
Abstract
Inflammatory responses play a critical role in ischemic brain injury. MicroRNA-155 (miR-155) induces the expression of inflammatory cytokines, and acetylbritannilactone (ABL) exerts potent antiinflammatory actions by inhibiting expression of inflammation-related genes. However, the functions of miR-155 and the actual relationship between ABL and miR-155 in ischemia-induced cerebral inflammation remain unclear. In this study, cerebral ischemia of wild-type (WT) and miR-155−/− mice was induced by permanent middle cerebral artery occlusion (MCAO). pAd-miR-155 was injected into the lateral cerebral ventricle 24 h before MCAO to induce miR-155 overexpression. MCAO mice and oxygen-glucose deprivation (OGD)-treated BV2 cells were used to examine the effects of ABL and miR-155 overexpression or deletion on the expression of proinflammatory cytokines. We demonstrated that ABL treatment significantly reduced neurological deficits and cerebral infarct volume by inhibiting tumor necrosis factor-α (TNF-α) and interleukin-1 β (IL-1 β) expression in ischemic cerebral tissue and OGD-treated BV2 cells. Mechanistic studies suggested that the observed decrease in TNF-α and IL-1 β expression was attributable to the ABL-induced suppression of the expression of nuclear factor-kappa B (NF-κB) and Toll-like receptor 4 (TLR4). We further found that miR-155 promoted TNF-α and IL-1 β expression by upregulating TLR4 and downregulating the expression of suppressor of cytokine signaling 1 (SOCS1) and myeloid differentiation primary response gene 88 (MyD88), while ABL exerted an inhibitory effect on miR-155-mediated gene expression. In conclusion, miR-155 mediates inflammatory responses in ischemic cerebral tissue by modulating TLR4/MyD88 and SOCS1 expression, and ABL exerts its antiinflam-matory action by suppressing miR-155 expression, suggesting a novel miR-155-based therapy for ischemic stroke.
Similar content being viewed by others

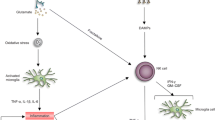

Introduction
Ischemic stroke is a leading cause of death and permanent disability that affects millions of people worldwide and remains a major challenge to public health. In addition to the primary hypoxic damage and secondary processes, such as excitotoxicity, inflammatory events that occur during cerebral ischemia are critical for the pathogenesis of tissue damage in ischemic stroke (1,2). In ischemic brain injury, molecules released from injured tissue, blood vessels and necrotic cells activate Toll-like receptors (TLRs) and induce production of proinflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α). These cytokines cause postischemic inflammation in the brain, subsequently leading to exacerbated primary brain damage (3,4). Previous work has demonstrated that activation of TLRs either by microbial or endogenous ligands results in an inflammatory response by delivering a signal from the intracellular TIR (Toll/IL-1 receptor) domain of TLR through a series of intracellular adaptor molecules, including MyD88, interleukin-1 receptorassociated kinase (IRAK) and TNF receptor associated factor 6 (TRAF6) (5,6). For example, IRAK and TRAF6 are recruited to the TLR complex via interaction with MyD88 (7) and activate nuclear factor-kappa B (NF-κB), a transcription factor that regulates the expression of a wide array of genes involved in inflammatory responses, as well as the mitogenactivated protein kinases (MAPKs), such as extracellular signal-regulated kinase (ERK), p38 and c-jun N-terminal kinase (JNK) (8,9). However, the precise mechanism of cerebral ischemia-induced expression of proinflammatory cytokines is still not fully understood.
The expression of inflammation-related genes can be regulated not only by transcription factors at the transcriptional level but also by microRNAs (miRNAs, miRs) at the posttranscriptional level. Emerging evidence suggest that miRNAs are involved in regulating several aspects of inflammation (10,11) and the response to cerebral ischemia (12). Among various miRNAs, miR-155 is an inflammation-related miRNA. Its expression is induced in inflammatory macrophages (11,13), as well as in the ischemic cerebral cortex by middle cerebral artery occlusion (MCAO) surgery (14) and, in turn, it enhances proinflammatory cytokine expression in macrophages by regulating the NF-κB signaling pathway (11,15). In addition, miR-155 can exert both pro- and antiinflammatory effects by targeting different mediators of inflammatory signaling, such as SHIP1, SOCS1, SMAD2 and TAB2 (16,17). Given the importance of miR-155 in the development of postischemic brain inflammation, understanding the role of miR-155 and the mechanism of miR-155 actions in ischemic cerebral injury will provide novel insights into ischemic stroke therapy.
Pharmacological alleviation of inflammatory response is one of the most promising avenues for stroke therapy. Acetylbritannilactone (ABL) is a new antiinflammatory compound isolated from Inula britannica L., a traditional Chinese medicinal herb (for its structure, see Figure 1A). Its chemical structure is different from ergolide, which was isolated previously (18). Several of the previous studies demonstrate that ABL inhibits the expression of inflammation-associated genes, such as the NOS and COX-2 genes, by reducing IκB-α phosphorylation and degradation, inhibiting NF-κB activation and blocking the binding of active NF-κB to the target gene promoters in RAW 264.7 macrophages and vascular smooth muscle cells (VSMCs) (19–21). In addition, ABL also can suppress PDGF-induced DNA synthesis and cell proliferation, subsequently leading to apoptosis in proliferative VSMCs via the induction of a higher ratio of Bax/Bcl-2, activation of caspase-9/-3 and the cleavage of the endogenous substrate Poly (ADP-ribose) polymerase (22). Although ABL exhibits potent antiinflammatory and proapoptotic effects in macrophages and VSMCs, the effects of ABL on cerebral ischemia have not been evaluated. Simultaneously, it also remains unclear whether antiinflammation effects of ABL on cerebral ischemia are mediated by miR-155. In this study, we investigated whether and how ABL exerts a protective effect on focal cerebral ischemia-induced inflammation by regulating miR-155 expression.

