases C and A2b for secretion via secretory lysosomes (32). Although viral infections and environmental factors are thought to trigger a bout of inflammation in patients with autoinflammatory diseases, continued production of IL-1 seems to drive chronic autoinflammation. For example, steady-state mRNA levels of IL-1β, caspase-1, IL-1α, IL-6 and TNFα are elevated in blood monocytes from patients with neonatal-onset multisystem inflammatory disease (NOMID) compared with cells from healthy subjects but decrease with treatment with anakinra (23). The likely mechanism is therefore IL-1 induction of caspase-1, which results in IL-1 induction of itself. With increased levels of IL-1, IL-1 then induces other cytokines. Regardless of the mechanism of increased IL-1β release, what characterizes these diseases as autoinflammatory is the rapid and sustained response to a therapeutic reduction in IL-1β activity.
Classes of Autoinflammatory Diseases
Autoinflammatory diseases fall into different classes. But as stated above, what characterizes these diseases as autoinflammatory is the rapid and sustained response to a therapeutic reduction in IL-1β activity. As shown in Table 1, the classic autoinflammatory diseases are hereditary. These include NOMID, the Muckle-Wells syndrome (MWS), familial cold-induced autoinflammatory syndrome (FCAS), hyperimmunoglobulin D syndrome (HIDS), TNF receptor-associated periodic syndrome (TRAPS) and familial Mediterranean fever (FMF). Although these diseases are rare, the clinical manifestations are common to nearly all inflammatory diseases as well as infectious diseases. As shown in Table 2, there are several diseases for which there is no known genetic basis. Nevertheless, each responds to anakinra or anti-IL-1β. For example, systemic-onset juvenile idiopathic arthritis (SJIA) responds to anakinra (1,2), and canakinumab (3,4) is highly effective. Adult-onset Still disease is also highly responsive to anakinra and is standard of therapy clinically (5–8). Table 2 also indicates several other chronic inflammatory diseases and the responses to IL-1 blockade by anakinra. Many are also responsive to canakinumab or rilonacept. Of these, Schnitzler syndrome is highly responsive to canakinumab and identifies IL-1β as causal in the disease (9–13). Schnitzler syndrome is similar to multiple myeloma as well as pre-multiple myeloma syndromes. Pre-multiple myeloma syndromes are also known as smoldering myeloma or also indolent myeloma. They respond to anakinra (45). An acute disease, which responds to anakinra, is macrophage activation syndrome (46–55). This life-threatening syndrome is likely under diagnosed. In a reassessment of the original anakinra trial in septic shock (56), 5% of the patients enrolled exhibited evidence of macrophage activation syndrome. The 28-day all-cause mortality was reduced by 62% (p < 0.006) in patients treated with anakinra compared with placebo-treated patients.
Gout and Type 2 Diabetes
Gouty Arthritis
Gout and Type 2 diabetes mellitus are also autoinflammatory diseases but hardly rare conditions. As stated above, important criteria for characterizing a disease as autoinflammatory is that on blocking IL-1, patients experience a rapid and sustained cessation of symptoms as well as reductions in biochemical, hematological and functional markers of their disease. In addition, the disease is not due to any T- or B-cell dysfunction. Gout is a perfect example. Acute as well as recurrent attacks of refractory gouty arthritis respond rapidly to anakinra (15,57–59) as well as to rilonacept (17,60) and canakinumab (14,16,61). As shown in Table 3, anakinra is effective in several joint and muscular diseases. Anakinra is used off-label but canakinumab is approved in Europe for gout. The mechanism by which uric acid crystals induce IL-1β secretion has been studied. Uric acid crystals alone do not stimulate human blood monocytes to release IL-1β (62,63). Rather, triggering of a toll-like receptor (TLR) such as TLR2 or TLR4 (62–64) is required. Fatty acids also function to provide a priming signal (63).
Patients with recurrent attacks of gouty arthritis unable to use colchicine and other standards of therapy often require steroids to control disease flares. When treated with anakinra, canakinumab or rilonacept, a rapid, sustained and remarkable reduction in pain as well as objective signs of reduced inflammation have been observed (14–17,57,58,65–69). The effect of IL-1 blockade appears to be superior to that of steroids and results in prolonged periods without flares. The likely mechanism for urate crystal-induced inflammation in the joint is the release of active IL-1β (63). Given the characteristic neutrophilic infiltration in gouty joints, it is also likely that the IL-1β precursor is processed extracellularly by neutrophilic enzymes (70). Pyrophosphate crystal arthritis is highly responsive to anakinra (66,68). Pyrophosphate crystal arthritis is highly responsive to anakinra (66,68).
Osteoarthritis
In humans, subcutaneous anakinra improved pain and swelling in an aggressive form of erosive osteoarthritis (71). Anakinra has also been injected intraarticularly in patients with knee osteoarthritis (72,73), but the benefit did not extend beyond 1 month (73), which may be because of the brief duration in the joint space. Systemic treatment of osteoarthritis with an antibody to the IL-1 receptor was carried out and a modest improvement was reported, particularly in those patients with high pain levels at enrollment (74).
Type 2 Diabetes
Studies of the role of IL-1β in the pathogenesis of Type 2 diabetes reported that high concentrations of glucose-stimulated IL-1β production from the β cell itself (75), thus implicating a self-destructive role of IL-1β autoinflammation by the β cell (76). Moreover, IL-1β increases the deposition of amyloid, which contributes further to β-cell loss (77). Indeed, gene expression for IL-1β is 100-fold higher in β cells from Type 2 diabetes patients (76) compared with non-diabetic patients. Thus, in Type 2 diabetes, there is progressive loss of the β cells because of IL-1-mediated inflammation, which may also underlie the mechanism of insulin resistance (78).
Clinical proof of a role for IL-1 in the pathogenesis of Type 2 diabetes can be found in the randomized, placebo-controlled study of anakinra for 13 wks, in which there was improved insulin production and glycemic control associated with decreased C-reactive protein (CRP) and IL-6 levels (79). Unexpectedly, in the 39 wks after the treatment, anakinra responders used 66% less insulin to obtain the same glycemic control compared with baseline requirements (80). This observation suggests that blocking IL-1β, even for a short period, restores the function of β cells or possibly allows for partial regeneration. These findings of anakinra treatment in Type 2 diabetes patients have been confirmed using anti-IL-1β monoclonal antibodies: Xoma gevokizumab (81), Novartis canakinumab (82) and Lilly LY2189102 (83). Gevokizumab treatment also reduced the fatigue in Type 2 diabetes patients (84), as did anakinra in the Sjögren syndrome (85). Anakinra has also been tested in obese nondiabetic patients with metabolic syndrome (78). There was a decrease in CRP and circulating leukocytes; the disposition index increased significantly after anakinra treatment, reflecting improved β-cell function (78).
Thus, Type 2 diabetes emerges as a chronic inflammatory disease, in which IL-1 progressively destroys the insulin-producing β cells or renders the β cell nonfunctional (76). The IL-1β can come from the β cell itself, but also from blood monocytes that infiltrate the islet. Obese individuals are at high risk for Type 2 diabetes, and caspase-1-dependent IL-1β production has been demonstrated in macrophages isolated from human fat (86). IL-1α and IL-1β exert the same toxic effect on the insulin-producing β cells.
A large body of preclinical data reveals that IL-1 plays a role in the progression of atherosclerosis (87–89). Because Type 2 diabetes increases the risk of cardiovascular events, blocking IL-1β activity in these patients may also reduce the incidence in myocardial infarction and stroke. The largest trial of an anticytokine is CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study), which tested whether canakinumab reduces cardiovascular events in Type 2 diabetes patients with high CRP levels despite optimal statin therapy (90). The rationale for CANTOS is based on the consistent decrease in CRP levels observed with anakinra (79), canakinumab (91) or gevokizumab (81).
Role of IL-1 Heart Disease
With several reports on the benefit of blocking IL-1 in mouse models of acute myocardial infarction (92–94), two clinical trials have been performed in patients with acute ST-elevated myocardial infarction (STEMI) (95,96). Both were randomized, placebo-controlled trials; anakinra (100 mg subcutaneously) or placebo was started within 24 h following standard of therapy for STEMI and continued for 14 consecutive days. Anakinra reduced the rise in CRP levels 72 h after the infraction, and 12 wks later, the anakinra-treated patients exhibited a statistically significant reduction in heart failure. It was concluded that blocking IL-1 during the immediate time of the infarct reduces the adverse effects on postinfarct remodeling.
Two other trials evaluated the role of IL-1 in patients with heart failure despite treatment with standard therapies. The first trial assessed a 2-wk course of anakinra (100 mg/d) on oxygen consumption and exercise tolerance in patients with classic systolic heart failure. Statistically significant improvements in these parameters were observed as well as a fall in CRP and IL-1β levels (13). A second trial was performed in patients with preserved ejection fraction heart failure. This condition is difficult to treat, and there is a predominance of women with preserved ejection fraction heart failure. Similar to the study in patients with systolic heart failure, there was significant improvement in inflammatory markers and physiologic parameters (97).
