
Abstract
Homeobox D10 (HoxD10) gene plays a critical role in cell differentiation and morphogenesis during development. However, the function of HoxD10 in tumor progression remains largely unknown. We demonstrate that the expression of HoxD10 is commonly downregulated in gastric cancer tissues (n = 33) and cell lines (n = 8) relative to normal stomach tissues. Functionally, reexpression of HoxD10 results in significant inhibition of cell survival, induction of cell apoptosis, and impairment of cell migration and invasion. Moreover, ectopic expression of HoxD10 suppresses gastric tumor growth in a mouse xenograft model. To identify target candidates of HoxD10, we performed cDNA microarray and showed that HoxD10 regulates multiple downstream genes including IGFBP3. Reintroduction of HoxD10 transcriptionally upregulates IGFBP3, activates caspase 3 and caspase 8, and subsequently induces cell apoptosis. Methylation specific PCR revealed that HoxD10 promoter DNA was hypermethylated in gastric cancer cell lines. Additionally, 5-aza demethylation treatment could transiently reactivate the expression of HoxD10 in gastric cancer cells. HoxD10 promoter methylation frequently was detected in gastric cancer tissues obtained from endoscopic biopsies (85.7%, 24/28) and surgically resected samples (82.6%, 57/69). Intestinal metaplasia tissues showed a 60% methylation rate (18/30), but no detectable methylation in normal stomach tissues (0%, 0/10). Taken together, our results suggest that HoxD10 functions as a candidate tumor suppressor in gastric cancer, which is inactivated through promoter hypermethylation.
Similar content being viewed by others


Introduction
Gastric cancer is the second leading cause of cancer-related death worldwide (1,2). Most intestinal-type gastric cancers are thought to develop from precancerous lesions (atrophic gastritis and intestinal metaplasia). Both genetic and epigenetic alterations contribute to this progression (2–4). The transcriptional silencing of tumor suppressor genes (TSGs) by promoter methylation is a major epigenetic event in the origin of many cancers, including gastric cancer (5–7). Thus, identification of novel TSGs inactivated by promoter methylation will be of great importance in understanding the progression of gastric cancer, and could be utilized as biomarkers for the early detection of cancer (7–9). We and others have found that zinc finger of the cerebellum (ZIC1), fructose-1, 6-bisphosphatase-1 (FBP1), chromodomain helicase DNA binding protein 5 (CHD5), protocadherin 10, and Klotho genes were silenced epigenetically in gastric cancer (10–14). In the current study, we demonstrate HoxD10 as another important TSGs which undergoes epigenetic modification.
The homeobox (Hox) superfamily genes, including HoxD10, encode transcription factors that control cell differentiation and morphogenesis during development (15,16). In addition to their roles in development, numerous Hox genes (HoxB13, HoxA5 and HoxC6) have been found to be expressed aberrantly in a variety of solid tumors, including breast, colon and prostate cancers (16–20). Emerging evidence suggests that the expression of Hox genes is under epigenetic control (19–22). For example, HoxA5 is suppressed in breast cancer through promoter methylation, and its suppression is correlated with the loss of p53 expression (20). Studies also demonstrate that CpG islands (CpGIs) in the promoters of Hox genes are commonly methylated in lung cancer (21,22). The dysregulation of Hox genes may affect various pathways that play critical roles in tumorigenesis and cancer metastasis (19). Evidence shows that ectopic expression of HoxB13 in prostate and colon cancer cells can suppress tumor growth through downregulating of β-catenin-TCF pathway (17,23). It has been shown that HoxD10 expression is reduced in both breast and endometrial tumors (24). Overexpression of HoxD10 impairs breast tumor cells motility and invasiveness significantly, which indicates that HoxD10 may serve as a tumor suppressor (25). However, the functional role of HoxD10 in tumorigenesis, and the biological significance of its epigenetic control in gastric cancer have not been defined.