ABL decreases inflammatory responses induced by focal cerebral ischemia. (A) The chemical structure of ABL. 1-O-acetylbritannilactone(R)-4([3aS,4S,7aR]-4-hydro><y-6-methyl-3-methylene-2-oxo-2, 3, 3a, 4, 7, 7a-hexahydrobenzofuran-5-yl) pentyl acetate. (B) Neurological deficit scores were assessed 24 h after MCAO. n = 10 per group. (*P< 0.05 versus MCAO group; **P < 0.01 versus MCAO group, Mann-Whitney Utest). (C) left, Representative TTC-stained coronal sections of mouse brain, showing cerebral infarction volume in each group of mice; right, Bar graphs show the infarct volume (%) of each group of mice. Data are means ± SD (n = 6 per group) *P< 0.05 versus MCAO group. (D) left, The protein levels of TNF-α and IL-1 β in brain tissues of sham, MCAO and MCAO + ABL-treated groups were measured by Western blotting; right, Band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group), *P < 0.05 versus sham group; #P< 0.05 versus MCAO group. (E) mRNA levels of TNF-α and IL-1 β in brain tissues of sham, MCAO and MCAO + ABL-treated groups were determined by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P < 0.05 versus sham group; #P < 0.05 versus MCAO group. (F) Bar graphs show the protein concentrations of TNF-α and IL-1 β in brain tissues of sham, MCAO, and MCAO + ABL-treated groups, as measured by ELISA. Data are presented as means ± SD (n = 6 per group);. *P < 0.05 versus sham group; #P < 0.05 versus MCAO. (G) Immunohistochemical staining for TNF-α and IL-1 β in brain tissues of sham, MCAO and MCAO + ABL-treated groups. Scale bar = 20 µmol/L. Bars represent the number of TNF-α- and IL-1 β-positive cells. n = 5 per group; *P < 0.05 versus sham group; #P < 0.05 versus MCAO group. (H) BV2 cells were treated with or without ABL (100 µmol/L) for 24 h prior to OGD. TNF-α and IL-1 β were detected by Western blotting, and band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD from three independent experiments; *P < 0.05 versus con group; #P < 0.05 versus OGD group.
Materials and Methods
Experimental Animals
Adult male CD1 mice (25–30 g) and male C57/BL6 mice (25–30 g) were purchased from the Vital River Laboratory Animal Technology Co., Ltd., Beijing, China. miR-155−/− mice (25–30 g) were purchased from the Jackson Laboratory (Bar Harbor, MN, USA). All mice were given at least 3 d to acclimatize ahead of any experimentation. During this period, all mice had free access to food and water. Animal houses were maintained in a light-dark (12:12 h) cycle with humidity of 60% ± 5% and ambient temperature of 22 ± 3°C. All experimental procedures were approved by Hebei Medical University Ethics Committee as well as the institutional animal care and use committee.
Animal Surgery for Middle Cerebral Artery Occlusion
A standard model of permanent right middle cerebral artery occlusion (MCAO) was established by following the surgical procedures as previously described (23). In brief, mice were anesthetized by intraperitoneal injection of chloral hydrate (10%). The common carotid artery (CCA) were exposed and isolated. The middle cerebral artery (MCA) was occluded by inserting a heparin-dampened monofilament nylon suture (Beijing Sunbio Biotech Co., Ltd., Beijing, China) into the internal carotid artery (ICA), and advancing it further until it closed the origin of the MCA. Sham-operated mice underwent the same surgical procedures except for filament insertion. Animal body temperature was monitored and maintained at 36.5°C to 37.5°C during the experiment.
Drug Administration
The ABL (Hebei Medical University, Shijiazhuang, China) with purity of more than 99.5% was freshly dissolved in 0.9% NaCl including 1% Tween-80 prior to the treatments. The drug or solvent was injected intraperitoneally 30 min prior to MCAO operation.
Adenovirus Expression Vector and Lateral Cerebral Ventricle (LV) Injection
Adenoviruses harboring a DNA fragment encompassing the Mus musculus (mmu)-miR-155 gene (pAd-miR-155) were Invitrogen products (Thermo Fisher Scientific Inc., Waltham, MA, USA). Briefly, DNA oligos encoding the mmumiR-155 (13MR0026-1F 5′-TGCTG TTAATGCTAATTGTGATAGGGGTGTTTTGGCCACTGACTGACACCCCTATCAATTAGCATTAA-3′; 13MR0026-1R 5′-CCTGTTAATGCTAATTGATAGGGGTGTCAGTCAGTGGCCAAAACACCCCT ATCACAATTAGCATTAAC-3′) were cloned into pcDNA6.2-GW/EmGFP-miR, then pDONR221 gateway entry vector by BP recombination reaction, and finally pAD/CMV-DEST by LR recombination reaction.
The stereotactic injections of adenoviral construct (pAd or pAd-miR-155) into LV were performed at coordinates −0.5 mm anteroposterior, ± 1.5 mm mediolateral, and −2.0 to −2.5 mm dorsoventral from the bregma 24 h prior to MCAO.
Experimental Groups and Treatments
Experiment 1. All CD1 mice were randomly divided into five groups (n = 195, 39 mice in each group): (1) sham group: animals received sham operation and equal volume of 0.9% NaCl; (2) MCAO group: animals received MCAO and equal volume of 0.9% NaCl; (3) three ABL treatment groups: animals underwent MCAO and were treated with a high dose of ABL (30 mg/kg), middle dose of ABL (20 mg/kg) or low dose of ABL (10 mg/kg).
Experiment 2. All C57/BL6 mice were randomly divided into eight groups (n = 264, 33 mice in each group): virus groups: 10 µL (2 × 109 plaque-forming units/mL) of adenoviral construct (pAd or pAd-miR-155) was injected stereotactiinto LV of C57/BL6 mice 24 h before MCAO, 20 mg/kg ABL or 0.9% NaCl was injected intraperitoneally 30 min before MCAO; KO groups: 20 mg/kg ABL or 0.9% NaCl was injected intraperitoneally in miR-155−/−mice or wild-type (WT) C57/BL6 mice 30 min before MCAO operation.
Neurological Deficit Score
A neurological test was administered by an investigator blinded to the experimental groups at 24 h after MCAO following a modified scoring system developed by Longa et al. as follows: 0, no deficits; 1, difficulty in fully extending the contralateral forelimb; 2, inability to extend the contralateral forelimb; 3, mild circling to the contralateral side; 4, severe circling; 5, falling to the contralateral side (n = 10 in each group). The higher the neurological deficit score, the more severe impairment of motor motion injury.
Brain Infarct Volume
Infarct volume of brain was measured 24 h after MCAO. After the brains (n = 6 in each group) were dissected, each brain sample was sliced into five slices with 2-mm thickness, incubated by a 2% solution of 2, 3, 5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, St Louis, MO, USA) at 37°C for 15 min, followed by fixation with 4% paraformaldehyde. With TTC staining, all normal tissue was stained in dark red, while the infarct area was stained in pale gray color. TTC-stained slices were then photographed and analyzed using Image-Pro Plus 5.1 software (Media Cybernetics Inc., Bethesda, MD, USA), and the infarct volumes were calculated as follows: percentage hemisphere lesion volume (% HLV) = {[total infarct volume − (volume of intact ipsilateral hemisphere − volume of intact contralateral hemisphere)]/contralateral hemisphere volume} × 100% (24).