Autoimmune Hearing Loss
In autoimmune inner ear disease, there is either sudden onset or progressive loss in hearing. Patients are usually treated with glucocorticoids; however, individuals not responding to glucocorticoid therapy have elevated IL-1β in the circulation, and peripheral blood monocytes release more IL-1β than monocytes from unaffected subjects (98). The pathogenic mechanisms for this disease remains unknown. Patients with this condition who are treated with corticosteroids and respond to the steroids are associated with an increase in circulating levels of the soluble form of the IL-1 decoy receptor type 2 (99); however, patients who do not respond to steroids do not exhibit the rise in the decoy receptor. At present, there is no effective treatment for patients who become unresponsive to steroids. In a phase I/II open-label trial of anakinra, subjects received 100 mg anakinra by subcutaneous injection for 84 d. Ten subjects completed the 84 d of treatment and 7 showed audiometric improvement (100). Circulating IL-1β levels decreased and correlated with improved hearing. The interpretation of these studies is that hearing loss due to this condition is reversible.
Patients with CAPS exhibit neurologic abnormalities such as aseptic leptomenigitis, thus reflecting IL-1-mediated inflammation in the brain. In a study of CAPS patients, 54% had sensorineural deafness (101). Treatment with either anakinra or canakinumab resulted in a complete resolution of symptoms (101–103). Children with severe CAPS show manifestations of elevated intracranial pressure and are believed to be mentally slow or even retarded. However, both mental and hearing impairment are reversed on treatment with anakinra (23,102,104–107) but also with specific neutralization of IL-1β with canakinumab (103,108–111). Meniere disease can also be characterized by progressive sensorineural hearing loss and is also associated with dysregulation of IL-1β (98,99,112).
Why No IL-1β?
Although the activity of IL-1β is such that a devastating systemic inflammatory state can be rapidly controlled with IL-1β blockade, there is no consistently statistically significant increase in the circulating levels of IL-1β between dangerously ill individuals and healthy subjects. There is one reliable clue to a disease that is likely mediated by IL-1β. Humans have been injected intravenously with IL-1, and even at doses of 1 or 2 ng/kg, IL-1β increases the peripheral white blood cell count (113). Assuming a blood volume of 5,000 mL, the maximal plasma level of IL-1 at these doses is <0.5 pg/mL. After 2 h, the rise in white blood cells is due to an absolute neutrophilia with a greater percentage of band forms (25%). The rapid rise in neutrophils is associated with an increase in circulating granulocyte colony-stimulating factor (G-CSF). The subcutaneous route also resulted in a severalfold increase in neutrophils. IL-1β at 3 ng/kg has been injected intravenously during a 15-min infusion into subjects with melanoma but with normal bone marrow function. Within 1 h, the neutrophil counts increased progressively, reaching peak levels at 4 h (113). During the first hour, monocytes and lymphocytes were decreased. In some studies, daily injections of IL-1 increased circulating neutrophils after each injection. In humans injected with endotoxin, neutrophilia is a prominent observation, and similar to IL-1, peak levels of neutrophils including band forms occur at 4 h (114). Importantly, the endotoxin-induced neutrophilia is, in part, IL-1 mediated as a coinfusion of anakinra, which which reduces peak neutrophil counts by >50% (114). In both the juvenile (22) and adult Still disease (36), the prominent neutrophilia decreases rapidly with anakinra. Similarly, a fall in the neutrophilia characterizes the response to anakinra in patients with NOMID (23) or FCAS (115). A specific antibody to IL-1β reverses the neutrophilia that characterizes CAPS (111). Thus, neutrophilia is a consistent biological marker for IL-1β-mediated neutrophilia. Increases in platelets were also observed in patients with normal marrow reserves after one, two or seven injections of IL-1 (116). In patients injected with seven daily doses of 100 ng/kg IL-1α, there was a mean increase of 73% in platelet counts after 15 d (117).
IL-1 And Cancer
Role of IL-1β in Multiple Myeloma
In the microenvironment of the bone marrow, IL-1β produced by myeloma precursor plasma cells stimulates the stromal cells to release much IL-6, which in turn promotes the survival and expansion of the pre-myeloma cells (118). It was reasoned that in the indolent stages of multiple myeloma, blocking IL-1β would reduce IL-6 activity (45). Patients with smoldering or indolent myeloma at high risk for progression to multiple myeloma were selected with the clinical objective of slowing or preventing progression to active disease. During 6 months of treatment with anakinra, there were decreases in CRP in most but not all patients, which paralleled a decrease in myeloma cell proliferation. After 6 months of anakinra, a low dose of dexamethasone was added. Of the 47 patients that received anakinra with dexamethasone, progression-free disease lasted over 3 years and in 8 patients over 4 years (45). Compared to historical experience, the findings indicate a significant failure to progress to active disease. Given the increasing incidence of multiple myeloma in the aging population, an option of anti-IL-1β as an early intervention treatment in the indolent stages of multiple myeloma might have a significant impact on this fatal cancer.
The Case for Treating Cancer with Anti-IL-1α
Sterile inflammation resulting from cell death is due to the release of cell contents, which are normally inactive and sequestered within the cell; fragments of cell membranes from dying cells also contribute to sterile inflammation. Endothelial cells undergoing stress-induced apoptosis release membrane microparticles, which become vehicles for proinflammatory signals. A population of large microparticles from endothelial cells contains nuclear fragments and histones and represents a class of inflammatory apoptotic bodies. These large apoptotic bodies contain full-length IL-1α precursor as well as a processed mature form of the cytokine (119). In vitro, these inflammatory apoptotic bodies induce monocyte chemotactic protein-1 and IL-8 chemokine secretion, which is independent of IL-1β. In vivo, the endothelium-derived particles induce neutrophilic infiltration into the peritoneal cavity of mice that was prevented by IL-1 receptor blockade. Thus, nonphagocytosed endothelial large apoptotic bodies are inflammatory, provide a vehicle for IL-1α and, therefore, constitute a unique mechanism for sterile inflammation.
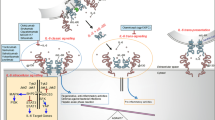
Most sterile inflammation resulting from ischemic damage starts with the release of the IL-1α precursor from necrotic cells (Figure 1). The IL-1α precursor is active (120). Membrane IL-1α is also highly active (121,122), and IL-1α is also present on platelets (123). In several diseases responsive to anakinra, it remains unclear if the disease is mediated by IL-1α or IL-1β activity. From experimental models, supernatants from cells undergoing a necrotic cell death induce local inflammation, which is initially due to IL-1α and not IL-1β (124). Although anakinra will prevent the activity of IL-1α and IL-1β, there is a need to selectively block IL-1α but not IL-1β. The rationale for this concept is based on a model of inflammatory bowel disease in mice in which IL-1α mediates the inflammation, whereas IL-1β mediates healing (125).

Initiation of sterile inflammation by IL-1α following an ischemic event. ①, In the necrotic area, dying cells lose membrane integrity. ②, Dying cells release their contents including the IL-1α precursor. Anti-IL-1α antibodies neutralize IL-1α at this step. ③,IL-1α binds to IL-1R type I (IL-1RI) on nearby resident fibroblasts, epithelial cells or in the brain astrocytes, releasing chemokines and establishing a chemokine gradient. Anakinra or anti-IL-1RI prevents this step. The chemokine gradient facilitates the passage to blood neutrophils into the ischemic area. ④, Capillaries in the ischemic tissues express the intra-cellular adhesion molecule-1 (ICAM-1). Circulating blood neutrophils roll on the endothelium, adhere to ICAM-1 and enter the ischemic tissue via diapedesis. ⑤, The number of neutrophils increases into the area of the necrotic event; the presence of local IL-1 prolongs the survival of neutrophils at this step. ⑥, Neutrophil proteases cleave the extracellular IL-1α precursor into mature, more active forms. ⑦, Neutrophils scavenge dying cells and release proteases that contribute to the destruction of penumbral cells. Image reproduced (no permission needed) from Dinarello et al. (137): Charles A Dinarello, Anna Simon, Jos W M van der Meer. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews Drug Discovery. 2012;11:633-52.

