
Abstract
High mobility group box 1 (HMGB1) is an evolutionarily ancient protein that is present in one form or another in all eukaryotes. It fundamentally resides in the nucleus but translocates to the cytosol with stress and is subsequently released into the extracellular space. HMGB1 global knockout mice exhibit lethal hypoglycemia, whereas tissues and cells from conditional knockout or knock-in mice are born alive without apparent significant functional deficit. An aberrant response to targeted stress in the liver, pancreas, heart or myeloid cells is consistent with a protective role for HMGB1 in sustaining nuclear homeostasis and enabling other stress responses, including autophagy. Under some conditions, HMGB1 is not required for liver and heart function. Many challenges remain with respect to understanding the multiple roles of HMGB1 in health and disease.
Similar content being viewed by others

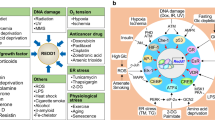

High Mobility Group Box 1 (HMGB1) and Stress
HMGB1 translocates from the nucleus under stress conditions (1). Within the nucleus, it serves as a highly abundant and conserved protein, binding and bending DNA, promoting access to various transcriptional factors within chromatin and inhibiting apoptosis (2). These roles are redundant because knockout animals are born alive, albeit with a markedly limited longevity. In initial reports, knockout of the protein was associated with the generation of a metabolically crippled animal that dies in the early postnatal period with hypoglycemia and, in inbred strains, is late embryonic lethal (3). HMGB1 is thus essential for life. HMGB1−/− mice die shortly after birth because of downregulation of the glucocorticoid receptor and inability to use glycogen stored in the liver (3). In contrast, glucose administration prolongs survival of HMGB1−/− mice, which die before reaching sexual maturity (3). Double knockout of HMGB1 and HMGB2 in mice or zebrafish embryos shows a significant deficiency in Wnt signaling and posterior digit development (4). Both endogenous and exogenous HMGB1 are required for pre-implantation embryo development in the mouse (5). As expected, injection of HMGB1 siRNA into the zygote increases apoptosis (5). In addition, overexpression of HMGB1 in cardiac tissue by transgenic knock-in methods significantly increases animal survival and protects mice against myocardial infarction by enhancing angiogenesis and cardiac function (6). The metabolic causes of this syndrome are unclear but have been variously attributed to alteration in gluconeogenesis within the liver or, alternatively, a defect in essential neonatal autophagy.
We and others have studied the selective ablation of HMGB1 in targeted tissues and found that, without stress, these animals appear to live normally without health impairments. This result suggests that HMGB1 available in trans is sufficient to provide its regulatory function to knockout cells, or alternatively, that it is redundant without stress within individual cells for basal metabolic functions. In contrast, an aberrant response to targeted stress is observed in the liver, pancreas or myeloid cells, consistent with a protective role for HMGB1 (7–9) as well as unpublished results of studies on natural killer and dendritic cells. Interestingly, a recent study from RF Schwabe and colleagues indicates that HMGB1 is not required for autophagy, mitochondrial function and organ function in vivo (10). In this study, the authors created mice with HMGB1 conditional knockout in the liver and heart, and the protein expression level of HMGB1 after gene depletion in livers (Hmgb1Δhep mice) decreased by 72%. Although the precise mechanism remains to be fully elucidated, Hmgb1Δhep mice exhibit normal mitochondrial structure, function and glucose metabolism under normal conditions (10). Induced stress needs to be assessed in RF Schwabe’s models in both the heart (for example, myocardial infarction or contusion) or the liver (for example, ischemia/reperfusion (I/R), acetaminophen or tumorigenesis).
