
Abstract
The mammalian immune system and the nervous system coevolved under the influence of cellular and environmental stress. Cellular stress is associated with changes in immunity and activation of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome, a key component of innate immunity. Here we show that α7 nicotinic acetylcholine receptor (α7 nAchR)-signaling inhibits inflammasome activation and prevents release of mitochondrial DNA, an NLRP3 ligand. Cholinergic receptor agonists or vagus nerve stimulation significantly inhibits inflammasome activation, whereas genetic deletion of α7 nAchR significantly enhances inflammasome activation. Acetylcholine accumulates in macrophage cytoplasm after adenosine triphosphate (ATP) stimulation in an α7 nAchR-independent manner. Acetylcholine significantly attenuated calcium or hydrogen oxide-induced mitochondrial damage and mitochondrial DNA release. Together, these findings reveal a novel neurotransmitter-mediated signaling pathway: acetylcholine translocates into the cytoplasm of immune cells during inflammation and inhibits NLRP3 inflammasome activation by preventing mitochondrial DNA release.
Similar content being viewed by others


Introduction
Neural circuits control multiple organ systems to fine-tune function and maintain homeostasis, which is crucial for survival of multicellular organisms. During infection or tissue injury, detection of the presence of pathogens or damaged host cells activates the immune system to initiate inflammatory responses. This step is essential for combating invading pathogens and promoting tissue repair. Failure to resolve inflammation, however, contributes to tissue damage and organ dysfunction. Nonresolving inflammation underlies the pathogenesis of acute and chronic diseases, including rheumatoid arthritis, atherosclerosis, cancer, obesity and insulin resistance (1). Previously, we identified a neural circuit, termed the “inflammatory reflex,” which regulates the function of the innate immune system (2,3). This prototypical regulatory pathway requires the vagus nerve, splenic CD4+ T cells that relay neural signals and produce acetylcholine, and α7 nAchR expressed in splenic macrophages and other cytokine-producing immune cells (4). Stimulation of the vagus nerve increases acetylcholine levels in the spleen, prevents excessive proinflammatory cytokine production during endotoxemia and experimental sepsis (4), and reduces disease severity in human rheumatoid arthritis (5). A key unsolved question is the molecular mechanism by which cholinergic neural signals regulate inflammatory responses in immune cells.
Extracellular high-mobility group box 1 (HMGB1) released by activated immune cells or damaged tissue cells is a cytokine-like mediator of inflammation during sterile and infectious injury (6). Administration of neutralizing anti-HMGB1 monoclonal antibody (mAb), or HMGB1 antagonists, significantly reduce the severity of experimental inflammatory disease, promotes bacterial clearance during Pseudomonas aeruginosa infection and prevents memory impairment in sepsis survivors (6–9). The molecular mechanism of HMGB1 release by activated immune cells depends on activation of inflammasomes. These are multiprotein complexes that cleave procaspase-1 to mediate the proteolytic maturation of interleukin (IL)-1 β and IL-18, and pyroptosis, a form of proinflammatory programmed cell death (10–16). Genetic deletion of inflammasome components or pharmacological inhibition of caspase activity inhibits HMGB1 release (10–15).
We and others have previously established that vagus nerve stimulation or cholinergic receptor agonists significantly inhibit HMGB1 release and prevent the systemic accumulation of HMGB1 during inflammation (17,18). Other findings indicate that the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome mediates HMGB1 release during endotoxemia or bacteremia (10–12) and that cytosolic oxidized mitochondrial DNA activates the NLRP3 inflammasome (19,20). Accordingly, here we postulated that cholinergic agonists inhibit NLRP3 inflammasome activation by preventing mitochondrial DNA release.