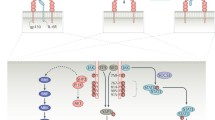
Role of IL 1β in sterile inflammation. ①, Following an ischemic event, cells undergo hypoxic damage, lose membrane integrity, and the dying cell releases cell content (see Figure 1). ②, The preformed IL-1α precursor is released. Anti-IL-1α antibodies neutralize IL-1α at this step. ③, IL-1α binds to IL-1 receptor type I (IL-1RI) on nearby resident macrophages. Anakinra or anti-IL-1RI antibodies prevent IL-1α activity at this step. ④, Triggered by IL-1α binding to the IL-1RI, resident macrophages synthesize inflammatory genes as well as the IL-1β precursor. ⑤, Intracellular processing of the IL-1β precursor by caspase-1. Caspase-1 inhibitors prevent the processing of IL-1β. ⑥, Active secretion of active IL-1β. Antibodies to IL-1β or rilonacept neutralize IL-1β in the extracellular compartment at this step. ⑦, With breakdown of the vascular integrity in the necrotic area, IL-1β gains access to the vascular compartment. ⑧, IL-1β binds to IL-1RI on capillaries and induces vascular cell adhesion molecule-1 (VCAM-1). ⑨, Blood monocytes roll along the endothelium and bind to VCAM-1, followed by emigration into the ischemic tissue via diapedesis. Increasing numbers of monocytes become a source of greater production of IL-1β. ⑩, Opening of the endothelial junction resulting in capillary leak with the passage of plasma proteins into the ischemic area. ⑪, Platelet-derived IL-1α binds to the endothelial IL-1RI and induces ICAM-1. ⑫, Large numbers of neutrophils enter the tissue space, and the presence of local IL-1 prolongs the survival of neutrophils. ⑬, Release of neutrophil proteases. ⑭, IL-1β precursor released into the extracellular space is cleaved by serine proteases generating active IL-1β. Natural inhibitors of serine proteases such as a1-antitrypsin prevent the extracellular processing of the IL-1β precursor. ⑮, Increasing numbers of neutrophils surround the necrotic area, scavenging dead cells and debris. ⑯, Damaging neutrophilic proteases attack and injure penumbral cells, increasing the loss of function. Blocking IL-1 cannot restore the necrotic tissue but reduces the loss of the penumbral cells. Image modified (no permission needed) from Dinarello et al. (137): Charles A Dinarello, Anna Simon, Jos W M van der Meer. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature Reviews Drug Discovery. 2012;11:633-52.
Human Anti-IL-1α
Patients with autoimmune diseases as well as healthy subjects have circulating antibodies to IL-1α, as discovered by Klaus Bendtzen (126,127). A fully human monoclonal anti-human IL-1α has been isolated from human B-lymphocytes and developed for neutralizing IL-1α for human disease. The monoclonal antibody is being tested in Type 2 diabetes, cancer, cancer cachexia, leukemia, psoriasis, vascular disease and scarring acne vulgaris.
Similar to sterile inflammation after an ischemic event, tumors undergoing a necrotic cell death are also a cause of sterile inflammation, which in mice is due to IL-1α, but not IL-1β or TLR (128). In human cancers, inflammation mediated by IL-1α will have a negative influence on survival because of increased angiogenesis, metastasis, metabolic syndrome and immunosuppression. Anti-IL-1α monotherapy was administered to patients refractory to antitumor therapies and losing weight (18). Dual-energy X-ray absorptiometry (DEXA) was used to assess LBM. Plasma IL-6 levels and circulating CD14-positive monocytes declined. Twenty-one patients were evaluated with DEXA, and 71% (15/21) showed increases in LBM (p < 0.001). Patients with increased LBM had a median survival of 19.3 months versus 6.6 months for individuals with LBM loss (18).
Are These Data Consistent with Properties of IL-1α?
In the early 1990s, either IL-1β or IL-1α was administered to patients with suppressed bone marrow due to chemotherapy to stimulate hematopoiesis; although effective, picomolar concentrations of IL-1α were toxic with fever, severe fatigue, loss of appetite, myalgias and frank hypotension (129). The association of noncancer chronic inflammation with loss of LBM is well established, and IL-1 can directly induce muscle protein breakdown (130).
The rationale for blocking IL-1α in any disease should not be on the basis of elevated circulating serum levels, since IL-1α is not released from living cells and is active as an integral cell surface protein. IL-1α is also present on platelets, which may account for the systemic effects such as elevated IL-6. In fact, the longstanding reports of platelet involvement in metastasis, which would include platelet-endothelial cell interaction (123) may now be, in part, understood by neutralization of IL-1α. The findings in the Hong study (18) are the first clinical evidence that endogenous IL-1α-induced IL-6 contributes to the thrombocytosis in cancer (129). Additional possible mechanisms of action with neutralization of IL-1α include decreased angiogenesis (131) and decreased immunosuppression (132). IL-1α neutralization also includes direct antitumor properties by inhibition of tumor growth. With the IL-1α precursor present in noncancerous as well as in most cancerous cells and given the broad inflammatory properties of IL-1α, no one mechanism accounts for the clinical improvements in the study (18).
Immunosuppression of Cancer
Cytokine-mediated inflammation has a role in the immunosuppression of cancer (133). From the above studies, IL-1α-mediated systemic inflammation is also a debilitating aspect of cancer. Many tumors produce IL-1α, which promotes angiogenesis and tumor growth (134). Therefore, blocking a cytokine in cancer is a therapeutic opportunity, particularly because anticytokines are without symptoms or physiologic side effects, and tumors are unlikely to develop resistance to a cytokine blockade. Although blocking IL-1 with anakinra reduces the progression of smoldering myeloma into overt myeloma (45), patients with epithelial cancers are rarely treated with a specific anticytokine such as IL-1-blocking agents (135). A prevailing but misunderstood concept is that blocking IL-1 would contribute to the immunosuppression of cancer and be contraindicated (136). Anti-IL-1α therapy demonstrated that treatment reduces systemic inflammation, since a fall in circulating IL-6 levels remains one of the most consistent observations of blocking IL-1 (137). The source of the inflammatory trigger is likely the tumor itself, since all cancer cells of epithelial cell origin contain IL-1α in its precursor form. Inflammation is also due to invading stromal cells into the tumor microenvironment. As tumors outgrow their vascular supply, they become necrotic, and the IL-1α precursor is readily released and triggers local production of chemokines, which facilitate an influx of neutrophils and monocytes (137). Unlike precursors of IL-1β, the IL-1α precursor is fully active. Neutralization of local IL-1α likely reduces the infiltration of tumor-associated macrophages and myeloid-derived suppressor cells, which contribute to the immunosuppression of cancer mediated by inflammation (133). The study also reports a reduction in fatigue, which is consistent with the use of anakinra in patients with inflammatory diseases unrelated to cancer (137).
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Dinarello CA, Renfer L, Wolff SM. (1977) Human leukocytic pyrogen: purification and development of a radioimmunoassay. Proc. Natl. Acad. Sci. U. S. A. 74:4624–7.
Murphy PA, Chesney PJ, Wood WB Jr. (1974) Further purification of rabbit leukocyte pyrogen. J. Lab. Clin. Med. 83:310–22.
Kawakami M, Pekala PH, Lane MD, Cerami A. (1982) Lipoprotein lipase suppression in 3T3-L1 cells by an endotoxin-induced mediator from exudate cells. Proc. Natl. Acad. Sci. U. S. A. 79:912–6.
Dinarello CA, et al. (1986) Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J. Exp. Med. 163:1433–50.
Dinarello CA. (1984) Interleukin-1 and the pathogenesis of the acute-phase response. N. Engl. J. Med. 311:1413–8.
Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. (1989) Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 2:244–7.
Buchan G, et al. (1988) Interleukin-1 and tumour S52 necrosis factor mRNA expression in rheumatoid arthritis: prolonged production of IL-1 alpha. Clin. Exp. Immunol. 73:449–55.
Dayer JM, Beutler B, Cerami A. (1985) Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J. Exp. Med. 162:2163–8.
Saklatvala J. (1986) Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature. 322:547–9.
Cerami A. (2011) The value of failure: the discovery of TNF and its natural inhibitor erythropoietin. J. Intern. Med. 269:8–15.
Alten R, et al. (2011) Efficacy and safety of the human anti-IL-1beta monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, phase II, dose-finding study. BMC Musculoskelet. Disord. 12:153.
Abbate A, Canada JM, Van Tassell BW, Wise CM, Dinarello CA. (2014) Interleukin-1 blockade in rheumatoid arthritis and heart failure: a missed opportunity? Int. J. Cardiol. 171:e125–6.
Van Tassell BW, et al. (2012) Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 7:e33438.
So A, et al. (2010) Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 62:3064–76.
So A, De Smedt T, Revaz S, Tschopp J. (2007) A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9:R28.
Schlesinger N, et al. (2011) Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann. Rheum. Dis. 70:1264–71.
Terkeltaub R, et al. (2009) The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann. Rheum. Dis. 68:1613–7.
Hong DS, et al. (2014) MABp1, a first-in-class true human antibody targeting interleukin-1alpha in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 15:656–66.
Netea MG, et al. (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 113:2324–35.
Okamoto M, et al. (2009) Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 285:6477–88.
Levandowski CB, et al. (2013) NLRP1 haplotypes associated with vitiligo and autoimmunity increase interleukin-1beta processing via the NLRP1 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 110:2952–6.
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. (2005) Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 201:1479–86.