Immunohistochemistry
For immunohistochemical staining, brain tissues were removed and immediately immersed in 4% paraformaldehyde with phosphate-buffered saline (PBS; 0.01 mol/L, pH 7.4) for 24 h at 4°C and then dehydrated in a graded series of alcohols and embedded in paraffin. Brain sections (5 µm thick) were blocked in 3% H2O2 to eliminate endogenous peroxidase activity and 3% normal goat serum, and then incubated overnight with antimouse NF-κB (1:100, Cell Signaling Technology, Danvers, MA, USA), TNF-α (1:100, Abcam, Cambridge, MA, USA), TLR-4 (1:100, Abgent, a WuXi AppTec company, Suzhou, Jiangsu Province, China), and IL-1β (1:100, Abcam) rabbit polyclonal antibodies in 0.01 mol/L PBS overnight at 4°C. After a PBS wash, the sections were incubated with secondary antibody at 37°C for 30 min. Immunohistochemical staining was visualized by use of a diaminobenzidine kit (Zhongshan Goldenbridge Biotechnology, Beijing, China) according to the manufacturer’s instructions. Meanwhile, five visual fields of ischemic region from the infarct area were selected and the immunoreactive cells were counted under a 400× light microscope. The average number was used for statistical analysis and represented the NF-κB-, TNF-α-, TLR-4-, and IL-1 β-immunopositive cells (n = 5 in each group).
Western Blotting
All proteins were obtained from the cortex of the ischemic hemisphere using a total protein extraction kit from Applygen Technologies Inc. (Beijing, China) following the manufacturer’s protocols. The protein concentrations of the extracts were determined using a BCA Protein Assay reagent kit (Novagen, Madison, WI, USA), while 50 µg of proteins (n = 6 in each group) was separated by SDS/PAGE electrophoresis and transferred to PVD membranes. The membranes were blocked with 5% nonfat milk in 0.01 mol/L PBS at room temperature for 1 h and then incubated overnight at 4°C with the primary antibodies, including 1:1000 anti-TNF-α (Abcamab9739), 1:500 anti-IL-1β (Abcamab9787), 1:1000 anti- NF-κB p56 (Cell Signaling Technology, 3034), 1:1000 antiTLR-4 (CABGENT, AP96850), 1:1000 anti-MyD88 (Abcam, ab2068), 1:1000 anti-I-κB (Cell Signaling Technology, 9242), 1:1000 anti-TAB2 (Abcamab172412), 1:500 anti-SOCS1 (Abcamab62584), 1:1000 anti-p-Akt (Cell Signaling Technology, 4060), 1:2000 anti-p-ERK (Cell Signaling Technology, 4370), 1:1000 anti-p-JNK (Cell Signaling Technology, 9255), 1:1000 anti-p-p38 (Cell Signaling Technology, 4511), 1:1000 anti-Ac-NF-κB (Affinity, Cincinnati, OH, USA, AF1017) and 1:1000 anti-p-NF-κB (Cell Signaling Technology, 3039). Polyclonal rabbit anti-β-actin antibody (1:1000, Sigma-Aldrich) was used for internal control. After three washes with TPBS, all membranes were incubated with IRDye 800-conjugated goat anti-rabbit or anti-mouse secondary antibodies for 1 h at room temperature. Signals were developed using Super Signal West Pico Chemi-luminescent Substrate (Thermo Scientific [Thermo Fisher Scientific]).
Quantitative Real-Time Polymerase Chain Reaction
Total RNA from ischemic region cortex was extracted by TRIzol (Invitrogen [Thermo Fisher Scientific]) and 1 µg of RNA was subjected to reverse transcription using first-strand cDNA synthesis kit (Invitrogen [Thermo Fisher Scientific]) according to the manufacturer’s instructions. Quantitative RT-PCR of mRNAs was performed using a Platinum SYBR Green qPCR SuperMix UDG Kit (Invitrogen [Thermo Fisher Scientific]). A GAPDH endogenous control was used for normalization. All RT-PCR experiments were run on a ABI 7700 FAST system, and the relative amount of transcripts was calculated using the 2−ΔΔCt formula (n = 6 in each group). qRT-PCR was used to analyze the levels of TNF-α, IL-1 β, TLR-4, NF-κB, SOCS1 and MyD88 mRNA at 24 h after MCAO. Mouse primer sequences are as follows: TLR-4, forward 5′-CTCACAGATAGCCTG GCCAATC-3′ and reverse 5′-CCATC TCACAAGGCATGTCCAG-3′; IL-1β, forward 5′-GCAACTGTTCCTGAACTCAA CT-3′ and reverse 5′-ATCTTTTGGG GTCCGTCAACT-3 ′; TNF-α, forward 5′-CACCACCATCAAGGACTCAA-3′ and reverse 5′-AGGCAACCTGACCAC TCTCC-3′; NF-κB, forward 5′-ACAGA CCCAGGAGTGTTCACAGA-3′ and reverse 5′-CATGGACACACCCTG GTTCAG-3′; SOCS1, forward 5′-GGTTG TAGCAGCTTGTGTC-3′ and reverse 5′-AATGAAGCCAGAGACCCTC-3′; MyD88, forward 5′-ATGTTCTCCATACCC TTG-3′ and reverse 5′-ACTGCTTTCC ACTCTGGC-3′; GAPDH, forward 5′-CGTGTTCCTACCCCCAATGT-3′ and reverse 5′-TGTCATCATACTTGG CAGGT TTCT-3′.
Quantitative RT-PCR (qRT-PCR) for the quantification of miR-155 and primary transcript (pri)-miR-155 was performed according to the manufacturer’s protocol (Applied Biosystems [Thermo Fisher Scientific]) in two steps using specific primers for mmu-miR-155 (assay ID: 001806) and mmu-pri-miR-155 (assay ID: Mm03306395) with RnU6b (assay ID: 001093) as a normalization control.
Enzyme-Linked Immunosorbent Assay
Tissue samples were taken from the ischemic region of the brain (n = 6 in each group). Protein levels of TNF-α and IL-1β were quantified using an enzyme-linked immunosorbent assay (ELISA) kit (Ray Biotech, Norcross, GA, USA) according to the manufacturer’s protocol. The absorbance was measured with a microplate reader (Tecan SPECTRAFluor Plus, Tecan, Grödig, Austria).
BV2 Culture and Treatment
The murine BV2 microglial cell line was obtained from Xiehe Medical University (Beijing, China). The cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 100 µg/mL streptomycin, 100 U/mL penicillin (Hyclone, Logan, UT, USA), and 2 mmol/L glutamine (Invitrogen [Thermo Fisher Scientific]) at 37°C in a humidified atmosphere containing 5% CO2. BV2 cells were pretreated by ABL (100 µmol/L) for 24 h and then stimulated with different treatments.
Oxygen-Glucose Deprivation
BV2 cells were grown in complete media supplemented with glucose (4.5 g/mL) for 18 h in normoxic conditions (5% CO2 and 21% O2). To initiate oxygen-glucose deprivation (OGD), BV2 cells were exposed to DMEM without serum or glucose in a humidified atmosphere containing 95% nitrogen and 5% CO2 at 37°C for 4 h in an incubator (Serico CB, Binder GmBH, Tultingen, Germany). Control cells were incubated for 4 h in 5% CO2 and 21% O2 in a media identical to the OGD media except for the addition of glucose.