Materials and Methods
Reagents
Lipopolysaccharides (LPS; Escherichia coli, 0111:B4), acetylcholine and adenosine triphosphate (ATP) were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA). Anti-mouse IL-1β (sc-1251), anti-mouse caspase-1 (sc-514) and anti-human caspase-1 (sc-622) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse anti-HMGB1 mAb IgG2b 2G7 (noncommercial antibody) is originally from Critical Therapeutic (Boston, MA, USA), available upon request. Plasmids expressing procaspase-1, ASC, NLRP3 or α7 nAchR were purchased from Origene (Rockville, MD, USA). Mitotracker green and Mitotracker deep red were purchased from Invitrogen/Life Technologies (Carlsbad, CA, USA). Mito-chondrial isolation and purification kits were from Qiagen (Germantown, MD, USA). Choline/acetylcholine assay kits were from Abcam (Cambridge, MA, USA). Murine macrophage colony-stimulating factor (GM-CSF) and human macrophage colony-stimulating factor (M-CSF) were purchased from Peprotech (Rocky Hill, NJ, USA).
Mice and Vagus Nerve Stimulation
Mice deficient for the α7 nicotinic receptor (C57BL/6 background) and WT littermates were purchased from The Jackson Laboratory (Bar Harbor, ME, USA; B6.129S7-Chrna7tm1Bay, number 003232). Mice were housed at 25°C on a 12-h light/dark cycle and acclimatized for at least 1 wk before conducting experiments. For vagus nerve stimulation, mice were anesthetized with ketamine (100 mg/kg intraperitoneally [IP]) and xylazine (10 mg/kg IP). A midline cervical incision was made and the left cervical vagus nerve was isolated and placed on a bipolar platinum electrode (Plastics One, Roanoke, VA, USA). Electrical stimulation was delivered at 1 V, 2 ms and 5 Hz for 2.5 min by a stimulation module (STM100A) controlled by the AcqKnowledge software (Biopac Systems, Aero Camino Goleta, CA, USA). Three hours after vagus nerve stimulation, endotoxin (LPS from Escherichia coli, 0111:B4; Sigma-Aldrich) was injected (10 mg/kg IP). Mice were euthanized 2.5 h later and serum was obtained for determination of IL-1β, IL-18 and IL-6 concentration by enzyme-linked immunosorbent assay (ELISA). Vagus nerve studies were acute, nonsurvival procedures where the animal remained anesthetized the entire period. All experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research, North Shore-LIJ Health System.
Cell Isolation and Culture
Peritoneal mouse macrophages. Peritoneal mouse macrophages were isolated as described previously (15). Briefly, mice were injected intraperitoneally with 4% of sterile thioglycollate solution 3–5 d before macrophage isolation. Mice were euthanized with CO2 and were then injected intraperitoneally with 11.6% of sterile sucrose solution. Peritoneal lavage was collected and filtered through a cell strainer with a pore size of 40 μm. Cells were spinned down at 400g for 15 min. The cell pellets were then resuspended in macrophage culture medium (RPMI medium 1640 supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin).
Bone marrow-derived dendritic cells. Bone marrow-derived dendritic cells (BMDCs) were generated in vitro from bone marrow cells from 6- to 8-wk-old mice as previously described (15). Briefly, femurs and tibias were flushed with RPMI-1640 to release the bone marrow cells in RPMI-1640 supplemented with 10% heat-inactivated fetal calf serum, 100 mg/mL penicillin, 100 mg/mL streptomycin and 5 × 10−5 mol/L mercaptoethanol (20 ng/mL murine GM-CSF). On d 3, the supernatants were gently removed and replaced with the same volume of media. On d 6, the nonadherent cells were collected.
Human macrophages. Primary blood mononuclear cells were isolated by density-gradient centrifugation. Then cells were collected and cultured in RPMI 1640 medium with 10% heat-inactivated human serum. After 2 h incubation at 37°C, adherent cells were detached with 10 mmol/L ethylenedi-aminetetraacetic acid (EDTA), and then resuspended (106 cells/mL) in medium supplemented with human macrophage colony-stimulating factor (20 ng/mL) and cultured for 7 d.
Cell stimulation. The 106 peritoneal mouse macrophages or M-CSF-differentiated human macrophages plated in 12-well plates were primed with ultrapure LPS (500 ng/mL) for 4 h and then pulsed with ATP (5 mmol/L) for 30 min.