In 1999, Haichao Wang and colleagues (11) made breakthrough progress in uncovering the extracellular role of HMGB1 in inflammation and infection. They demonstrated that HMGB1 functions as a late lethal mediator in sepsis, a systemic inflammatory response syndrome resulting from microbial infection. In 2002, Marco Bianchi and colleagues indicated that HMGB1 is a damage-associated molecular pattern (DAMP) molecule and demonstrated that HMGB1 released from necrotic cells, but not apoptotic cells, triggers the inflammatory response (12). We now know that HMGB1 can be actively secreted by immune cells or passively released by dead, dying or injured cells (13). Extracellular HMGB1 has multiple functions and is involved in several processes such as inflammation, immunity, migration, invasion, proliferation, differentiation, antimicrobial defense and tissue regeneration (14). Notably, the extracellular activities of HMGB1 depend on its form (for example, reduced or oxidized, full-length or cleaved, single or partner), receptor types (for example, positive or negative) and downstream signaling (15,16).
HMGB1 and Autophagy
Although autophagy inhibition prevents HMGB1 release, secretion and degradation, HMGB1 itself regulates autophagy at multiple levels (17–25). For example, nuclear HMGB1 functions as a transcriptional cofactor, regulating the expression of heat shock protein β-1 (26), which in turn sustains dynamic intracellular trafficking during autophagy and mitophagy (26). Cytosolic HMGB1 competes with B-cell lymphoma 2 (BCL2) for interaction with Beclin1/Atg6 by forming intra-molecular disulfide bridge (C23/45) of HMGB1, which in turn promotes generation of Beclin1-mediated autophagosomes (27). The interaction between HMGB1 and Beclin1 is positively regulated by unc-51-like kinase 1 (ULK1) (28), mitogen-activated protein kinase (MAPK) (27) and nucleus accumbens-1 (NAC1) (29), but negatively regulated by p53 (30), α-synuclein (31) and lysosomal thiol reductase (32). Extracellular HMGB1 in its reduced form promotes autophagy through binding to the receptor for advanced glycation end products (RAGE) (19), which may contribute to lactate production and glutamine metabolism to sustain tumor growth (33). HMGB1-mediated autophagy increases chemoresistance in cancer cells such as colon cancer, pancreatic cancer, osteosarcoma, leukemia, gastric cancer, retinoblastoma and ovarian cancer (22,28,34–41). In addition, HMGB1-mediated autophagy in vitro or in vivo prevents polyglutamine aggregates in Huntington disease (42), systemic inflammation during sepsis (9,43), N-methyl-d-aspartate-induced excitotoxicity (44) and hepatic I/R injury (45,46) and sustains T-cell survival in myositis (47). Microtubule-associated protein light chain 3 (LC3) is now widely used to monitor autophagy. Interestingly, Hmgb1Δhep mice crossed with GFP-LC3 mice exhibits GFP-LC3 puncta formation in liver tissue after starvation for 24 h, suggesting an HMGB1-independent role in the regulation of autophagy in vivo (10). Of note, overexpression of GFP-LC3 in vivo or in vitro may lead to easy incorporation of LC3 into protein aggregates, which is independent of autophagy. For example, GFP-LC3 puncta are observed in hepatocytes from mice bearing an Atg5 depletion or Atg7 liver-specific knockout (48–50). Thus, GFP-LC3 mice present some limitations in the interpretation of LC3 puncta formation and localization.
HMGB1 and Mitochondria
HMGB1 expressed in human umbilical vein endothelial cell mitochondria is important for fission and fusion processes occurring during human endothelial cell Toxoplasma gondii infection (51). Recombinant human HMGB1 has been shown to exert cytotoxic activity on malignant tumor cells by promoting the formation of giant mitochondria (52). We demonstrated that HMGB1 is required for mitochondrial quality control and ATP production in immortalized mouse embryonic fibroblasts and cancer cells, which may have elevated HMGB1 expression (26). The metalloproteinase Zmpste24 is responsible for diverse human progeroid syndromes, which are characterized by signs of premature aging (53). Zmpste24−/− mice exhibit altered metabolic pathways, including mitochondrial dysfunction and aberrant HMGB1 expression (53). Conditional knockout of HMGB1 in the liver accelerates mitochondrial injury and reactive oxygen species and alanine aminotransferase (ALT) production (8) at 6 h of liver I/R injury. However, there is no alteration of ALT level at 90 min of liver I/R injury with conditional knockout of HMGB1 in the liver (9). Thus, intracellular HMGB1 may have a time-dependent role in the stress response to liver I/R injury. Intracellular HMGB1 in general is an antiapoptotic protein in yeast and mammalian cells in response to several apoptotic stimuli such as ultraviolet radiation, CD95 or tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) ligation as well as Bax activation (2,54). In addition, gene chip microarray studies demonstrate that knockout of HMGB1 in mouse embryonic fibroblasts significantly impairs metabolic pathways, including those involved in fructose, mannose, galactose, glycolysis and purine metabolism (55). These findings suggest a role of HMGB1 in the regulation of mitochondrial structure and function.