Cell Transfection
BV2 cells were transfected with indicated plasmids using Lipofectamine 2000 (Invitrogen [Thermo Fisher Scientific]) according to the manufacturer’s instructions. After transfection for 24 h, BV2 cells were pretreated with ABL (100 µmol/L) for 24 h and then exposed to OGD for 4 h. 1 µmol/L of the mature mimic miR-155 (Dharmacon Research Inc., Lafayette, CO, USA), respectively, 2 µmol/L miR-155 inhibitor miRNA (Dharmacon) was transiently transfected into BV2 cells using Lipofectamine 2000 reagent (Invitrogen [Thermo Fisher Scientific]) according to the manufacturer’s protocol. Twenty hours after transfection, the cells were treated with ABL and OGD as mentioned above.
Reporter Gene Assays
miR-155 (BIC) promoter (−1500 ± 10 bp) was constructed using PGL3 plasmids. BV2 cells were seeded in a 24-well plate and grown for 24 h before transfection with indicated plasmids. Cells were transfected using Lipofectamine 2000 (Invitrogen [Thermo Fisher Scientific]) according to the manufacturer’s instructions. Cells were then harvested and luciferase activities were measured using a Dual Luciferase Assay Kit (Promega, Madison, WI, USA). Specific promoter activity was expressed as the relative activity ratio of firefly luciferase to Renilla luciferase. All promoter constructs were evaluated in a minimum of three separate wells per experiment.
Statistical Analysis
All data were analyzed using proper tools from SPSS 13.0 package, while all quantitative data were expressed as mean ± SD. Statistical comparisons were conducted using one-way analysis of variance (ANOVA) followed by SNK and LSD tests for intergroup comparisons. For neurological deficit, Mann-Whitney U test was applied for comparisons between groups. Differences with P < 0.05 were considered statistically significant.
All supplementary materials are available online at https://doi.org/www.molmed.org.
Results
ABL Decreases Inflammatory Responses Induced by Focal Cerebral Ischemia
The protective effect of ABL on ischemic brain injury was evaluated by assessment of neurological deficit scores and cerebral infarct volumes of MCAO mice at 24 h after ischemia. The results showed that ABL treatment dose dependently reduced ischemia-induced neurological deficits and cerebral infarct volumes as determined by neurological testing (Figure 1B; *P < 0.05) and TTC staining (Figure 1C; *P < 0.05). The ABL-treated group showed significantly reduced neurological deficits at a dose of 20 mg/kg of ABL and significantly decreased infarct volume at a dose of 10 mg/kg of ABL. To assess whether the protective effect of ABL against cerebral ischemic injury is related to its antiinflammatory properties, we examined some cytokines closely related to the proinflammatory response. Western blotting analysis showed a significant increase in TNF-α and IL-1β in ischemic cerebral tissues 24 h after MCAO. ABL treatment markedly suppressed ischemia-induced upregulation of TNF-α and IL-1β in ischemic cerebral tissues, and 20 mg/kg and 30 mg/kg of ABL were more effective than 10 mg/kg of ABL (Figure 1D), suggesting that ABL dose-dependently inhibits inflammation induced by MCAO. Consistent with the inhibitory effect of ABL on protein expression, ABL also significantly inhibited the mRNA expression of TNF-α and IL-1β induced by MCAO, as assessed by qRT-PCR (Figure 1E). The protein concentrations of TNF-α and IL-1β in brain tissues 24 h after MCAO were measured by ELISA, and the results showed a similar trend to those observed in the qRT-PCR analysis (Figure 1F). In addition, the antiinflammatory effect of ABL was further verified by immunohistochemical staining for TNF-α and IL-1β, which showed that upregulation of TNF-α and IL-1β by MCAO also was suppressed with the different doses of ABL, as shown by the number of IL-1β- and TNF-α-positive cells (Figure 1G left and right).
Because microglia are the primary immune competent cells enriched in brain tissue and because they respond rapidly to brain insults by changing their morphology and cytokine production (25), we sought to examine the effect of ABL on the activation of BV2 microglial cells after OGD. As shown in Figure 1H (left and right), the levels of TNF-α and IL-1β were increased after 4 h of OGD, and 100 µmol/L of ABL significantly reduced the TNF-α and IL-1β levels in BV2 cells under OGD for 4 h. This result is consistent with the inhibitory effect of ABL on ischemia-induced inflammation in MCAO mice.
ABL Decreases Inflammatory Responses via Inhibiting the TLR4/NF-κB Signaling Cascades
Because TLR4 mediates microglial inflammatory responses, which is important for microglial activation (26), we sought to determine whether the TLR signaling pathway mediates the expression of proinflammatory cytokines in response to ischemic brain injury and whether ABL inhibits cerebral ischemia-induced inflammation by blocking the TLR signaling pathway. As shown in Figures 2A and Figure 2B, the expression levels of TLR4 and NF-κB were obviously increased after 24 h of MCAO, as determined by Western blotting and the number of NF-κB- and TLR4-positive cells in immunohistochemical staining. MCAO provoked a significant decrease in the expression of I-κB and TAB2 (see Figure 2A). However, the protein levels of MyD88 and SOCS1 were unchanged after 24 h of MCAO. Compared to the MCAO group, TLR4 and NF-κB expression were significantly decreased in the three ABL-treated groups, and the inhibitory effect of 30 mg/kg of ABL on TLR4 and NF-κB expression was more significant than that of 10 mg/kg and 20 mg/kg of ABL (Figure 2A right). By contrast, ABL dose-dependently increased the expression of I-κB, MyD88 and SOCS1; these protein expression levels were much higher in MCAO mice treated with 30 mg/kg of ABL than those in control mice. A similar result was observed in OGD-treated BV2 cells (Figure 2C), showing that ABL reduced NF-κB and TLR4 levels significantly, but increased levels of I-κB, MyD88 and SOCS1. These results indicate that ABL decreases inflammatory responses induced by MCAO and OGD by suppressing NF-κB and TLR4 expression and promoting the expression of MyD88, SOCS1 and I-κB, which are involved in the TLR signaling pathway and negatively regulate inflammatory responses.

ABL decreases inflammatory responses via inhibiting the TLR4/NF-κB signaling cascades. (A) Total proteins were extracted from brain tissues of sham, MCAO and MCAO + ABL-treated mice and analyzed by Western blotting for TLR4, NF-κB, I-κB, MyD88, SOCS1 and TAB2, and band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group), *P < 0.05 versus sham group; #P < 0.05 versus MCAO group. (B) Immunohistochemical staining for NF-κB and TLR4 in brain tissues of sham, MCAO, and MCAO + ABL-treated groups. Scale bar = 20 µmol/L. Bars represent the number of NF-κB- and TLR4-positive cells. n = 5 per group; *P < 0.05 versus sham group; #P < 0.05 versus MCAO group. (C) BV2 cells were treated with or without ABL (100 µmol/L) for 24 h prior to OGD. TLR4, NF-κB, I-κB, MyD88, SOCS1 and TAB2 were detected by Western blotting, and band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD from three independent experiments; *P< 0.05 versus con group; #P< 0.05 versus OGD group.