Goldbach-Mansky R, et al. (2006) Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 355:581–92.
Lachmann HJ, et al. (2009) In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J. Exp. Med. 206:1029–36.
Livneh A, et al. (1999) MEFV mutation analysis in patients suffering from amyloidosis of familial Mediterranean fever. Amyloid. 6:1–6.
Chae JJ, et al. (2003) Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell. 11:591–604.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29:301–5.
Aksentijevich I, et al. (2002) De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 46:3340–8.
de Koning HD, et al. (2006) Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann. Rheum. Dis. 65:542–4.
Gattorno M, et al. (2007) Pattern of interleukin-1beta secretion in response to lipopolysaccharide and ATP before and after interleukin-1 blockade in patients with CIAS1 mutations. Arthritis Rheum. 56:3138–48.
Solle M, et al. (2001) Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 276:125–32.
Andrei C, et al. (2004) Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: Implications for inflammatory processes. Proc. Natl. Acad. Sci. U. S. A. 101:9745–50.
Quartier P, et al. (2011) A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis. 70:747–54.
Ruperto N, et al. (2012) Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 367:2396–406.
Ruperto N, et al. (2012) A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 64:557–67.
Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. (2005) Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 52:1794–803.
Kalliolias GD, Georgiou PE, Antonopoulos IA, Andonopoulos AP, Liossis SN. (2007) Anakinra treatment in patients with adult-onset Still’s disease is fast, effective, safe and steroid sparing: experience from an uncontrolled trial. Ann. Rheum. Dis. 66:842–3.
Laskari K, Tzioufas AG, Moutsopoulos HM. (2011) Efficacy and long-term follow-up of IL-1R inhibitor anakinra in adults with Still’s disease: a case-series study. Arthritis Res. Ther. 13:R91.
Iliou C, Papagoras C, Tsifetaki N, Voulgari PV, Drosos AA. (2013) Adult-onset Still’s disease: clinical, serological and therapeutic considerations. Clin. Exp. Rheumatol. 31:47–52.
de Koning HD, Schalkwijk J, van der Meer JW, Simon A. (2011) Successful canakinumab treatment identifies IL-1beta as a pivotal mediator in Schnitzler syndrome. J. Allergy Clin. Immunol. 128:1352–4.
de Koning HD, et al. (2013) Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann. Rheum. Dis. 72:1634–8.
Pesek R, Fox R. (2014) Successful treatment of Schnitzler syndrome with canakinumab. Cutis. 94:E11–12.
Szturz P, et al. (2011) Schnitzler syndrome: diagnostics and treatment. Klin. Onkol. 24:271–7.
Vanderschueren S, Knockaert D. (2013) Canakinumab in Schnitzler syndrome. Semin. Arthritis Rheum. 42:413–6.
Lust JA, et al. (2009) Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84:114–22.
Bruck N, et al. (2011) Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J. Clin. Rheumatol. 17:23–7.
Durand M, Troyanov Y, Laflamme P, Gregoire G. (2011) Macrophage activation syndrome treated with anakinra. J. Rheumatol. 37:879–80.
Kahn PJ, Cron RQ. (2013) Higher-dose Anakinra is effective in a case of medically refractory macrophage activation syndrome. J. Rheumatol. 40:743–4.
Kelly A, Ramanan AV. (2008) A case of macrophage activation syndrome successfully treated with anakinra. Nat. Clin. Pract. Rheumatol. 4:615–20.
Loh NK, Lucas M, Fernandez S, Prentice D. (2012) Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Intern. Med. J. 42:1358–62.
Simon DW, Aneja R, Carcillo JA, Halstead ES. (2014) Plasma exchange, methylprednisolone, IV immune globulin, and now anakinra support continued PICU equipoise in management of hyperferritinemia-associated sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome/secondary hemophagocytic lymphohistiocytosis syndrome. Pediatr. Crit. Care Med. 15:486–8.
Schulert GS, Grom AA. (2014) Pathogenesis of macrophage activation syndrome and potential for cytokine-directed therapies. Annu. Rev. Med. 2014, Nov 5 [Epub ahead of print].
Mohr V, et al. (2014) Urticaria, fever, and hypofibrinogenemia. Arthritis Rheumatol. 66:1377.
Tayer-Shifman OE, Ben-Chetrit E. (2013) Refractory macrophage activation syndrome in a patient with SLE and APLA syndrome — successful use of PET-CT and Anakinra in its diagnosis and treatment. Mod. Rheumatol. 2013, Oct 21 [Epub ahead of print].
Ravelli A, Grom AA, Behrens EM, Cron RQ. (2012) Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 13:289–98.
Opal SM, et al. (1997) Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial: the Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 25:1115–24.
Singh D, Huston KK. (2009) IL-1 inhibition with anakinra in a patient with refractory gout. J. Clin. Rheumatol. 15:366.
Gratton SB, Scalapino KJ, Fye KH. (2009) Case of anakinra as a steroid-sparing agent for gout inflammation. Arthritis Rheum. 61:1268–70.
Ghosh P, Cho M, Rawat G, Simkin PA, Gardner GC. (2013) Treatment of acute gouty arthritis in complex hospitalized patients with anakinra. Arthritis Care Res. 65:1381–4.
Schumacher HR Jr, et al. (2012) Rilonacept (interleukin-1 trap) for prevention of gout flares during initiation of uric acid-lowering therapy: results from a phase III randomized, double-blind, placebo-controlled, confirmatory efficacy study. Arthritis Care Res. 64:1462–70.
Cronstein BN, Sunkureddi P. (2013) Mechanistic aspects of inflammation and clinical management of inflammation in acute gouty arthritis. J. Clin. Rheumatol. 19:19–29.
Giamarellos-Bourboulis EJ, et al. (2009) Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 beta by mononuclear cells through a caspase 1-mediated process. Ann. Rheum. Dis. 68:273–8.
Joosten LA, et al. (2010) Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1beta production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 62:3237–48.
Duff GW, Atkins E, Malawista SE. (1983) The fever of gout: urate crystals activate endogenous pyrogen production from human and rabbit mononuclear phagocytes. Trans. Assoc. Am. Physicians. 96:234–45.
McGonagle D, et al. (2007) Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann. Rheum. Dis. 66:1683–4.
McGonagle D, Tan AL, Madden J, Emery P, McDermott MF. (2008) Successful treatment of resistant pseudogout with anakinra. Arthritis Rheum. 58:631–3.
Terkeltaub R. (2010) Update on gout: new therapeutic strategies and options. Nat. Rev. Rheumatol. 6:30–8.
Announ N, Palmer G, Guerne PA, Gabay C. (2009) Anakinra is a possible alternative in the treatment and prevention of acute attacks of pseudogout in end-stage renal failure. Joint Bone Spine. 76:424–6.
Schlesinger N, et al. (2012) Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann. Rheum. Dis. 71:1839–48.
Joosten LA, et al. (2009) Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 60:3651–62.
Bacconnier L, Jorgensen C, Fabre S. (2009) Erosive osteoarthritis of the hand: clinical experience with anakinra. Ann. Rheum. Dis. 68:1078–9.
Chevalier X, et al. (2005) Safety study of intraarticular injection of interleukin 1 receptor antagonist in patients with painful knee osteoarthritis: a multicenter study. J. Rheumatol. 32:1317–23.
Chevalier X, et al. (2009) Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 61:344–52.
Cohen SB, et al. (2011) A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL 1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 13:R125.
Maedler K, et al. (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110:851–60.
Donath MY, Shoelson SE. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11:98–107.
Masters SL, et al. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11:897–904.
van Asseldonk EJ, et al. (2011) Treatment with anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J. Clin. Endocrinol. Metab 96:2119–26.
Larsen CM, et al. (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356:1517–26.
Larsen CM, et al. (2009) Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 32:1663–8.
Cavelti-Weder C, et al. (2012) Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care. 35:1654–62.
Rissanen A, Howard C, Botha J, Thuren T. (2011) IL-1beta antibody (canakinumab) improves insulin secretion rates in subjects with impaired glucose tolerance and type 2 diabetes. Diabetes. 60 Suppl 1A.
Sloan-Lancaster J, et al. (2013) Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1beta antibody, in patients with type 2 diabetes. Diabetes Care. 36:2239–46.
Cavelti-Weder C, et al. (2011) Inhibition of IL-1beta improves fatigue in type 2 diabetes. Diabetes Care. 34:e158s.
Norheim KB, Harboe E, Goransson LG, Omdal R. (2012) Interleukin-1 inhibition and fatigue in primary Sjogren’s syndrome: a double blind, randomised clinical trial. PLoS One. 7: e30123.
Stienstra R, et al. (2011) The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell. Metab. 12:593–605.
Kamari Y, et al. (2007) Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis. 195:31–8.
Duewell P, et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 464:1357–61.
Libby P, Ridker PM, Hansson GK. (2009) Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 54:2129–38.
Ridker PM, Thuren T, Zalewski A, Libby P. (2011) Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 162:597–605.