ELISA
Levels of IL-1β, IL-18 and IL-6 in the culture medium or mouse serum samples were determined using quantitative ELISA kits (R&D Systems, Minneapolis, MN, USA) with reference to standard curves of purified recombinant proteins at various dilutions.
Western Blot Analysis
Proteins from cell-free supernatants were extracted by methanol/chloroform precipitation as described previously (15). Briefly, cell culture supernatants were precipitated by the addition of an equal volume of methanol and 0.25 volumes of chloroform and then were vortexed and centrifuged for 10 min at 20,000g. The upper phase was discarded and 500 μL methanol was added to the interphase. This mixture was centrifuged for 10 min at 20,000g, and the protein pellet was dried at 55°C, resuspended in Laemmli buffer and boiled for 5 min at 99°C. Cell extracts were prepared as described previously. Samples were separated by 4–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE) or 4–20% native PAGE and were transferred onto polyvinylidene fluoride membranes.
Reconstituted Inflammasome in HEK293A Cells
For the human NLRP3 inflammasome reconstitution assays, 0.2 × 106 HEK293A cells were seeded onto 24-well plates in cell culture media lacking antibiotics. Then, 24 h later, cells were transfected with plasmids expressing pro-IL-1β and inflammasome components (Origene), such as ASC, procaspase-1 and NLRP3 by using lipofectamine 2000. These modified cells were then cotransfected with either plasmids that express α7 nAchR or empty plasmids (Origene). The total amount of DNA was standardized to 600 ng per well by using a control plasmid. The transfection efficiency was monitored by using a GFP-encoding plasmid and fluorescent microscopy, and the transfection rate was observed to be around 70%. Cell lysates were harvested 24 h later and analyzed for caspase-1 cleavage by Western blot analysis. Non-transfected HEK293A cells do not express detectable procaspase-1. Caspase-1 activation was standardized by levels of procaspase-1 in cell lysates of transfected HEK293A cells.
Acetylcholine Assay
Mouse peritoneal macrophages were primed with LPS for 3 h. And cells were stimulated with ATP (2.5 mmol/L) in the presence or absence of extracellular acetylcholine (1 mmol/L) for the indicated time. Then cells were washed two times with precooled 1× phosphate-buffered saline (PBS) to get rid of extracellular acetylcholine. Cell lysates were obtained, and total choline and acetylcholine levels were assessed by using a choline/acetylcholine assay kit (Abcam).
Flow Cytometry Analysis
Mitochondrial perturbation was assessed by flow cytometry as described previously (23,24) with some modifications. Briefly, BMDCs were primed with LPS (200 ng/mL) in 15-mL tubes for 2 h and then stained with Mitotracker green and Mitotracker deep red at 100 nmol/L for 30 min at 37°C. The nonadherent cells were collected and stimulated by ATP (1 mmol/L) for 15 min. Cells were quickly transferred on ice for fluorescence-activated cell sorter analysis.
Mitochondrial DNA Release Assay
The mtDNA levels in cytosol were measured as described previously (20). Briefly, 1 × 107 peritoneal macrophages were homogenized with a dounce homogenizer in the presence of protease inhibitor and then were subjected to centrifugation at 700g for 10 min at 4°C. Protein concentration and volume of the supernatant were normalized, followed by centrifugation at 10,000g for 30 min at 4°C for the production of a supernatant corresponding to the cytosolic fraction. DNA was isolated from 200 μL of the cytosolic fraction by using a DNeasy Blood & Tissue kit (Qiagen).
In the cell-free system, purified mitochondria were stimulated with CaCl2 or H2O2 (0.5 mmol/L) as described previously (23). Briefly, the purified mitochondria (120 mg protein per mL) were incubated with either 200 μmol/L CaCl2 or 0.5 mmol/L H2O2 in the presence or absence of different doses of acetylcholine for 15 min at room temperature and were immediately pelleted by centrifugation (10 min, 7,000g) at 4°C. DNA was isolated from 200 μL mitochondrial supernatant by using a DNeasy Blood & Tissue kit.