As transcription factors, Hox proteins can regulate the expression of multiple downstream genes. Genome-wide analysis has revealed that genes including BMP7, FGFR2 and PDGFRA are regulated transcriptionally by HoxC6 gene in prostate cancer cell lines (26). It was accepted that Hox genes could regulate the promoter of genes by binding consensus elements TTAT, TAAT and TTAC. Combined with microarray profiling, DNA binding site analysis and chromatin immunoprecipitation (Chip) analysis identified that five genes are directly regulated by HoxC8 in mouse embryo fibroblast cells (27). These candidate genes have been implicated in oncogenesis, cell proliferation and cell apoptosis. Thus, identification of downstream targets of HoxD10 genes in the cancer setting may shed light on the understanding of the mechanisms on tumorigenesis.
In this study, we provide evidence that promoter hypermethylation contributes to the downregulation of HoxD10 in gastric cancer, and these methylation events frequently occur in precancerous lesions and gastric cancer tissues. Ectopic expression of HoxD10 suppresses tumor growth in vivo and in vitro. Additionally, HoxD10 regulates multiple downstream genes which participate in carcinogenesis by microarray analysis. Thus, our results suggest that HoxD10 potentially functions as a tumor suppressor that is inactivated through promoter hypermethylation in gastric cancer.
Materials and Methods
Cell Culture and Demethylation Treatment
Eight gastric cancer cell lines (AGS, BGC823, HGC27, MGC803, MKN28, MKN45, NCI-N87 and SGC7901) were obtained from Riken Gene Bank (Tsukuba, Japan) and American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and incubated at 5% CO2, 37°C and 95% humidity.
Tumor cells (AGS, MKN28, MKN45 and NCI-N87) were treated for 72 h with 5 μmol/L 5-aza-2′-deoxycytidine (5-Aza) (Sigma, St Louis, MO, USA) for induction of demethylation. 5-Aza was replenished every 24 h. An equivalent concentration of the vehicle (DMSO) was used as the control. In addition, cells were cultured for another 48 h after withdrawal of the treatment.
Clinical Samples
Ninety-three tissue biopsies, including gastric cancer (n = 33), intestinal metaplasia (IM) (n = 30), and normal stomach (n = 33), were obtained from endoscopic samples from Sir Run Run Shaw Hospital and Second Affiliated Hospital in Zhejiang University. In addition, another 69 surgically resected gastric cancer and adjacent nontumor tissues were obtained as described previously (11,12). Gastric cancers are classified according to Lauren Standard. The IM pathological status was evaluated by the updated Sydney system. All patients gave written informed consent, and the study protocol was approved by the Clinical Research Ethics Committee of Sir Run Run Shaw Hospital of Zhejiang University. All specimens were frozen immediately in liquid nitrogen and stored at -80° C until further processing.
Total RNA and Genomic DNA Extraction, Bisulphite Treatment
Total RNA and genomic DNA were extracted with Trizol Reagent (Invitrogen) following manufacturer’s instruction. The concentrations of RNA and DNA were quantified by Nanodrop 1000 (Nanodrop, Wilmington, DE, USA). Genomic DNA was bisulphite-treated with Zymo DNA Modification Kit (Zymo Research, Orange, CA, USA) according to the protocol provided by the manufacture.
RT-PCR and Quantitative Real-Time PCR
Reverse transcription reaction was performed using 1μg of total RNA with Reverse Transcription System (Promega, Madison, WI, USA). The expression levels of HoxD10 were determined by conventional polymerase chain reaction (RT-PCR) with GoTaq polymerase (Promega) and quantitative RT-PCR (qPCR) using SYBR Green Master Mix Kit (TaKaRa) in an ABI 7500 PCR system. Glyceraldehyde-3-phosohate dehydrogenase (GAPDH) was used as an internal control, and the expression levels of HoxD10 mRNA were determined using the 2−ΔΔCt method. All primer sequences are listed in Supplementary Table 1.
Methylation Specific PCR (MSP) Analysis
Promoter methylation of HoxD10 was determined by methylation specific PCR (MSP) using above bisulfite modified genomic DNA as the template. MSP was carried out for 40 cycles with an annealing temperature at 60° C as described previously (10). Methylation (M) primers were: F 5′-TTATT TATGG GGATT CGGGT TC, R 5′-AAAAA AACCG AATCC AAAAC AAACG, and Unmethylation (U) primers were: F 5′-ATTTA TTTAT GGGGA TTTGG GTTT, R 5′-AAAAA AACCA AATCC AAAAC AAACA.