Ridker PM, et al. (2012) Effects of interleukin-1beta inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: a phase IIb randomized, placebo-controlled trial. Circulation. 126:2739–48.
Abbate A, et al. (2008) Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 117:2670–83.
Abbate A, et al. (2010) Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur. J. Heart Fail. 12:319–22.
Toldo S, et al. (2013) Interleukin-1beta blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp. Physiol. 98:734–45.
Abbate A, et al. (2010) Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am. J. Cardiol. 105:1371–77, e1371.
Abbate A, et al. (2013) Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am. J. Cardiol. 111:1394–400.
Van Tassell BW, et al. (2014) Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 113:321–7.
Pathak S, Goldofsky E, Vivas EX, Bonagura VR, Vambutas A. (2011) IL-1beta is overexpressed and aberrantly regulated in corticosteroid nonresponders with autoimmune inner ear disease. J. Immunol. 186: 1870–9.
Vambutas A, et al. (2009) Alternate splicing of interleukin-1 receptor type II (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with AIED. PLoS One. 4:e5293.
Vambutas A, et al. (2014) Early efficacy trial of anakinra in corticosteroid-resistant autoimmune inner ear disease. J. Clin. Invest. 124:4115–22.
Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. (2010) Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 74:1267–70.
Goldbach-Mansky R. (2011) Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr. Rheumatol. Rep. 13:123–31.
Kuemmerle-Deschner JB, et al. (2011) Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann. Rheum. Dis. 70:2095–102.
Klein AK, Horneff G. (2011) Improvement of sensoneurinal hearing loss in a patient with Muckle-Wells syndrome treated with anakinra. Klin. Padiatr. 222:266–8.
Lepore L, et al. (2011) Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J. Pediatr. 157:310–315, e311.
Miyamae T, et al. (2011) Effect of anakinra on arthropathy in CINCA/NOMID syndrome. Pediatr. Rheumatol. Online J. 8:9.
Neven B, et al. (2011) Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 62:258–67.
Mueller SM, Itin P, Haeusermann P. (2011) Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 223:113–8.
Kuemmerle-Deschner JB, et al. (2011) Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res. Ther. 13:R34.
Kone-Paut I, et al. (2011) Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with canakinumab: results of a double-blind placebo-controlled randomized withdrawal study. Arthritis Res. Ther. 13:R202.
Lachmann HJ, et al. (2009) Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 360:2416–25.
Furuta T, et al. (2011) Association of interleukin-1 gene polymorphisms with sudden sensorineural hearing loss and Meniere’s disease. Int. J. Immunogenet. 38:249–54.
Ogilvie AC, et al. (1996) IL-1 beta does not cause neutrophil degranulation but does lead to IL- 6, IL-8, and nitrite/nitrate release when used in patients with cancer. J. Immunol. 156:389–94.
Granowitz EV, et al. (1993) Hematological and immunomodulatory effects of an interleukin-1 receptor antagonist coinfusion during low-dose endotoxemia in healthy humans. Blood. 82:2985–90.
Hoffman HM, et al. (2004) Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist prevents. Lancet. 364:1779–85.
Tewari A, Buhles WC Jr, Starnes HF Jr. (1990) Preliminary report: effects of interleukin-1 on platelet counts. Lancet. 336:712–4.
Janik JE, et al. (1996) Phase II trial of interleukin 1 alpha and indomethacin in treatment of metastatic melanoma. J. Natl. Cancer Inst. 88:44–9.
Lust JA, Donovan KA. (1999) The role of interleukin-1 beta in the pathogenesis of multiple myeloma. Hematol. Oncol. Clin. North Am. 13:1117–25.
Berda-Haddad Y, et al. (2011) Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc. Natl. Acad. Sci. U. S. A. 108:20684–9.
Kim B, et al. (2013) The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front. Immunol. 4:391.
Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER. (1985) Identification of a membrane-associated interleukin-1 in macrophages. Proc. Natl. Acad. Sci. U. S. A. 82:1204–8.
Kaplanski G, et al. (1994) Interleukin-1 induces interleukin-8 from endothelial cells by a juxacrine mechanism. Blood 84:4242–8.
Kaplanski G, et al. (1993) Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood. 81:2492–5.
Rider P, et al. (2011) IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187:4835–43.
Bersudsky M, et al. (2014) Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut. 63:598–609.
Bendtzen K, Svenson M, Jonsson V, Hippe E. (1990) Autoantibodies to cytokines: friends or foes? Immunol. Today. 11:167–9.
Svenson M, Poulsen LK, Fomsgaard A, Bendtzen K. (1989) IgG autoantibodies against interleukin 1 alpha in sera of normal individuals. Scand. J. Immunol. 29:489–92.
Chen CJ, et al. (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13:851–6.
Smith JW 2nd, et al. (1993) The effects of treatment with interleukin-1 alpha on platelet recovery after high-dose carboplatin. N. Engl. J. Med. 328:756–61.
Baracos V, Rodemann HP, Dinarello CA, Goldberg AL. (1983) Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin-1): a mechanism for the increased degradation of muscle proteins during fever. N. Engl. J. Med. 308:553–8.
Voronov E, et al. (2003) IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 100:2645–50.
Smith CJ, et al. (2012) Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 58:384–9.
Balkwill FR, Mantovani A. (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin. Cancer Biol. 22:33–40.
Krelin Y, et al. (2007) Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67:1062–71.
Dinarello CA. (2010) Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 29:317–29.
Dinarello CA. (2014) Interleukin-1alpha neutralisation in patients with cancer. Lancet Oncol. 15:552–3.
Dinarello CA, Simon A, van der Meer JW. (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11:633–52.
Alenazi A, Al Sonbul A, Al Jumaah S, Al Mehaidib A, Al-Mayouf SM. (2012) A retrospective review of autoinflammatory diseases in Saudi children at a rheumatology clinic. Ann. Saudi Med. 32:43–8.
Calligaris L, Marchetti F, Tommasini A, Ventura A. (2008) The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever. Eur. J. Pediatr. 167:695–6.
Chae JJ, et al. (2006) The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. U. S. A. 103:9982–7.
Meinzer U, et al. (2011) Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin. Arthritis Rheum. 41:265–71.
Mitroulis I, Papadopoulos VP, Konstantinidis T, Ritis K. (2008) Anakinra suppresses familial Mediterranean fever crises in a colchicine-resistant patient. Neth. J. Med. 66:489–91.
Moser C, et al. (2009) Successful treatment of familial Mediterranean fever with anakinra and outcome after renal transplantation. Nephrol. Dial. Transplant. 24:676–8.
Ozen S, Bilginer Y, Aktay Ayaz N, Calguneri M. (2011) Anti-interleukin 1 treatment for patients with familial Mediterranean fever resistant to colchicine. J. Rheumatol. 38:516–8.
Petropoulou AD, Robin M, Socie G, Galicier L. (2011) Transmission of familial Mediterranean fever mutation after bone marrow transplantation and successful treatment with anakinra. Transplantation. 90:102–3.
Stankovic Stojanovic K, et al. (2012) Dramatic beneficial effect of interleukin-1 inhibitor treatment in patients with familial Mediterranean fever complicated with amyloidosis and renal failure. Nephrol. Dial. Transplant. 27:1898–901.
Bilginer Y, Ayaz NA, Ozen S. (2010) Anti-IL-1 treatment for secondary amyloidosis in an adolescent S55 with FMF and Behcet’s disease. Clin. Rheumatol. 29:209–10.
Soriano A, Verecchia E, Afeltra A, Landolfi R, Manna R. (2013) IL-1beta biological treatment of familial Mediterranean fever. Clin. Rev. Allergy Immunol. 45:117–30.
Akgul O, Kilic E, Kilic G, Ozgocmen S. (2013) Efficacy and safety of biologic treatments in Familial Mediterranean Fever. Am. J. Med. Sci. 346:137–41.
Estublier C, Stankovic Stojanovic K, Bergerot JF, Broussolle C, Seve P. (2013) Myositis in a patient with familial Mediterranean fever and spondyloarthritis successfully treated with anakinra. Joint Bone Spine. 80:645–9.
Ben-Zvi I, Livneh A. (2014) Colchicine failure in familial Mediterranean fever and potential alternatives: embarking on the anakinra trial. IMAJ. 16:271–3.
Hedrich CM, et al. (2012) “Mutation negative” familial cold autoinflammatory syndrome (FCAS) in an 8-year-old boy: clinical course and functional studies. Rheumatol. Int. 32:2629–36.
Metyas SK, Hoffman HM. (2006) Anakinra prevents symptoms of familial cold autoinflammatory syndrome and Raynaud’s disease. J. Rheumatol. 33:2085–7.
Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. (2004) Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthr. Rheumat. 50:607–12.
Hawkins PN, Lachmann HJ, McDermott MF. (2003) Interleukin-1 receptor antagonist in the Muckle-Wells syndrome. N. Engl. J. Med. 348:2583–4.