The levels of mtDNA encoding cytochrome c oxidase 1 were measured by quantitative real-time PCR with the same volume of the DNA solution. The following primers were used: mouse cychrome c oxidase I forward, 5′-GCCCCAGATA TAGCATTCCC-3′, and reverse, 5′-GTTCA TCCTGTTCCTGCTCC-3′.
Statistical Analysis
Data are expressed as mean ± standard deviation (SD) of at least three independent experiments (n = 3-5). One-way analysis of variance and Student t test were used for comparison among all different groups. A p value <0.05 was considered statistically significant.
All supplementary materials are available online at https://doi.org/www.molmed.org .
Results
Cholinergic Agonists Inhibit Inflammasome Activation
To test the hypothesis that cholinergic neural signals inhibit NLRP3 inflammasome activation, LPS-primed mouse peritoneal macrophages were stimulated with ATP in the presence or the absence of different concentrations of acetylcholine. Acetylcholine exposure dosedependently inhibited ATP-induced activation of caspase-1, cleavage of pro-IL-1β and HMGB1 release (Figure 1A). Addition of acetylcholine failed to inhibit the expression of pro-IL-1β in the cell lysates (Figure 1A), indicating that inhibition of IL-1β production by acetylcholine is due to the suppression of inflammasome activity, rather than LPS-induced priming. Next, inflammasome activation was assessed in human macrophages exposed to acetylcholine. Human macrophages did not secrete detectable amounts of IL-1β after LPS stimulation (Figure 1B). However, ATP subsequent stimulation of LPS-primed human macrophages rapidly induced IL-1β secretion, which was dose dependently inhibited by acetylcholine. In contrast, addition of acetylcholine failed to attenuate LPS-induced IL-6 production, which does not depend on inflammasome activation (Figure 1B). To investigate whether cholinergic neural signals inhibit inflammasome activation in vivo, mice were subjected to vagus nerve stimulation or sham operation. Three hours later, the animals were challenged with lethal doses of LPS. We observed that vagus nerve stimulation significantly reduced serum levels of IL-1β and IL-18, as compared with that of sham operation (Figure 1C); IL-6 was not inhibited by vagus nerve stimulation (Figure 1C). Finally, inflammasome activity was assessed in LPS-primed mouse peritoneal macrophage-activated ATP in the presence or absence of GTS-21, an α7 nAchR agonist. GTS-21 dose-dependently inhibited ATP-induced IL-1β cleavage (Figure 1D). Together, these observations indicate that cholinergic agonists inhibit inflammasome activation in vitro and in vivo.

Cholinergic neural signals inhibit inflammasome activation. LPS-primed peritoneal mouse macrophages (A) or human macrophages (B) were stimulated with ATP in the presence or the absence of different dose of acetylcholine (Ach) for 30 min. (C) Mice were subjected to vagus nerve stimulation (VNS) or sham operation. At 3 h later, all mice were injected intraperitoneally with LPS (10 mg/kg). And serum samples were collected 2.5 h later. (D) LPS-primed peritoneal mouse macrophages were stimulated with ATP in the presence or the absence of different dose of GTS-21 for 15 min. (D) Mice were injected intraperitoneally with LPS (10 mg/kg) alone or LPS (10 mg/kg) and GTS-21 (4 mg/kg). And serum samples were collected 2.5 h later. IL-1β cleavage, HMGB1 release and caspase-1 activation were assessed by Western blot analysis. Levels of IL-1β, IL-18 or IL-6 in cell culture medium or serum samples were measured by ELISA. Blots are representative of three independent experiments. Data shown are means ± SD. #p < 0.05 versus nonstimulated or sham-operated groups.
α7 nAchR Signaling Inhibits the NLRP3 Inflammasome Activation
The major target of GTS-21 is a7 nAchR, which is expressed in macrophages and dendritic cells (21). Together with our previous findings that α7 nAchR is an important regulator of inflammation and an essential component of the inflammatory reflex (22), we reasoned that cholinergic agonists might inhibit the NLRP3 inflammasome activation through interaction with α7 nAchR. To test this hypothesis, we challenged both α7 nAchR knockout (α7 KO) mice and wild-type (WT) controls with a lethal dose of LPS. Notably, the serum levels of IL-1β and IL-18 were significantly higher in α7 KO compared with that of WT mice (Figure 2A). The production of IL-6, however, was not significantly altered in α7 KO mice (Figure 2A). Genetic deletion of α7 nAchR also significantly increased IL-1β cleavage in peritoneal mouse macrophages in vitro (Figure 2B). These results indicate that α7 nAchR negatively regulates inflammasome activation.