Plasmid Construction and Cell Transfection
The HoxD10 expression plasmid was constructed by cloning of the full-length HoxD10 open reading frame into the mammalian expression vector pcDNA3.1 with BamHI and XhoI restriction enzyme sites. The sequences were confirmed by DNA sequencing. Cells were cultured in 12-well plate for 24 h and transfected with pcDNA3.1-HoxD10 or empty vector pcDNA3.1 using Lipofectamine 2000 (Invitrogen). G418 resistant colonies were selected in transfected cells (AGS and MKN28) for 14 d in a 12-well plate. After confirming the transfection efficiency by RT-PCR and Western blot in the surviving colonies, cells were transferred into a 6-well plate, and continuously selected with G418 to generate stable cell lines.
Two DNA fragments (−2,251 to −2,073 for BS1, and −1,727 to −943 for BS2) of IGFBP3 were amplified by PCR using genomic DNA of HEK-293T cells as the template. The primer pairs were designed as follows, BS1 (Forward: GGGGT ACCCA TTCGG CACTG AACAA G, Reverse: GAAGA TCTAG CCTGG ACTGA CCACT G) and BS2 (Forward: GGGGT ACCGG CACTC CATTG TTCTT, Reverse: GAAGA TCTGA ATAAT AAAGA CAATA AACTG G). PCR products were digested with KpnI and BglII enzymes and cloned into upstream of pGL3-promoter vector (Promega) to generate the recombinant plasmid of promoter-luciferase reporter gene. All constructs were confirmed by DNA sequencing.
Colony Formation Assay
AGS and MKN28 cells were cultured for 24 h in a 12-well plate (1.5 × 105/well) and transfected with pcDNA3.1 or pcDNA3.1-HoxD10. Forty hours later, the transfectants were replated in 6-well and selected with G418 resistant colonies for 14–16 d. Surviving colonies were stained with gentian violet after methanol fixation and visible colonies were counted. The experiments were performed in triplicate.
Quantitative Chromatin Immunoprecipitation (ChIP) Analysis
ChIP analysis was determined by the Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology Inc., Danvers, MA). The chromatin was immunoprecipitated with normal rabbit IgG (negative control) and HoxD10 monoclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Quantitative real-time PCR was performed using SYBR Green Master Mix Kit (Takara Otsu, Shiga, Japan). According to transcription factor binding site prediction software (PROMO search program; Algorithmics and Genetics Group [ALGGEN], Polytechnic University of Catalonia, Barcelona, Spain; http://alggen.lsi.upc.es/), five potential HoxD10 binding sites (H1-H5) were localized in the upstream 2.3-kb sequence of IGFBP3 gene. Four pairs of primers (A1-A4) were designed to detect different DNA fragments in the promoter regions of IGFBP3 (A1 for H1 and H2, A2 for H3, A3 for H4 and A4 for H5, respectively) (Supplementary Table 2). The ChIP analysis results were expressed as fold change compare to rabbit IgG normalized to 1. The results were considered as positive HoxD10-binding sequences when the fold change greater than three-fold from triplicate experiments.
Dual-Luciferase Reporter Assays
5 × 104 Cells were cultured in 24-well plate for 24 h and transfected with pcDNA3.1-HoxD10 or empty vector pcDNA3.1, pGL3-promoter vector or pGL3-BS (IGFBP3) and pRL-TK vector (Promega) containing reference control Renilla using Fugene HD (Roche, Penzberg, Germany). The activities of Firefly and Renilla luciferase were determined 30 h after the transfection by the dual-luciferase reporter assays (Promega) following the protocol of the manufacture. The relative fold of luciferase activity was expressed as firefly/renilla ratio. The experiments were performed in triplicate.
Cell Proliferation Assay (MTS)
Cell proliferation was detected with Non-Radioactive Cell Proliferation Assay with MTS reagents (Promega). Stable transfected cells (1 × 104/well) were plated into 96-well plates for 6, 24, 72 and 120 h respectively. After incubation with CellTiter 96 Aqueous One Solution reagent for 1 h, the absorbance was measured at 490 nm according to the manufacturer’s instruction.