ABL Inhibits miR-155 Expression in Ischemic Cerebral Tissue and BV2 Microglial Cells
miR-155 is an inflammation-related miRNA and enhances proinflammatory cytokine expression (13). To examine whether miR-155 is required for cerebral inflammatory responses induced by MCAO and whether the inhibitory effect of ABL on cerebral inflammatory responses is related to its regulation of miR-155, we examined miR-155 expression in ischemic cerebral tissue of MCAO mice and OGD-treated BV2 cells. The results showed miR-155 was upregulated in ischemic cerebral tissues after 24 h of MCAO. Importantly, ABL treatment dose-dependently suppressed the expression of miR-155 induced by MCAO (Figure 3A). The results from MCAO mice were verified further with OGD-treated BV2 cells, which showed that the miR-155 upregulation induced by OGD was abolished by treating BV2 cells with 100 µmol/L of ABL (Figure 3B). In parallel with these results, measurements of luciferase activity showed that OGD treatment significantly increased the transcriptional activity of the miR-155 promoter compared with OGD-untreated BV2 cells (Figure 3C). These results indicate that miR-155 is implicated in the cerebral inflammation induced by MCAO, and that ABL has an inhibitory effect on the miR-155 expression induced by MCAO.

ABL inhibits miR-155 expression in the ischemic cerebral tissue and BV2 microglial cells. (A) miR-155 at brain tissues of sham, MCAO and MCAO + ABL-treated mice was examined by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P< 0.05 versus sham group; #P< 0.05 versus MCAO group. (B) BV2 cells were treated with or without ABL (100 µmol/L) for 24 h prior to OGD. miR-155 at BV2 cells of con, con + ABL, OGD and OGD + ABL-treated groups was examined by qRT-PCR. Bars represent means ± SD from three independent experiments; *P< 0.05 versus con group; #P< 0.05 versus OGD group. (C) BV2 cells were transfected with miR-155 (BIC) promoter and PGL3 plasmids, and then treated with or without ABL (100 µmol/L) for 4 h prior to OGD, miR-155 promoter activity was detected by dual luciferase assay. Data are presented as the relative activity ratio of firefly luciferase to Renilla luciferase. *P< 0.05 versus con group.
ABL Suppresses miR-155-Mediated Inflammatory Responses in Ischemic Cerebral Tissue and BV2 Microglial Cells
Next, the role of miR-155 in ischemic brain injury induced by MCAO was evaluated by comparing the cerebral infarction volume of miR-155−/− and WT mice. We found that knockout (KO) of miR-155 obviously reduced the neurological deficit (see Figure 1B) and cerebral infarct volume (see Figure 1C) induced by MCAO, suggesting that deletion of miR-155 protects against ischemic brain injury induced by MCAO. Western blotting analysis showed that the upregulation of TNF-α and IL-1β by MCAO in the ischemic cerebral tissues of miR-155−/− mice was reduced markedly compared with WT mice, and ABL treatment further reduced TNF-α and IL-1β expression in miR-155−/− mice (Figures 4A, B). A similar result was obtained by qRT-PCR, which showed that knockout of miR-155 obviously reduced the TNF-α and IL-1β mRNA levels after MCAO surgery (Figure 4C). We then introduced pAd-miR-155 into WT mice through an LV injection to overexpress miR-155. We found miR-155 levels were dramatically increased in the brain after 24 h of delivery of pAd-miR-155, and miR-155 was widely expressed in the whole mouse brain, especially in the cortex and hippocampus (Supplementary Figure 1). Measurements of protein expression showed that miR-155 overexpression markedly increased the levels of TNF-α and IL-1β in ischemic cerebral tissue of MCAO mice, and that the up-regulation of these two cytokines induced by miR-155 overexpression was slightly reduced by ABL treatment relative to that of miR-155 overexpression alone (Figures 4D, E). The qRT-PCR results revealed significant parallel alterations in the mRNA expression of these genes (Figure 4F). Furthermore, using cultured BV2 microglial cells, we examined the effect of miR-155 overexpression or knockdown on the expression of TNF-α and IL-1β. The results showed that overexpression of miR-155 significantly increased the levels of TNF-α and IL-1β in OGD-treated BV2 cells (Figure 4G). Conversely, siRNAmediated knockdown of miR-155 was accompanied by a marked reduction in TNF-α and IL-1β compared with the nonsilencing control siRNA, and ABL treatment further decreased the expression levels of these two cytokines (Figure 4H). These results clearly suggest that miR-155 mediates the expression of TNF-α and IL-1β in ischemic cerebral tissue.

ABL suppresses miR-155-mediated inflammatory responses in the ischemic cerebral tissue and BV2 microglial cells. (A) Total proteins were extracted from brain tissues of MCAO WT and miR-155−/− mice treated with or without ABL (20 mg/kg) and analyzed by Western blotting for TNF-α and IL-1 β. (B) Band intensities in panel A that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group); *P < 0.05 versus WT + MCAO; #P < 0.05 versus miR-155KO + MCAO. (C) mRNA levels of TNF-α and IL-1 β in brain tissues of MCAO WT and miR-155−/− mice treated with or without ABL (20 mg/kg) were detected by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P< 0.05 versus WT + MCAO; #P< 0.05 versus miR-155KO + MCAO. (D) WT mice were infected with pAd or pAd-miR-155 and treated with or without ABL (20 mg/kg). TNF-α and IL-1 β 24 h after MCAO were detected by Western blotting. (E) Band intensities in panel D that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group); *P< 0.05 versus pAd + MCAO; #P< 0.05 versus pAd-miR155 + MCAO. (F) mRNA levels of TNF-α and IL-1 β in brain tissues of pAd- or pAd-miR-155-infected MCAO mice treated with or without ABL (20 mg/kg) were detected by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P< 0.05 versus pAd + MCAO; #P< 0.05 versus pAd-miR155 + MCAO. (G) BV2 cells were infected with pAd-miR-155 or si-miR-155, and then treated with or without ABL (100 µmol/L) for 24 h prior to OGD. TNF-α and IL-1 β were detected by Western blotting. (H) Band intensities in panel G that were normalized to β-actin are represented by bar graphs as the means ± SD from three independent experiments; *P< 0.05 versus pAd + OGD; #P< 0.05 versus si-Con + OGD; **P< 0.05 versus pAd-miR155 + OGD; ##P< 0.05 versus si-miR155 + OGD.