α7 nAchR negatively regulates inflammasome activation. (A) LPS-primed peritoneal mouse macrophages from either WT or α7 nAchR knockout (a7 KO) mice were stimulated with ATP in the presence or the absence of acetylcholine for 30 min. (B) WT and a7 KO mice were injected intraperitoneally with LPS (10 mg/kg). Serum samples were collected 2.5 h later. (C) HEK293A cells were transfected with plasmids as indicated. At 24 h after transfection, cell lysates were harvested. IL-1 β cleavage and caspase-1 activation were assessed by Western blot analysis. Serum levels of IL-1 β, IL-18 or IL-6 were measured by ELISA. Blots are representative of three independent experiments. Data shown are means ± SD.
To further investigate the role of α7 nAchR in NLRP3 inflammasome activation, we reconstituted the functional NLRP3 inflammasome in nonimmune HEK293A cells by transfection of these cells with plasmids that encode inflammasome components NLRP3, ASC and procaspase-1 (15). Western blot analysis reveals that expression of these inflammasome components in HEK293A cells induces cleavage of procaspase-1; this is indicative of a functional inflammasome (Figure 2C). Coexpression of α7 nAchR in these genetically modified cells significantly inhibited the cleavage of procaspase-1. Addition of a selective α7 nAchR agonist (PNU-28879) further decreased activation of caspase-1 (Figure 2C). Collectively, these findings establish that α7 nAchR signaling inhibits NLRP3 inflammasome activation.
α7 nAchR Signaling Attenuates ATP-Induced Mitochondrial Perturbation
Prior work implicated mitochondrial perturbation in NLRP3 inflammasome activation (23). ATP stimulation mobilizes Ca2+ from the endoplasmic reticulum or extracellular space to the cytoplasm, and the resultant increase in mitochondrial permeability has been linked to the NLRP3 inflammasome activation (24). α7 nAchR is expressed in the mitochondrial outer membrane in nonneuronal cells, including monocytes, B lymphocytes and hepatocytes (25,26). To confirm that α7 nAchR is localized in mitochondria, we transfected HEK293 cells with Myctagged α7 nAchR and then stained the genetically modified cells with the anti-Myc antibodies and a mitochondrial marker. Indeed, we observed the localization of Myc-tagged α7 nAchR in mitochondria (Supplementary Figure S1). We next hypothesized that α7 nAchR signaling attenuates ATP-induced mitochondrial perturbation. To test this, we incubated primary BMDCs with MitoTrack green and MitoTrack deep red to stain total mitochondria and mitochondria with normal membrane potential, respectively. Consistent with previous studies, LPS and ATP stimulation resulted in robust increase of cells that had lost mito-Track deep red staining, indicative of mitochondrial perturbation in these cells (Figure 3). Genetic deletion of α7 nAchR significantly enhanced ATP-induced mitochondrial perturbation in LPS-prime BMDCs (Figure 3A). Further, acetylcholine dose-dependently inhibited ATP-induced mitochondrial perturbation in WT, but not in a7 nAchR knockout (a7 KO), BMDCs (Figures 3B–E), indicating that α7 nAchR signaling attenuates ATP-induced mitochondrial perturbation.

Cholinergic neural signals attenuate ATP-induced mitochondrial perturbation. (A,C) LPS-primed mouse BMDCs from WT or a7 KO mice were stimulated by ATP (B,D) LPS-primed mouse BMDCs from WT mice were stimulated by ATP in the presence or the absence of absence of acetylcholine. (C) LPS-primed mouse BMDCs from WT mice were stimulated by ATP in the presence or the absence of acetylcholine. Mitochondrial perturbation was assessed by flow cytometry analysis by using Mitotracker green and Mitotracker deep red. Upper panels (A,B) show the representative graph; lower panels (C-E) show means ± SD. #p < 0.05 versus ATP-stimulated alone.