Xenograft Tumor Model
Animal experimental protocols were approved by the Committee of Animal Ethics, Zhejiang University. Female athymic nude mice (nu/nu) (3 to 4 wksold) were purchased from Shanghai Laboratory Animal Co Ltd (SLAC, Shanghai, China). MKN28 cells (7 × 106 cells) transfected with pcDNA3.1 or pcDNA3.1-HoxD10 were injected subcutaneously into the flank of mice (n = 6 each group). After 6 d of implantation of tumor cells, tumor volume was assessed every 3 d. Tumor volume was calculated according to the following formula: V = L×W 2/2 where V, volume (mm3); L, biggest diameter (mm); W, smallest diameter (mm). The mice were euthanized and the tumors were weighed at the end of the experiments.
Cell Migration and Invasion Assay
AGS cells transfected with pcDNA3.1 vector or pcDNA3.1-HoxD10 were used for cell migration and invasion assays. Cell migration was assessed by modified Boyden transwell chambers assay (Corning, NY, USA). Briefly, 5 × 104 cells/well were plated into 250 μL of 5% FBS medium in the upper chamber, and 500 μL of medium containing 15% FBS were added to the lower chamber. The cells were incubated for 24 h. The nonmigratory cells in the upper chamber were removed with a cotton swab. The cells on the bottom of the membrane were fixed and stained with DAPI. The number of visible cells was counted by fluorescence microscope in five random high power fields.
For evaluation of cell invasion, QCM 24-Well Collagen-Based Cell Invasion Assay (Millipore, Bedford, MA, USA) was used. Briefly, invaded cells on the bottom of the membrane were incubated with Cell Stain Solution, and 100 μL of the dye mixture was transferred to a 96-well microtiter plate for colorimetric measurement at 560 nm.
Cell Apoptosis and Cell Cycle Analysis
Cell apoptosis assays were performed using the annexin V/PI kit (Invitrogen) by flow cytometry analysis (FCA). Briefly, stably transfected cells (AGS, MKN28) were suspended in annexinbinding buffer, Alexa Fluor 488 annexin V and PI working solution were added in sequence. The stained cells were finally analyzed by FACScan flow cytometry (BD, Franklin Lakes, NJ, USA). Meanwhile, 2 × 105 seeded cells were exposed to ultraviolet light to induce apoptosis as a positive control.
Cell cycle distribution was detected by the Cycletest Plus DNA Reagent kit (BD). Briefly, transfected cells were harvested and washed in PBS. Cellular DNA was stained with 125 μg/mL propidium iodide for 20 min at 4° C in dark. The cells then were sorted by FACS Calibur, and cell-cycle distribution was determined using the ModFit LT software (Verity Software House Topsham, ME, USA).
Western Blot Analysis
Total proteins were extracted from stably transfected cells using RAPI lysis buffer. Lysates were resolved on SDSPAGE gel and transferred to PVDF membranes (Millipore). Primary antibodies were used as follows: HoxD10 (1:500; Santa Cruz Biotechnology Inc.), IGFBP3 (1:500; Santa Cruz Biotechnology Inc.), procaspase 3 (1:1000; Cell Signaling Technology Inc.), cleaved caspase 3 (1:1000; Cell Signaling Technology Inc.), procaspase 8 (1:1000; Cell Signaling Technology Inc.), cleaved caspase 8 (1:1000; Cell Signaling Technology Inc.) and β-actin (1:2500, Multisciences Biotech, Hangzhou, China). The blots were developed using a chemiluminescence with Las-4000 Imaging System (Fujifilm, Tokyo, Japan).
cDNA Microarray
Total RNA was extracted from stably transected MKN28 cells with pcDNA3.1-HoxD10 or pcDNA3.1 vector, and reverse transcribed to cDNA. Labeled samples were hybridized to 41,000 probes and transcripts based on Affymetrix platform in duplicate. The microarray data was performed using Agilent Feature Extraction software. We selected log2 ratio ≥ or ≤−1 as the threshold for upregulation or downregulation. The gene expression profiles were to their functions (cell proliferation, apoptosis, migration and angiogenesis, for example). Nine representatives of target genes were verified with qPCR. All primers for qPCR are listed in Supplementary Table 1.