ABL Prohibits miR-155-Mediated Inflammatory Responses by Regulating TLR4/MyD88 and SOCS1 Expression
Because we found that miR-155 regulated the expression of TNF-α and IL-1β in ischemic cerebral tissue, and the increased expression of TNF-α and IL-1β was decreased by ABL treatment via inhibition of the TLR4/NF-κB signaling cascades, we sought to determine the mechanism by which miR-155 and ABL modulate the inflammatory responses induced by cerebral ischemia. As shown in Figure 5A, marked expression of TLR4 was observed in ischemic cerebral tissue of WT mice at 24 h after MCAO, and this expression was obviously reduced in miR-155−/− mice (Figure 5B, group 3 versus group 1), suggesting that deletion of miR-155 protects against TLR4 upregulation induced by MCAO. TLR4 expression was not further decreased in miR-155−/− mice regardless of ABL treatment (Figure 5B, group 4 versus group 3), most likely due to the deletion of miR-155. As expected, the expression levels of MyD88 and SOCS1, negative regulators of TLR-riggered inflammatory responses, were upregulated by ABL in WT mice after MCAO (Figure 5B). MyD88 and SOCS1 expression levels were further increased in ischemic cerebral tissue of miR-155−/− mice regardless of ABL treatment (see Figure 5B). The effect of miR-155 deletion on TLR4, MyD88 and SOCS1 expression was further verified by qRT-PCR of the mRNA expression of these genes (Figure 5C). To further determine whether the expression of MyD88 and SOCS1 is regulated by miR-155 and ABL, miR-155 was overexpressed in WT mice by injecting pAd-miR-155 into the LV. The results showed that TLR4 expression was significantly increased in ischemic cerebral tissue of miR-155-overexpressing mice compared with pAd-infected mice (Figure 5E, group 3 versus group 1), and ABL treatment reduced TLR4 expression regardless of whether miR-155 was overexpressed (Figure 5E, group 2 versus group 1; group 4 versus group 3). Accordingly, miR-155 overexpression was reduced, while ABL treatment increased the expression of MyD88 and SOCS1 (Figure 5E). In support of this finding, we also showed significant parallel alterations in the mRNA expression of these genes by qRT-PCR (Figure 5F). In keeping with the in vivo data, miR-155 overexpression and knockdown in OGD-treated BV2 cells, promoted and inhibited TLR4 expression, but inhibited and promoted MyD88 and SOCS1 expression (Figure 5G). The effects of ABL on the expression of TLR4, MyD88 and SOCS1 in OGD-treated BV2 cells, where miR-155 was overexpressed or knocked down, were similar to those observed in miR-155−/− mice and miR-155-overexpressing mice after MCAO (Figure 5H). These results suggest that miR-155 mediates inflammatory responses by regulating TLR4/MyD88 and SOCS1 expression.

ABL prohibits miR-155-mediated inflammatory responses through regulating the TLR4/MyD88 and SOCS1 expression. (A) Total proteins were extracted from brain tissues of MCAO WT and miR-155™/™ mice treated with or without ABL (20 mg/kg) and analyzed by Western blotting for TLR4, MyD88 and SOCS1. (B) Band intensities in panel A that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group); *P < 0.05 versus WT + MCAO. (C) mRNA levels of TLR4, MyD88 and SOCS1 in brain tissues of MCAO WT and miR-155−/− mice treated with or without ABL (20 mg/kg) were detected by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P < 0.05 versus WT + MCAO; #P < 0.05 versus miR-155KO + MCAO. (D) WT mice were infected with pAd or pAd-miR-155 and treated with or without ABL (20 mg/kg). TLR4, MyD88 and SOCS1 24 h after MCAO were detected by Western blotting. (E) Band intensities in panel D that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group); *P< 0.05 versus pAd + MCAO; #P< 0.05 versus pAd-miR155 + MCAO. (F) mRNA levels of TLR4, MyD88 and SOCS1 in brain tissues of pAd- or pAd-miR-155-infected MCAO mice treated with or without ABL (20 mg/kg) were detected by qRT-PCR. Bars represent means ± SD (n = 6 per group); *P < 0.05 versus pAd + MCAO; #P < 0.05 versus pAd-miR-155 + MCAO. (G) BV2 cells were infected with pAd-miR-155 or si-miR-155, and then treated with or without ABL (100 µmol/L) for 24 h prior to OGD. TLR4, MyD88 and SOCS1 were detected by Western blotting. (H) Band intensities in panel G that were normalized to β-actin are represented by bar graphs as the means ± SD from three independent experiments; *P < 0.05 versus pAd + OGD; #P < 0.05 versus si-Con + OGD; **P < 0.05 versus pAd-miR155 + OGD; ##P < 0.05 versus si-miR155 + OGD.
ABL Does Not Act Directly on Proinflammatory Signaling
Because it is known that many signaling proteins, including p38 MAPK, JNK and PI3K/Akt, are important downstream effector molecules that participate in the MyD88-dependent TLR4 signaling cascades that can lead to NF-κB activation (27,28), we sought to examine whether miR-155 and ABL might also affect the activation of these signaling molecules. The results shown in Figure 6A demonstrate an obvious increase in the levels of p-Akt, p-ERK, p-JNK and p-NF-κB in ischemic cerebral tissue of miR-155-overexpressing mice after MCAO compared with pAd-infected mice (see Figure 6A). Conversely, miR-155 knockout abrogated the activation effect of cerebral ischemia on these signaling molecules (see Figure 6A), indicating that miR-155 is involved in cerebral ischemia-induced phosphorylation and activation of Akt, ERK, JNK and NF-κB. However, phosphorylation of p38 MAPK was not affected by miR-155 overexpression or knockout under brain ischemic conditions induced by MCAO. Interestingly, deletion of miR-155 markedly suppressed NF-κB acetylation, but the acetylation of NF-κB was not significantly affected by miR-155 overexpression (see Figure 6A). Next, we used the cultured BV2 microglial cells to assess the effect of ABL on the OGD-induced phosphorylation of Akt, ERK, JNK and p38. The results showed that ABL treatment did not obviously affect the phosphorylation of Akt, ERK, JNK and p38, or NF-κB acetylation induced by OGD (Figure 6B). These findings suggest that ABL exerts its inhibitory action on the activation of proinflammatory signaling cascades by suppressing miR-155 expression, but it does not act directly on proinflammatory signaling molecules.

ABL does not act directly on proinflammatory signaling. (A) p-Akt, p-ERK, p-JNK, p-p38, p-NF-κB and Ac-NF-κB in brain tissues of pAd-miR-155-infected mice and miR-155−/− mice treated with or without ABL (20 mg/kg) were detected by Western blotting, and band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD (n = 6 per group); *P < 0.05 versus pAd + MCAO; #P < 0.05 versus WT + MCAO. (B) BV2 cells were treated with or without ABL (100 µmol/L) for 24 h prior to OGD. p-Akt, p-ERK, p-JNK, p-p38 and Ac-NF-κB were detected by Western blotting, and band intensities that were normalized to β-actin are represented by bar graphs as the means ± SD from three independent experiments; *P < 0.05 versus con group.