Conclusion and Future Research Directions
Several genetic animal models of HMGB1 have recently been created to examine the physiological and pathological roles of HMGB1 in health and disease. The phenotype of HMGB1 conditional knockout mice is complex and even paradoxical. The understanding of the tissue-specific role of HMGB1 remains largely unknown. Most models, particularly under stress conditions, require further characterization to determine which features of human disease they include. In addition, further insight into the mechanisms and contributions of the key events of the HMGB1 signaling pathway hold the keys to furthering our understanding of HMGB1 function.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Li G, Tang D, Lotze MT. (2013) Ménage à Trois in stress: DAMPs, redox and autophagy. Semin. Cancer Biol. 23:380–90.
Lange SS, Mitchell DL, Vasquez KM. (2008) High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. U. S. A. 105:10320–5.
Calogero S, et al. (1999) The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22:276–80.
Itou J, et al. (2011) HMGB factors are required for posterior digit development through integrating signaling pathway activities. Dev. Dyn. 240:1151–62.
Cui XS, Shen XH, Kim NH. (2008) High mobility group box 1 (HMGB1) is implicated in preimplantation embryo development in the mouse. Mol. Reprod. Dev. 75:1290–9.
Kitahara T, et al. (2008) High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc. Res. 80:40–6.
Kang R, et al. (2014) Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 146:1097–107.
Huang H, et al. (2014) Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology. 59:1984–97.
Yanai H, et al. (2013) Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc. Natl. Acad. Sci. U. S. A. 110:20699–704.
Huebener P, et al. (2014) High-mobility group box 1 is dispensable for autophagy, mitochondrial quality control, and organ function in vivo. Cell Metab. 19:539–47.
Wang H, et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–51.
Scaffidi P, Misteli T, Bianchi ME. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–5.
Bianchi ME. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81:1–5.
Lotze MT, Tracey KJ. (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5:331–42.
Andersson U, Tracey KJ. (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–62.
Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. (2013) HMGB1 in cancer: good, bad, or both? Clin. Cancer Res. 19:4046–57.
Tang D, et al. (2010) Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190:881–92.
Liu L, et al. (2011) HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 25:23–31.
Tang D, et al. (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 29:5299–310.
Dupont N, et al. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30:4701–11.
Thorburn J, et al. (2009) Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16:175–83.
Zhan Z, et al. (2012) Autophagy-mediated HMGB1 release antagonizes apoptosis of gastric cancer cells induced by vincristine via transcriptional regulation of Mcl-1. Autophagy. 8:109–21.
Zhang Y, et al. (2012) Tanshinone IIA sodium sulfonate facilitates endocytic HMGB1 uptake. Biochem. Pharmacol. 84:1492–500.
Li W, et al. (2011) EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem. Pharmacol. 81:1152–63.
Kawano A, et al. (2012) Regulation of P2X7-dependent inflammatory functions by P2X4 receptor in mouse macrophages. Biochem. Biophys. Res. Commun. 420:102–7.
Tang D, et al. (2011) High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 13:701–11.
Tang D, et al. (2010) Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190:881–92.
Huang J, et al. (2012) HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 72:230–8.
Cheng P, et al. (2013) The novel BH-3 mimetic apogossypolone induces Beclin-1- and ROS-mediated autophagy in human hepatocellular carcinoma [corrected] cells. Cell Death Dis. 4:e489.
Livesey K, et al. (2012) p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 72:1996–2005.