Acetylcholine Accumulates in Cytoplasm upon ATP Stimulation
Although it is known that acetylcholine and other α7 nAchR agonists abolish Ca2+ or hydrogen oxide-induced cytochrome c release from isolated mitochondria (25), the mechanism for extracellular acetylcholine translocation into cytoplasm has remained unknown. One possibility is that extracellular acetylcholine accumulates in the cytoplasm of immune cells upon ATP stimulation. To test this, LPS-primed mouse peritoneal macrophages were stimulated with ATP in the presence of extracellular acetylcholine (1 mmol/L), and acetylcholine levels in cell lysates were subsequently measured. We observed that ATP stimulation induced increased acetylcholine levels in the cell lysates (Figure 4A), indicating that extracellular acetylcholine accumulates in cytoplasm on ATP stimulation. This process does not depend on α7 nAchR, because genetic deletion of α7 nAchR did not block ATP-induced acetylcholine accumulation in the cell lysates (Figure 4B). Previous studies show that ATP stimulation could result in the formation of pannexin pores that increase permeability of cytoplasmic membrane to small molecules (<900 Da) (27). To determine whether pannexin is involved in ATP-induced acetylcholine accumulation in cell lysates, mouse macrophages were stimulated with ATP in the presence of Panx-10, a peptide that is reported to block the pannexin pores (27), or a control peptide. Notably, addition of Panx-10 did not significantly inhibit ATP-induced acetylcholine accumulation in cell lysates, as compared with that of control peptide (Figure 4C). Collectively, these results demonstrated that acetylcholine rapidly accumulates in the cytoplasm of immune cells on ATP stimulation in an α7 nAchRand pannexin-independent manner.

Acetylcholine translocates into cytoplasm upon ATP stimulation. (A) LPS-primed peritoneal mouse macrophages from WT mice were stimulated by ATP for an indicated time. (B) LPS-primed peritoneal mouse macrophages from WT or a7 KO mice were stimulated by ATP for an indicated time. (C) LPS-primed peritoneal mouse macrophages from WT mice were stimulated by ATP in the presence of an indicated dose of 10 Panx or scramble peptides. Intracellular acetylcholine (Ach) levels were assessed using a choline/acetylcholine assay kit.
α7 nAchR Signaling Prevents Mitochondrial DNA Release
Early studies indicate that mitochondrial DNA release into cytoplasm is associated with NLRP3 inflammasome activation (19,20). Oxidized mitochondrial DNA molecules function as NLRP3 ligands that induce the assembly and activation of the NLRP3 inflammasome (19). Accordingly, we reasoned that α7 nAchR signaling prevents mitochondrial DNA release into cytoplasm. Mouse macrophages were stimulated with ATP in the presence of different concentrations of acetylcholine. Then the cytoplasmic fraction was extracted, and mitochondrial DNA levels were assessed by using quantitative polymerase chain reaction (qPCR) with primers for the cytochrome c oxidase 1 gene. Indeed, acetylcholine dose-dependently inhibited ATP-induced mitochondrial DNA release into cytoplasm in WT macrophages (Figure 5A). However, acetylcholine did not significantly inhibit ATP-induced mitochondrial DNA release into cytoplasm in α7 nAchR knockout (α7 KO) macrophages, even at the higher concentration (Figure 5B). These data indicate that acetylcholine inhibits ATP-induced mitochondrial DNA release into cytoplasm in an α7 nAchR-dependent manner. Further, we asked whether acetylcholine could inhibit mitochondrial DNA release in isolated mitochondria in a cell-free system. Macrophage mitochondria were isolated and then challenged with Ca2+ or hydrogen oxide in the presence of different doses of acetylcholine. Mitochondrial DNA levels in supernatant were measured by qPCR. Addition of acetylcholine significantly inhibited Ca2+ or hydrogen oxide-induced mitochondrial DNA release (Figures 5C, D). Mitophagy is a selective form of autophagy by which injured mitochondria are degraded into auto-lysosomes. Recent studies have shown that activation of autophagy/mitophagy by inflammatory signals limits IL-1β production (20). To study the role of α7 nAchR signaling in regulation of autophagy, LPS-primed mouse macrophages were stimulated with ATP in the presence of different concentration of acetylcholine for 20 min. Exposure to acetylcholine did not alter the cellular levels of LC3B-II (Supplementary Figure S2), suggesting that α7 nAchR signaling prevents mitochondrial DNA release in an autophagy-independent manner.