Discussion
Our findings in this study are as follows: (1) ABL dose-dependently inhibits TNF-α and IL-1β expression in ischemic cerebral tissue of MCAO mice and OGD-treated BV2 cells. (2) ABL decreases inflammatory cytokine production by suppressing the expression of the proinflammatory signaling molecules NF-κB and TLR4. (3) miR-155 mediates TNF-α and IL-1β expression in the ischemic cerebral tissue of MCAO mice and OGD-treated BV2 cells, and ABL has an inhibitory effect on miR-155 expression. (4) miR-155 promotes TNF-α and IL-1β expression by upregulating TLR4 expression and downregulating SOCS1 and MyD88 expression, and ABL blocks the actions of miR-155. (5) ABL inhibits proinflammatory signaling cascades by suppressing miR-155 expression, but it does not act directly on proinflammatory signaling molecules.
In inflammation after cerebral ischemia, proinflammatory cytokines, IL-1β, IL-6 and TNF-α are released by necrotic and injured tissue. Inflammatory response occurs through the action of these cytokines (29,30). In the present study, we demonstrated that the mRNA expression levels of TNF-α and IL-1β in ischemic cerebral tissue of MCAO mice were significantly increased 24 h after MCAO compared with those in control mice, which is consistent with a previous observation that expression of proinflammatory cytokines, including IL-6, IL-1β and TNF-α, is elevated significantly in EDA+/+ mice after cerebral ischemia (31). Because previous studies have identified microglia as the major cellular mediator of injury (32,33) and the main source of IL-1β (34), we also tested the effect of OGD on the expression of proinflammatory cytokines in BV2 microglia cells. As expected, we found that the expressions of TNF-α and IL-1β in OGD-treated BV2 cells were similar to those seen in ischemic cerebral tissue. Importantly, ABL treatment significantly suppressed the ischemia-induced upregulation of TNF-α and IL-1β. Meanwhile, the cerebral infarction volume also was reduced after ABL treatment, indicating that ABL can efficiently reduce brain injury in a focal cerebral ischemia injury model by inhibiting proinflammatory cytokine expression and that the antiinflammation action of ABL in ischemic cerebral tissue is related to its inhibitory effect on microglia-mediated inflammation (31).
We next investigated how ischemia induces the production of proinflammatory cytokines. Because TLRs are known to recognize host-derived molecules released from injured tissues and cells (35) and to initiate the production of proinflammatory cytokines through the activation of NF-κB (36–38), we first examined the effect of ischemia on signaling transduction molecules that participate in the MyD88-dependent TLR4 signaling cascades. We showed that cerebral ischemia enhanced TLR4 and NF-κB, but not MyD88 and SOCS-1. Previous studies have demonstrated that MyD88 negatively regulates the inflammatory responses triggered by LPS and is essential for the induction of the inflammatory cytokines triggered by all TLRs (8). SOCS-1, a negative regulator of the cytokine signaling cascade, inhibits LPS-induced NF-κB and STAT1 activation in macrophages presumably by binding to IRAK (39). The above findings indicate that TLR4 and NF-κB play an important role in mediating the inflammatory response to cerebral ischemia, whereas ischemia do not significantly affect the expression of negative regulators of inflammation, MyD88 and SOCS1. Furthermore, to identify potential targets of ABL action, we determined the effects of ABL on the TLR-triggered proinflammatory signaling cascades. We found that ABL markedly inhibited the NF-κB and TLR4 expression induced by MCAO and OGD and increased the expression of MyD88, SOCS1 and I-κB. This is consistent with previous findings that ABL could suppress NF-κB activation and block the binding of active NF-κB to the target gene promoters (19–21). Our results suggest that ABL exerts its inhibitory effect on the ischemia-induced inflammation via abrogating the ischemia-induced upregulation of NF-κB and TLR4 and inducing the expression of MyD88, SOCS1 and I-κB.
Following focal cerebral ischemia, significant changes in the miRNA transcriptome were observed (40), indicating that miRNAs could play a pivotal role in regulating the complex cascade of molecular signaling associated with cerebral ischemia-mediated inflammation. A previous study has demonstrated that miR-15a contributes to the pathogenesis of ischemic vascular injury through direct inhibition of the antiapoptotic gene bcl-2 (41). Gain or loss of miR-15a significantly reduced or increased oxygen-glucose deprivation-induced cerebral vascular endothelial cell death, respectively. Another recent study showed that miR-497 was induced in mouse brain transient MCAO, and in vivo repression of miR-497 using antagomirs could effectively reduce MCAO-induced infarcts and improve brain damage with a corresponding increase in bcl-2 protein (42). In the present study, we first demonstrated that miR-155 expression was upregulated 25fold in ischemic cerebral tissues of MCAO mice compared with control mice, and a similar result was obtained in OGD-treated BV2 cells. These findings suggest that overexpression of miR-155 is responsible for postischemic inflammation induced by MCAO. Because miR-155 is known to enhance proinflammatory cytokine expression in macrophages by targeting several mediators of inflammatory signaling, such as SHIP1, SOCS1, SMAD2 and TAB2 (16,17), we determined which signaling transduction molecule in the TLR-triggered proinflammatory signaling cascades was targeted by miR-155. Our data showed that the expression of TLR4 was significantly decreased in ischemic cerebral tissue of miR-155−/− mice after MCAO, whereas enforced overexpression of miR-155 obviously increased TLR4 expression in ischemic cerebral tissue. Like the in vivo data, miR-155 overexpression and knockdown in OGD-treated BV2 cells, respectively, promoted and inhibited TLR4 expression, but inhibited and promoted SOCS1 and MyD88 expression. These data clearly indicate that SOCS1 and MyD88 may be important targets of miR-155, whereas TLR4 expression also appears to be indirectly regulated by miR-155. To further examine whether the inhibitory effect of ABL on ischemia-induced inflammation is related to its regulation of miR-155 expression, we determined the effect of miR-155 gain or loss on TNF-α and IL-1β expression, as well as whether ABL affected the expression of miR-155. As expected, overexpression or knockout of miR-155 significantly increased or reduced the expression of TNF-α, IL-1β and NF-κB in ischemic cerebral tissue or OGD-treated BV2 cells, respectively. Importantly, ABL treatment markedly suppressed the expression of miR-155 in ischemic cerebral tissues of MCAO mice and OGD-treated BV2 cells, indicating that miR-155 mediates ischemia-induced inflammation through regulating the TLR4/MyD88 and SOCS1 expression, and that ABL blocks proinflammatory signaling cascades by suppressing miR-155 expression.