Song JX, et al. (2014) HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: a process modulated by the natural autophagy inducer corynoxine B. Autophagy. 10:144–54.
Chiang HS, Maric M. (2011) Lysosomal thiol reductase negatively regulates autophagy by altering glutathione synthesis and oxidation. Free Radic. Biol. Med. 51:688–99.
Luo Y, et al. (2014) Cancer usurps skeletal muscle as an energy repository. Cancer Res. 74:330–40.
Livesey KM, et al. (2012) p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 72:1996–2005.
Liu L, et al. (2011) HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 25:23–31.
Zhao M, et al. (2011) HMGB1 regulates autophagy through increasing transcriptional activities of JNK and ERK in human myeloid leukemia cells. BMB Rep. 44:601–6.
Yang L, et al. (2012) Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk. Lymphoma. 53:315–22.
Kang R, et al. (2010) The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 17:666–76.
Zhang Y, et al. (2012) NAC1 modulates sensitivity of ovarian cancer cells to cisplatin by altering the HMGB1-mediated autophagic response. Oncogene. 31:1055–64.
Liu K, et al. (2014) MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy. 10:442–52.
Ni Z, et al. (2013) Natural Bcl-2 inhibitor (−)- gossypol induces protective autophagy via reactive oxygen species-high mobility group box 1 pathway in Burkitt lymphoma. Leuk. Lymphoma. 54:2263–8.
Min HJ, et al. (2013) Chaperone-like activity of high-mobility group box 1 protein and its role in reducing the formation of polyglutamine aggregates. J. Immunol. 190:1797–806.
Hagiwara S, et al. (2012) Infusion of a glucose solution reduces autophagy in the liver after LPS-induced systemic inflammation. Inflammation. 35:249–58.
Perez-Carrion MD, Cena V. (2013) Knocking down HMGB1 using dendrimer-delivered siRNA unveils its key role in nmda-induced autophagy in rat cortical neurons. Pharm. Res. 30:2584–95.
Shen M, et al. (2013) Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. 2013:461536.
Fang H, Liu A, Dahmen U, Dirsch O. (2013) Dual role of chloroquine in liver ischemia reperfusion injury: reduction of liver damage in early phase, but aggravation in late phase. Cell Death Dis. 4:e694.
Zong M, et al. (2014) A8.24 autophagy may contribute to glucocorticoid resistance in myositis patients by maitaining muscle T cells homeostasis. Ann. Rheum. Dis. 73 Suppl 1:A85–6.
Shvets E, Elazar Z. (2008) Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy. 4:1054–6.
Kuma A, Matsui M, Mizushima N. (2007) LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 3:323–8.
Komatsu M, et al. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169:425–34.
Stumbo AC, et al. (2008) Mitochondrial localization of non-histone protein HMGB1 during human endothelial cell-Toxoplasma gondii infection. Cell Biol. Int. 32:235–8.
Gdynia G, et al. (2010) Danger signaling protein HMGB1 induces a distinct form of cell death accompanied by formation of giant mitochondria. Cancer Res. 70:8558–68.
Peinado JR, et al. (2011) Proteomic profiling of adipose tissue from Zmpste24−/− mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol. Cell. Proteomics. 10:M111008094.
Brezniceanu ML, et al. (2003) HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 17:1295–7.
Krynetskaia N, Xie H, Vucetic S, Obradovic Z, Krynetskiy E. (2008) High mobility group protein B1 is an activator of apoptotic response to antimetabolite drugs. Mol. Pharmacol. 73:260–9.
Acknowledgments
The authors thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by the National Institutes of Health (grant R01CA160417 to D Tang and grant R01 CA181450 to HJ Zeh and MT Lotze) and a 2013 Pancreatic Cancer Action Network-AACR Career Development Award (grant 13-20-25-TANG).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Tang, D., Kang, R., Van Houten, B. et al. High Mobility Group Box 1 (HMGB1) Phenotypic Role Revealed with Stress. Mol Med 20, 359–362 (2014). https://doi.org/10.2119/molmed.2014.00063
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00063