Acetylcholine prevents mitochondrial DNA (mtDNA) release. LPS-primed peritoneal mouse macrophages from either WT (A) or a7 KO (B) mice were stimulated with ATP in the presence or the absence of acetylcholine for 15 min. DNA was isolated from the cytosolic fraction of macrophages. And then the levels of mtDNA were measured by quantitative real-time PCR. Purified mitochondria were incubated with either 200 μmol/L CaCl2 (C) or 0.5 mmol/L H2O2 (D) in the presence or absence of different dose of acetylcholine for 15 min and were immediately pelleted by centrifugation. Levels of mtDNA in the mitochondria-free supernatant were measured by quantitative real-time PCR. Data shown are means ± SD. #p < 0.05 versus stimulated groups.
Discussion
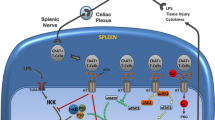
These findings establish an important mechanism of mitochondrial α7 nAchR signaling in regulation of the NLRP3 inflammasome activation that reduces mitochondrial DNA release. Inflammasomes are intracellular protein complexes that mediate maturation of IL-1β and IL-18 and execute the process of proinflammatory programmed cell death (termed “pyroptosis”). Unlike other types of inflammasomes, the NLRP3 inflammasome can be triggered by a broad range of endogenous and exogenous danger signals (28). Instead of binding NLRP3 directly, these stimuli activate the NLRP3 inflammasome by provoking intracellular potassium efflux and mitochondrial DNA release into cytoplasm (19,29). In this study, we found that stimulation of ATP results in a rapid influx of acetylcholine into the cytoplasm, enabling intracellular acetylcholine to interact with mitochondrial α7 nAchR and in turn prevent mitochondrial DNA release (Figure 6). Because oxidized mitochondrial DNA molecules bind and activate the NLRP3 inflammasome (19), our findings provide a mechanism for acetylcholine inhibition of ATP-induced NLRP3 inflammasome activation by preventing mitochondrial DNA release into cytoplasm. It is noteworthy that other NLRP3 inflammasome agonists, such as nigericin and adjuvant aluminum, have also been implicated in inducing the influx of acetylcholine into the cytoplasm through formation of pores in the cytoplasmic membrane and after lysosomal damage, respectively.

The mechanisms by which acetylcholine inhibits inflammasome activation. Extracellular ATP activates the NLRP3 inflammasome by provoking intracellular potassium efflux and mitochondrial DNA release into cytoplasm. Stimulation of ATP also results in rapid influx of acetylcholine into cytoplasm. There, acetylcholine prevents mitochondrial DNA release through α7 nAchR and subsequently inhibits the NLRP3 inflammasome activation.
The inflammasome-dependent cytokines IL-1β, IL-18 and HMGB1 orchestrate the inflammatory responses to infection and sterile tissue injury. However, deregulated production of these cytokines drives the pathogenesis of many inheritable or acquired diseases and disorders, such as obesity, type 2 diabetes, atherosclerosis, arthritis, colitis and sepsis (30). Interestingly, cholinergic neural signals also inhibit the production of inflammasome-independent cytokines, including tumor necrosis factor (31). This study reveals a novel pathway through which cholinergic agonists may modulate target cells by activating intracellular cholinergic receptors on mitochrondria. Although emerging evidence shows that many nonneuron cells (such as monocytes, B lymphocytes and hepatocytes) contain intracellular pools of nicotinic cholinergic receptors, the biological function of these intracellular cholinergic receptors had previously been elusive (25,26).