Rynne M, Maclean C, Bybee A, McDermott MF, Emery P. (2006) Hearing improvement in a patient with variant Muckle-Wells syndrome in response to interleukin 1 receptor antagonism. Ann. Rheum. Dis. 65:533–4.
Kuemmerle-Deschner JB, et al. (2011) NLRP3 E311K mutation in a large family with Muckle-Wells syndrome: description of a heterogeneous phenotype and response to treatment. Arthritis Res. Ther. 13:R196.
Kuemmerle-Deschner JB, et al. (2013) Treatment of Muckle-Wells syndrome: analysis of two IL-1-blocking regimens. Arthritis Res. Ther. 15:R64.
Stew BT, Fishpool SJ, Owens D, Quine S. (2013) Muckle-Wells syndrome: a treatable cause of congenital sensorineural hearing loss. B-ENT. 9:161–3.
Sabroe RA, et al. (2013) Muckle-Wells syndrome without mutation in exon 3 of the NLRP3 gene, identified by evidence of excessive monocyte production of functional interleukin 1beta and rapid response to anakinra. Clin. Exp. Dermatol.
Enriquez R, et al. (2013) Nephrotic syndrome and AA amyloidosis revealing adult-onset cryopyrin-associated periodic syndrome. Ren. Fail. 35:738–41.
Jeru I. (2011) Role of interleukin-1 in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy.
Lovell DJ, Bowyer SL, Solinger AM. (2005) Interleukin-1 blockade by anakinra improves clinical symptoms in patients with neonatal-onset multisystem inflammatory disease. Arthritis Rheum. 52:1283–6.
Sibley CH, et al. (2012) Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum. 64:2375–86.
Aubert P, et al. (2012) Homeostatic tissue responses in skin biopsies from NOMID patients with constitutive overproduction of IL-1beta. PLoS One. 7:e49408.
Balow JE Jr, et al. (2013) Microarray-based gene expression profiling in patients with cryopyrin-associated periodic syndromes defines a disease-related signature and IL-1-responsive transcripts. Ann. Rheum. Dis. 72:1064–70.
Sacre K, et al. (2008) Dramatic improvement following interleukin 1beta blockade in tumor necrosis factor receptor-1-associated syndrome (TRAPS) resistant to anti-TNF-alpha therapy. J. Rheumatol. 35:357–8.
Simon A, et al. (2004) Beneficial response to interleukin-1 receptor antagonist in TRAPS. Am. J. Med. 117:208–10.
Gattorno M, et al. (2008) Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 58:1516–20.
Kimberley FC, Lobito AA, Siegel RM, Screaton GR. (2007) Falling into TRAPS: receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res. Ther. 9:217.
Cantarini L, Lucherini OM, Cimaz R, Galeazzi M. (2010) Recurrent pericarditis caused by a rare mutation in the TNFRSF1A gene and with excellent response to anakinra treatment. Clin. Exp. Rheumatol. 28:802.
Andres M, Pascual E. (2013) Anakinra for a refractory case of intermittent hydrarthrosis with a TRAPS-related gene mutation. Ann. Rheum. Dis. 72:155.
Lucherini OM, et al. (2013) First report of circulating microRNAs in tumour necrosis factor receptor-associated periodic syndrome (TRAPS). PLoS One. 8:e73443.
Bodar EJ, van der Hilst JC, Drenth JP, van der Meer JW, Simon A. (2005) Effect of etanercept and anakinra on inflammatory attacks in the hyper-IgD syndrome: introducing a vaccination provocation model. Neth. J. Med. 63:260–4.
Cailliez M, et al. (2006) Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J. Inherit. Metab. Dis. 29:763.
Stoffels M, Simon A. (2011) Hyper-IgD syndrome or mevalonate kinase deficiency. Curr. Opin. Rheumatol. 23:419–23.
Simon A, van der Meer JW. (2007) Pathogenesis of familial periodic fever syndromes or hereditary autoinflammatory syndromes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292:R86–98.
Bodar EJ, et al. (2011) On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann. Rheum. Dis. 70:2155–8.
Shendi HM, Walsh D, Edgar JD. (2012) Etanercept and anakinra can prolong febrile episodes in patients with hyperimmunoglobulin D and periodic fever syndrome. Rheumatol. Int. 32:249–51.
Braun-Falco M, Kovnerystyy O, Lohse P, Ruzicka T. (2012) Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J. Am. Acad. Dermatol. 66:409–15.
Brenner M, Ruzicka T, Plewig G, Thomas P, Herzer P. (2009) Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br. J. Dermatol. 161:1199–201.
Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. (2005) Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology. 44:406–8.
Schellevis MA, et al. (2011) Variable expression and treatment of PAPA syndrome. Ann. Rheum. Dis. 70:1168–70.
Shoham NG, et al. (2003) Pyrin binds the PST-PIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. U. S. A. 100:13501–6.
Demidowich AP, et al. (2012) Brief report: genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum. 64:2022–7.
Pazyar N, Feily A, Yaghoobi R. (2012) An overview of interleukin-1 receptor antagonist, anakinra, in the treatment of cutaneous diseases. Curr. Clin. Pharmacol. 7:271–5.
Marzano AV, et al. (2013) Autoinflammatory skin disorders in inflammatory bowel diseases, pyoderma gangrenosum and Sweet’s syndrome: a comprehensive review and disease classification criteria. Clin. Rev. Allergy Immunol. 45:202–10.
Reddy S, et al. (2009) An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 360:2438–44.
Aksentijevich I, et al. (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 360:2426–37.
Sakran W, et al. (2013) Chronic recurrent multi-focal osteomyelitis and deficiency of interleukin-1-receptor antagonist. Pediatr. Infect. Dis. J. 32:94.
Arostegui JI, et al. (2007) NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 56:3805–13.
Punzi L, Gava A, Galozzi P, Sfriso P. (2011) Miscellaneous non-inflammatory musculoskeletal conditions: Blau syndrome. Best Pract. Res. Clin. Rheumatol. 25:703–14.
Ruiz Gomez A, et al. (2012) Clinical, genetic, S56 and therapeutic diversity in 2 patients with severe mevalonate kinase deficiency. Pediatrics. 129:e535–9.
Herlin T, et al. (2013) Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann. Rheum. Dis. 72:410–3.
Adam Z, et al. (2008) Schnitzler syndrome: report on a fourteen-year course of the disease and an overview of information on the disease. Vnitr. Lek. 54:1140–53.
Cascavilla N, Bisceglia M, D’Arena G. (2010) Successful treatment of Schnitzler’s syndrome with anakinra after failure of rituximab trial. Int. J. Immunopathol. Pharmacol. 23:633–6.
De Koning HD, et al. (2006) Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann Rheum Dis. 65:542–4.
Eiling E, Moller M, Kreiselmaier I, Brasch J, Schwarz T. (2007) Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J. Am. Acad. Dermatol. 57:361–4.
Gran JT, Midtvedt O, Haug S, Aukrust P. (2011) Treatment of Schnitzler’s syndrome with anakinra: report of three cases and review of the literature. Scand. J. Rheumatol. 40:74–9.
Lipsker D. (2010) The Schnitzler syndrome. Orphanet J. Rare Dis. 5:38.
Martinez-Taboada VM, Fontalba A, Blanco R, Fernandez-Luna JL. (2005) Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum. 52:2226–7.
Schuster C, et al. (2009) Schnitzler syndrome: response to anakinra in two cases and a review of the literature. Int. J. Dermatol. 48:1190–4.
Larocca CA, et al. (2012) Schnitzler’s syndrome associated with pancreatitis: a disease of IL-1 dysregulation. Clin. Rheumatol. 31:169–74.
Benitez Gutierrez L, Mellor Pita S, Tutor de Ureta P, Yebra Bango M. (2013) [Schnitzler’s syndrome: a case report and literature review of the response to treatment with anakinra]. Med. Clin. (Barc.) 140:427–8.
Jain T, Offord CP, Kyle RA, Dingli D. (2013) Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. 98:1581–5.
Simon A, et al. (2013) Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 68:562–8.
Launay D, et al. (2013) Effect of in vitro and in vivo anakinra on cytokines production in Schnitzler syndrome. PLoS One. 8:e59327.
Volz T, et al. (2012) Dermal interleukin-1 expression and effective and long-lasting therapy with interleukin-1 receptor antagonist anakinra in Schnitzler syndrome. Acta. Derm. Venereol. 92:393–4.
Furlan A, Botsios C, Ruffatti A, Todesco S, Punzi L. (2008) Antisynthetase syndrome with refractory polyarthritis and fever successfully treated with the IL-1 receptor antagonist, anakinra: a case report. Joint Bone Spine. 75:366–7.
Mesquida M, et al. (2014) Current and future treatments for Behcet’s uveitis: road to remission. Int. Ophthalmol. 34:365–81.
Kuemmerle-Deschner JB, et al. (2011) Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 63:840–9.