Because ERK, JNK, p38 and Akt signalings could participate in the TLR-triggered proinflammatory signaling cascades induced by cerebral ischemia, we examined the role of these signaling cascades in the TLR-triggered proinflammation by measuring their phosphorylation levels (28). Our results showed that ABL did not affect the phosphorylation level of ERK, JNK, p38 and Akt, suggesting that ABL modulates the expression of proinflammatory signaling molecules by suppressing miR-155 expression but does not act directly on the proinflammatory signaling molecules ERK, JNK, p38 and Akt.
Although the present study provides some novel insights into the protective action of ABL in focal cerebral ischemiainduced inflammatory responses, a major limitation of the present study is the lack of characterization of the targets of miR-155 in the context of ABL treatment, which has been included in the continuing efforts of our next project. Altogether, our results suggest that miR-155 might represent a potential therapeutic target of ABL in the prevention and treatment of inflammatory brain damage after cerebral ischemia.
Conclusion
Our current study indicates that miR-155 mediates inflammatory responses in the ischemic cerebral tissue by modulating TLR4/MyD88 and SOCS1 expression and that ABL exerts its antiinflammatory action by suppressing miR-155 expression, suggesting a novel miR-155-based intervention strategy for ischemic stroke.
Disclosure
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Macrez R, et al. (2011) Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 10:471–80.
Iadecola C, Anrather J. (2011) The immunology of stroke: from mechanisms to translation. Nat. Med. 17:796–808.
Jin R, Yang G, Li G. (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 87:779–89.
Morganti-Kossman MC, et al. (1997) Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol. Psychiatry. 2:133–6.
Blohmke CJ, et al. (2010) TLR5 as an antiinflammatory target and modifier gene in cystic fibrosis. J. Immunol. 185:7731–8.
Bsibsi M, Ravid R, Gveric D, van Noort JM. (2002) Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61:1013–21.
Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. (1997) MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 7:837–47.
Yang Y, et al. (2007) Lipopolysaccharide (LPS) regulates TLR4 signal transduction in nasopharynx epithelial cell line 5–8F via NF-κB and MAPKs signaling pathways. Mol. Immunol. 44:984–92.
Takeda K, Akira S. (2004) TLR signaling pathways. Semin. Immunol. 16:3–9.
Xiao C, Rajewsky K. (2009) MicroRNA control in the immune system: basic principles. Cell. 136:26–36.
O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. (2010) Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 10:111–22.
Ouyang YB, Stary CM, Yang GY, Giffard R. (2013) microRNAs: innovative targets for cerebral ischemia and stroke. Curr. Drug Targets. 14:90–101.
Nazari-Jahantigh M, et al. (2012) MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Invest. 122:4190–202.
Hunsberger JG, Fessler EB, Wang Z, Elkahloun AG, Chuang DM. (2012) Post-insult valproic acid-regulated microRNAs: potential targets for cerebral ischemia. Am. J. Transl. Res. 4:316–32.
Li T, et al. (2010) MicroRNAs modulate the non-canonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat. Immunol. 11:799–805.
Androulidaki A, et al. (2009) The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 31:220–31.
Ceppi M, et al. (2009) MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 106:2735–40.
Whan Han J, et al. (2001) Ergolide, sesquiterpene lactone from Inula britannica, inhibits inducible nitric oxide synthase and cyclo-oxygenase-2 expression in RAW 264.7 macrophages through the inactivation of NF-kappaB. Br. J. Pharmacol. 133:503–12.
Han M, Wen JK, Zheng B, Zhang DQ. (2004) Acetylbritannilatone suppresses NO and PGE2 synthesis in RAW 264.7 macrophages through the inhibition of iNOS and COX-2 gene expression. Life Sci. 75:675–84.
Liu YP, Wen JK, Zheng B, Zhang DQ, Han M. (2007) Acetylbritannilactone suppresses lipopolysaccharide-induced vascular smooth muscle cell inflammatory response. Eur. J. Pharmacol. 577:28–34.
Liu B, Han M, Wen JK. (2008) Acetylbritannilactone inhibits neointimal hyperplasia after balloon injury of rat artery by suppressing nuclear factor-κB activation. J. Pharmacol. Exp. Ther. 324:292–8.
Liu B, et al. (2011) Acetylbritannilactone induces G1 arrest and apoptosis in vascular smooth muscle cells. Int. J. Cardiol. 149:30–8.
Yang C, Zhang X, Fan H, Liu Y. (2009) Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 1282:133–41.
Simonyi A, et al. (2005) Polyphenols in cerebral ischemia: novel targets for neuroprotection. Mol. Neurobiol. 31:135–47.
Kreutzberg GW. (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19:312–8.
Olson JK, Miller SD. (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173:3916–24.
Meng Z, Yan Z, Deng Q, Gao DF, Niu XL. (2013) Curcumin inhibits LPS-induced inflammation in 209 rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol. Sin. 34:901–11.
Laird MH, et al. (2009) TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 85:966–77.
Wang Q, Tang XN, Yenari MA. (2007) The inflammatory response in stroke. J. Neuroimmunol. 184:53–68.
Kariko K, Weissman D, Welsh FA. (2004) Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J. Cereb. Blood Flow Metab. 24:1288–304.
Khan MM, et al. (2012) Alternatively-spliced extra domain A of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke. 43:1376–82.
Simard AR, Rivest S. (2007) Neuroprotective effects of resident microglia following acute brain injury. J. Comp. Neurol. 504:716–29.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. (2011) Physiology of microglia. Physiol. Rev. 91:461–553.
Davies CA, et al. (1999) The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J. Cereb. Blood Flow Metab. 19:87–98.
Medzhitov R. (2001) Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135–45.
Yamamoto M, et al. (2002) Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 420:324–9.
Fitzgerald KA, et al. (2001) Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 413:78–83.
Kawai T, Akira S. (2007) Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13:460–9.
Nakagawa R, et al. (2002) SOCS-1 participates in negative regulation of LPS responses. Immunity. 17:677–87.
Rink C, Khanna S. (2011) MicroRNA in ischemic stroke etiology and pathology. Physiol Genomics. 43:521–8.
Yin KJ, et al. (2010) Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J. Neurosci. 30:6398–408.
Yin KJ, et al. (2010) miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiol Dis. 38:17–26.
Acknowledgments
We thank Bin Zheng and Xinhua Zhang (Department of Biochemistry, Hebei Medical University) for their technical assistance. This work was supported by the National Natural Science Foundation of PR China (grant no. 81371287). DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Wen, Y., Zhang, X., Dong, L. et al. Acetylbritannilactone Modulates MicroRNA-155-Mediated Inflammatory Response in Ischemic Cerebral Tissues. Mol Med 21, 197–209 (2015). https://doi.org/10.2119/molmed.2014.00199
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00199