Conclusion
Here we show that ATP stimulation induces rapid influx of acetylcholine into cytoplasm, where the acetylcholine can engage mitochondrial cholinergic receptors. Importantly, we and others show that these intracellular nicotinic cholinergic receptors (especially the α7 nAchR) are biologically functional. Addition of acetylcholine or other α7 nAchR agonists, such as choline, markedly inhibits Ca2+ or hydrogen oxide-induced mitochondrial DNA or cytochrome C release in isolated mitochondria from macrophages or hepatocytes, respectively. These findings suggest two antiinflammatory drug targets: mitochondrial membrane receptors and plasma membrane acetylcholine transporters that regulate mitochondrial homeostasis and inflammasome activity.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Okin D, Medzhitov R. (2012) Evolution of inflammatory diseases. Curr. Biol. 22:R733–40.
Andersson U, Tracey KJ. (2012) Reflex principles of immunological homeostasis. Annu. Rev. Immunol. 30:313–35.
Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. (2012) Rethinking inflammation: neural circuits in the regulation of immunity. Immunol. Rev. 248:188–204.
Rosas-Ballina M, et al. (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 334:98–101.
Koopman FA, et al. (2012) Pilot study of stimulation of the cholinergic anti-inflammatory pathway with an implantable vagus nerve stimulation device in patients with rheumatoid arthritis. Arthritis Rheum. 64(10 Suppl):S195. Abstract No. 451.
Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–62.
Patel VS, et al. (2013) High mobility group box-1 mediates hyperoxia-induced impairment of Pseudomonas aeruginosa clearance and inflammatory lung injury in mice. Am. J. Respir. Cell. Mol. Biol. 48:280–7.
Wang H, et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–51.
Chavan SS, et al. (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol. Med. 18:930–7.
Willingham SB, et al. (2009) NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 183:2008–15.
Lu B, Wang H, Andersson U, Tracey KJ. (2013) Regulation of HMGB1 release by inflammasomes. Protein Cell. 4:163–7.
Lamkanfi M, et al. (2010) Inflammasomedependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185:4385–92.
Kayagaki N, et al. (2011) Non-canonical inflammasome activation targets caspase-11. Nature. 479:117–21.
Qin S, et al. (2006) Role of HMGB1 in apoptosis mediated sepsis lethality. J. Exp. Med. 203:1637–42.
Lu B, et al. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 488:670–4.
Hett EC, et al. (2013) Chemical genetics reveals a kinase-independent role for protein kinase R in pyroptosis. Nat. Chem. Biol. 9:398–405.
Wang H, et al. (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 10:1216–21.
Huston JM, et al. (2007) Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit. Care. Med. 35:2762–8.
Shimada K, et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 36:401–14.
Nakahira K, et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12:222–30.
Rosas-Ballina M, et al. (2009) The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol. Med. 15:195–202.
Wang H, et al. (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 421:384–8.
Zhou R, Yazdi AS, Menu P, Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature. 469:221–5.
Murakami T, et al. (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U. S. A. 109:11282–7.
Gergalova G, et al. (2012) Mitochondria express α7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: study on isolated mitochondria. PLoS One. 7:e31361.
Kalashnyk OM, Gergalova GL, Komisarenko SV, Skok MV. (2012) Intracellular localization of nicotinic acetylcholine receptors in human cell lines. Life Sci. 91:1033–7.
Gulbransen BD, et al. (2012) Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 18:600–4.
Rathinam VA, Vanaja SK, Fitzgerald KA. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13:333–2.
Muñoz-Planillo R, et al. (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 38:1142–53.
Strowig T, Henao-Mejia J, Elinav E, Flavell R. (2012) Inflammasomes in health and disease. Nature. 481:278–86.
Borovikova LV, et al. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 405:458–62.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Lu, B., Kwan, K., Levine, Y.A. et al. α7 Nicotinic Acetylcholine Receptor Signaling Inhibits Inflammasome Activation by Preventing Mitochondrial DNA Release. Mol Med 20, 350–358 (2014). https://doi.org/10.2119/molmed.2013.00117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2013.00117