Leslie KS, et al. (2006) Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Arch. Dermatol. 142:1591–7.
Thornton BD, Hoffman HM, Bhat A, Don BR. (2007) Successful treatment of renal amyloidosis due to familial cold autoinflammatory syndrome using an interleukin 1 receptor antagonist. Am. J. Kidney Dis. 49:477–81.
Ait-Abdesselam T, et al. (2011) Anakinra efficacy in a Caucasian patient with renal AA amyloidosis secondary to cryopyrin-associated periodic syndrome. Joint Bone Spine. 77:616–7.
Boyer EM, Turman M, O’Neil KM. (2011) Partial response to anakinra in life-threatening Henoch-Schonlein purpura: case report. Pediatr. Rheumatol. Online J. 9:21.
Picco P, et al. (2009) Successful treatment of idiopathic recurrent pericarditis in children with interleukin-1beta receptor antagonist (anakinra): an unrecognized autoinflammatory disease? Arthritis Rheum. 60:264–8.
Scott IC, Vijay Hajela V, Hawkins PN, Lachmann HJ. (2011) A case series and systematic literature review of anakinra and immunosuppression in idiopathic recurrent pericarditis. J. Cardiol. Cases. 4: e93–7.
Scardapane A, Brucato A, Chiarelli F, Breda L. (2013) Efficacy of an interleukin-1beta receptor antagonist (anakinra) in idiopathic recurrent pericarditis. Pediatr. Cardiol. 34:1989–91.
Seferovic PM, et al. (2013) Pericardial syndromes: an update after the ESC guidelines 2004. Heart Failure Rev. 18:255–66.
Camacho-Lovillo M, Mendez-Santos A. (2013) Successful treatment of idiopathic recurrent pericarditis with interleukin-1 receptor antagonist (Anakinra). Pediatr. Cardiol. 34:1293–94.
Paul SM, et al. (2005) Functional outcomes of treatment of neonatal onset multisystem inflammatory disease (NOMID) with anakina. Arthr. Rheumat. 52 Suppl:S536.
Gattorno M, et al. (2008) The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 58:1505–15.
Nigrovic PA, et al. (2011) Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 63:545–55.
DeWitt EM, et al. (2012) Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. (Hoboken). 64:1001–10.
Aelion JA, Odhav SK. (2004) Prompt response to treatment with anakinra in adult onset Still’s disease. Ann. Rheum. Dis. 63 Suppl 1:265.
Haraoui B, Bourrelle D, Kaminska E. (2004) Anakinra in the treatment of adult onset Still’s disease. Ann. Rheum. Dis. 63 Suppl 1:263.
Kotter I, et al. (2007) Anakinra in patients with treatment-resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin. Arthritis Rheum. 37:189–97.
Lahiri M, Teng GG. (2010) A case of refractory adult-onset Still’s disease treated with anakinra. Int. J. Rheum. Dis. 13:e36–41.
Lequerre T, et al. (2008) Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann. Rheum. Dis. 67:302–8.
Moulis G, Sailler L, Astudillo L, Pugnet G, Arlet P. (2010) May anakinra be used earlier in adult onset Still disease? Clin. Rheumatol. 29:1199–200.
Mylona E, et al. (2008) Acute hepatitis in adult Still’s disease during corticosteroid treatment successfully treated with anakinra. Clin. Rheumatol. 27:659–61.
Naumann L, et al. (2010) IL1-receptor antagonist anakinra provides long-lasting efficacy in the treatment of refractory adult-onset Still’s disease. Ann. Rheum. Dis. 69:466–7.
Raffeiner B, et al. (2011) Adult-onset Still’s disease with myocarditis successfully treated with the interleukin-1 receptor antagonist anakinra. Joint Bone Spine. 78:100–1.
Rudinskaya A, Trock DH. (2003) Successful treatment of a patient with refractory adult-onset Still’s disease with anakinra. J. Clin. Rheumatol. 9:330–2.
Tamaki H, et al. (2010) Marked effect and steroid-sparing ability of anakinra on a patient with refractory adult-onset Still’s disease. Mod. Rheumatol. 20:200–4.
Vasques Godinho FM, Parreira Santos MJ, Canas da Silva J. (2005) Refractory adult onset Still’s disease successfully treated with anakinra. Ann. Rheum. Dis. 64:647–8.
Vordenbaumen S, Ostendorf B, Sander O, Richter J, Schneider M. (2009) Continuation of anakinra despite the development of a pruritic rash in an otherwise refractory case of adult-onset Still’s disease. Ann. Rheum. Dis. 68:450–1.
Youssef J, Lazaro E, Blanco P, Viallard JF. (2008) Blockade of interleukin 1 receptor in Still’s disease affects activation of peripheral T-lymphocytes. J. Rheumatol. 35:2453–6.
Woo P. (2008) Anakinra treatment for systemic juvenile idiopathic arthritis and adult onset Still disease. Ann. Rheum. Dis. 67:281–2.
Teran A, et al. (2009) Adult-onset Still’s disease with liver failure requiring liver transplantation. Gastroenterol. Hepatol. 32:681–6.
Rinkin C, Van Durme C, Fautrel B, Malaise M. (2013) A rare case of arthritis and fever. Revue medicale de Liege. 68:423–7.
Jaqua NT, Finger D, Hawley JS. (2012) Adult-onset Still’s disease masquerading as sepsis in S57 an asplenic active duty soldier. Case Rep. Med. 2012:349521.
Giampietro C, et al. (2013) Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 65:822–6.
Behrens EM, Kreiger PA, Cherian S, Cron RQ. (2006) Interleukin 1 receptor antagonist to treat cytophagic histiocytic panniculitis with secondary hemophagocytic lymphohistiocytosis. J. Rheumatol. 33:2081–4.
Aronson IK, Worobec SM. (2010) Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol. Ther. 23:389–402.
Chou RC, Dinarello CA, Ferry JA, Dal Cin P. (2010) A 36-year-old woman with recurrent high-grade fevers, hypotension, and hypertriglyceridemia. Arthritis Care Res. 62:137–40.
Butin M, et al. (2013) Successful immunotherapy in life-threatening parvovirus B19 infection in a child. Pediatr. Infect. Dis. J. 32:789–92.
Delluc A, et al. (2008) Efficacy of anakinra, an IL1 receptor antagonist, in refractory Sweet syndrome. Ann. Rheum. Dis. 67:278–9.
Kluger N, Gil-Bistes D, Guillot B, Bessis D. (2011) Efficacy of anti-interleukin-1 receptor antagonist anakinra (Kineret(R)) in a case of refractory Sweet’s syndrome. Dermatology. 222:123–7.
Belani H, et al. (2013) Neutrophilic urticaria with systemic inflammation: a case series. JAMA Dermatol. 149:453–8.
Lipsker D, Perrigouard C, Foubert A, Cribier B. (2010) Anakinra for difficult-to-treat neutrophilic panniculitis: IL-1 blockade as a promising treatment option for neutrophil-mediated inflammatory skin disease. Dermatology. 220:264–7.
Aouba A, et al. (2010) Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood. 116:4070–6.
Courcoul A, Vignot E, Chapurlat R. (2014) Successful treatment of Erdheim-Chester disease by interleukin-1 receptor antagonist protein. Joint Bone Spine. 81:175–7.
Aubert O, et al. (2013) Favorable radiological outcome of skeletal Erdheim-Chester disease involvement with anakinra. Joint Bone Spine. 80:206–7.
Colina M, et al. (2010) Dysregulation of P2X7 receptor-inflammasome axis in SAPHO syndrome: successful treatment with anakinra. Rheumatology (Oxford). 49:1416–8.
Eleftheriou D, et al. (2011) Biologic therapy in refractory chronic non-bacterial osteomyelitis of childhood. Rheumatology (Oxford). 49:1505–12.
Wendling D, Prati C, Aubin F. (2012) Anakinra treatment of SAPHO syndrome: short-term results of an open study. Ann. Rheum. Dis. 71:1098–1100.
Stojanov S, et al. (2011) Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc. Natl. Acad. Sci. U. S. A. 108:7148–53.
Cantarini L, et al. (2012) Typical and severe tumor necrosis factor receptor-associated periodic syndrome in the absence of mutations in the TNFRSF1A gene: a case series. Rheumatol. Int. 32:4015–8.
Galeotti C, et al. (2008) IL-1RA agonist (anakinra) in the treatment of multifocal Castleman disease. J. Pediatr. Hematol. Oncol. 30:920–4.
Sparsa L, et al. (2012) Jessner-Kanof disease induced by leflunomide: a dermal variant of cutaneous lupus? Rheumatol. Int. 31:255–8.
Cohen S, et al. (2012) A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann. Rheum. Dis. 71:2059–61.
van de Veerdonk FL, Netea MG, Dinarello CA, van der Meer JW. (2011) Anakinra for the inflammatory complications of chronic granulomatous disease. Neth. J. Med. 69:95.
de Luca A, et al. (2014) IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc. Natl. Acad. Sci. U. S. A. 111:3526–31.
Leslie KS, Tripathi SV, Nguyen TV, Pauli M, Rosenblum MD. (2014) An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J. Am. Acad. Dermatol. 70:243–51.
Helmy A, et al. (2014) Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J. Cereb. Blood Flow Metab. 34:845–51.
Abramson SB, Amin A. (2002) Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology (Oxford). 41:972–80.
Bresnihan B, Cobby M. (2003) Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology (Oxford). 42 Suppl 2:ii22–8.
Bresnihan B, et al. (1998) Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 41:2196–204.
Botsios C, et al. (2007) Anakinra, a recombinant human IL-1 receptor antagonist, in clinical practice: outcome in 60 patients with severe rheumatoid arthritis. Reumatismo. 59:32–7.
Singh JA, et al. (2010) Biologics for rheumatoid arthritis: an overview of Cochrane reviews. Sao Paulo Med. J. 128:309–10.
Bao J, et al. (2011) Secondary failure to treatment with recombinant human IL-1 receptor antagonist in Chinese patients with rheumatoid arthritis. Clin. Rheumatol. 30:697–701.
Bresnihan B, Newmark RD, Robbins S, McCabe DP, Genant HK. (2000) Anakinra reduces the rate of joint destruction after 1 year of treatment in a randomized controlled cohort of patients with rheumatoid arthritis. Arthrit. Rheum. 43 Suppl S9:S289.
Bresnihan B, Newmark R, Robbins S, Genant HK. (2004) Effects of anakinra monotherapy on joint damage in patients with rheumatoid arthritis: extension of a 24-week randomized, placebo-controlled trial. J. Rheumatol. 31:1103–11.
Jiang Y, et al. (2000) A multicenter, double-blind, dose-ranging, randomized, placebo-controlled study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis: radiologic progression and correlation of Genant and Larsen scores. Arthritis Rheum. 43:1001–9.
Genant HK, et al. (2001) Treatment with anakinra reduces the rate of joint destruction and shows accelerated benefit in the second 6 months of treatment for patients with rheumatoid arthritis. Ann. Rheumat. Dis. 40 Suppl 1:169.
Cunnane G, Madigan A, Murphy E, FitzGerald O, Bresnihan B. (2001) The effects of treatment with interleukin-1 receptor antagonist on the inflamed synovial membrane in rheumatoid arthritis. Rheumatology (Oxford). 40:62–9.
Devine EB, Alfonso-Cristancho R, Sullivan SD. (2011) Effectiveness of biologic therapies for rheumatoid arthritis: an indirect comparisons approach. Pharmacotherapy. 31:39–51.
Wang SY, et al. (2011) Circulating Dickkopf-1 is correlated with bone erosion and inflammation in rheumatoid arthritis. J. Rheumatol. 38:821–7.
Turkstra E, Ng SK, Scuffham PA. (2011) A mixed treatment comparison of the short-term efficacy of biologic disease modifying anti-rheumatic drugs in established rheumatoid arthritis. Curr. Med. Res. Opin. 27:1885–97.
Nixon R, Bansback N, Brennan A. (2007) The efficacy of inhibiting tumour necrosis factor alpha and interleukin 1 in patients with rheumatoid arthritis: a meta-analysis and adjusted indirect comparisons. Rheumatology (Oxford). 46:1140–7.
Cohen SB, Strand V, Aguilar D, Ofman JJ. (2004) Patient- versus physician-reported outcomes in rheumatoid arthritis patients treated with recombinant interleukin-1 receptor antagonist (anakinra) therapy. Rheumatology (Oxford). 43:704–11.
Cohen SB. (2004) The use of anakinra, an interleukin-1 receptor antagonist, in the treatment of rheumatoid arthritis. Rheum. Dis. Clin. North. Am. 30:365–80.
Kavanaugh A. (2006) Anakinra (interleukin-1 receptor antagonist) has positive effects on function and quality of life in patients with rheumatoid arthritis. Adv. Ther. 23:208–17.
Cohen SB, Woolley JM, Chan W. (2003) Interleukin 1 receptor antagonist anakinra improves functional status in patients with rheumatoid arthritis. J. Rheumatol. 30:225–31.
Cohen SB, et al. (2004) A multicentre, double blind, randomised, placebo controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Ann. Rheum. Dis. 63:1062–8.
Fleischmann RM, et al. (2003) Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: a large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 48:927–34.
Jansen JP, Buckley F, Dejonckheere F, Ogale S. (2014) Comparative efficacy of biologics as monotherapy and in combination with methotrexate on patient reported outcomes (PROs) in rheumatoid arthritis patients with an inadequate response to conventional DMARDs: a systematic review and network meta-analysis. Health Qual. Life Outcomes. 12:102.
Pontifex EK, et al. (2011) Change in CD3 positive T-cell expression in psoriatic arthritis synovium correlates with change in DAS28 and magnetic resonance imaging synovitis scores following initiation of biologic therapy: a single centre, open-label study. Arthritis Res. Ther. 13:R7.
Jung N, et al. (2011) An open-label pilot study of the efficacy and safety of anakinra in patients with psoriatic arthritis refractory to or intolerant of methotrexate (MTX). Clin. Rheumatol. 29:1169–73.
Brown C, Toth A, Magnussen R. (2011) Clinical benefits of intra-articular anakinra for persistent knee effusion. J. Knee Surg. 24:61–5.
Brown CA, Toth AP, Magnussen B. (2011) Clinical benefits of intra-articular anakinra for arthrofibrosis. Orthopedics. 33:877.
Kraus VB, et al. (2012) Effects of intraarticular IL1-Ra for acute anterior cruciate ligament knee injury: a randomized controlled pilot trial (NCT00332254). Osteoarthritis Cartilage. 20:271–8.
Chauffier K, London J, Beaudouin C, Fautrel B. (2009) Indications of anakinra. Presse Med. 38:799–807.
Buonuomo PS, et al. (2011) Relapsing polychondritis: new therapeutic strategies with biological agents. Rheumatol. Int. 30:691–3.
Vounotrypidis P, Sakellariou GT, Zisopoulos D, Berberidis C. (2006) Refractory relapsing polychondritis: rapid and sustained response in the treatment with an IL-1 receptor antagonist (anakinra). Rheumatology (Oxford). 45:491–2.
Moore GH, Rootman DB, Roybal N, Goldberg RA. (2014) Orbital relapsing polychondritis: a unique presentation, complication, and treatment. Ophthal. Plast. Reconstr. Surg. 2014, Jul 28 [Epub ahead of print].
Rech J, et al. (2012) Adult-onset Still’s disease and chronic recurrent multifocal osteomyelitis: a hitherto undescribed manifestation of autoinflammation. Rheumatol. Int. 32:1827–9.
Haibel H, Rudwaleit M, Listing J, Sieper J. (2005) Open label trial of anakinra in active ankylosing spondylitis over 24 weeks. Ann Rheum Dis.
Bennett AN, et al. (2008) Sustained response to anakinra in ankylosing spondylitis. Rheumatology (Oxford). 47:223–4.
Tan AL, et al. (2004) Efficacy of anakinra in active ankylosing spondylitis: a clinical and magnetic resonance imaging study. Ann. Rheum. Dis. 63:1041–5.
So A. (2010) Epidemiology: gout—bad for the heart as well as the joint. Nat. Rev. Rheumatol. 6:386–7.
Diamantopoulos AP, Brodin C, Hetland H, Haugeberg G. (2012) Interleukin 1beta blockade improves signs and symptoms of chronic calcium pyrophosphate crystal arthritis resistant to treatment. J. Clin. Rheumatol. 18:310–11.
Molto A, Ea HK, Richette P, Bardin T, Liote F. (2012) Efficacy of anakinra for refractory acute calcium pyrophosphate crystal arthritis. Joint Bone Spine. 79:621–3.
Ottaviani S, et al. (2013) Efficacy of anakinra in calcium pyrophosphate crystal-induced arthritis: a report of 16 cases and review of the literature. Joint Bone Spine. 80:178–82.
Funck-Brentano T, et al. (2011) First observation of the efficacy of IL-1ra to treat tophaceous gout of the lumbar spine. Rheumatology (Oxford). 50:622–4.
Zong M, et al. (2014) Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann. Rheum. Dis. 73:913–20.
Latourte A, Frazier A, Briere C, Ea HK, Richette P. (2013) Interleukin-1 receptor antagonist in refractory haemochromatosis-related arthritis of the hands. Ann. Rheum. Dis. 72:783–4.
Acknowledgements
This work was supported by National Institutes of Health Grants AI-15614, CA-04-6934 and AR-45584.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Dinarello, C.A. An Expanding Role for Interleukin-1 Blockade from Gout to Cancer. Mol Med 20 (Suppl 1), S43–S58 (2014). https://doi.org/10.2119/molmed.2014.00232
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00232