Statistical Analysis
The differential HoxD10 mRNA expression between gastric cancer and normal tissues was analyzed by the Wilcoxon matched pairs test. The twotailed chi-square or Fisher exact tests were used for comparison of categorical variables. The probability of overall survival was calculated with the KaplanMeier method and differences between curves were evaluated with the Log-rank test. A cutoff of P < 0.05 applied for statistical significance.
All supplementary materials are available online at www.molmed.org .
Results
HoxD10 Is Downregulated in Gastric Cancer
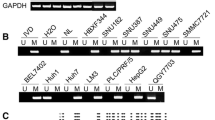
We first determined HoxD10 mRNA expression in 33 paired endoscopic biopsy samples from gastric cancer and normal stomach tissues (Supplementary Table 3). qPCR results showed that HoxD10 expression was downregulated significantly in tumor tissues relative to normal gastric tissues (P < 0.001) (Figure 1A). To further investigate the expression pattern of HoxD10 in gastric cancer, we examined HoxD10 expression in a panel of gastric cancer cell lines (n = 8) by PCR and Western blot analysis. Results demonstrated that HoxD10 expression was absent or reduced in all gastric cancer cell lines when compared to normal gastric tissues (Figure 1B and Supplementary Figure 1). Thus, these data suggest that HoxD10 might function as a tumor suppressor in gastric cancer.

Downregulation of HoxDWin gastric cancer. (A) HoxDWmRNA expression was determined by quantitative real-time PCR in 33 paired endoscopic biopsy samples from gastric cancer and normal stomach tissues. The data were described as relative log10 value, and analyzed by Wilcoxon matched pairs test. (B) The expression profile of HoxD10 was analyzed in eight gastric cancer cell lines, as well as in two of normal stomach tissues by conventional RT-PCR. GAPDH was used as the internal control. Normal: Normal stomach tissue.
HoxD10 Suppresses Proliferation, Migration and Invasion of Gastric Cancer Cells
To understand the potential function of HoxD10 in gastric cancer, AGS gastric cancer cells were transfected with pcDNA3.1-HoxD10 or pcDNA3.1 vector. The transfection efficiency was confirmed by RT-PCR and Western blot (Figure 2A). We observed that the number of surviving colonies formed on the plates was significantly reduced in HoxD10-transfected cells when compared to the control vector transfectants (P < 0.01) (Figure 2B). Similar results were reproduced in another gastric cell line MKN28 (Supplementary Figure 2). The cell proliferation inhibition of HoxD10 in AGS cells was confirmed by MTS assay, which showed significant reduction in growth in HoxD10-transfected cells (P < 0.05) (Figure 2C). In addition, ectopic expression of HoxD10 suppressed AGS cell migration (P < 0.01, Figure 2D) and cell invasion (P < 0.01, Figure 2E) significantly in the 24-transwell cell migration and collagen-based cell invasion system, respectively. Collectively, these results indicate that ectopic expression of HoxD10 inhibits gastric tumor cells survival, migration and invasion in vitro.

HoxD10 inhibits proliferation, and impairs cell migration and invasion of gastric cancer cells. (A) Overexpression of HoxD10 in AGS gastric cancer cells was confirmed by RT-PCR (left lane) and Western blot (right lane). GAPDH and β-actin were used as internal controls. (B) The monolayer colony formation assays. AGS cells were transfected with pcDNA3.1-HoxD10or pcDNA3.1 empty vector and selected with G418 for 14 to16 d. The numbers of survival colonies in each empty vector-transfected control were set to 100%, while HoxD10-transfected cells were presented as mean percentage ± SD from three individual experiments in right diagram. (C) The number of viability cells was measured by MTS cell proliferation assays. The assay was performed at 6, 24, 72 or 120 h in AGS cells transfected with pcDNA3.1-HoxD10or pcDNA3.1 empty vector. Experiments were performed in triplicate. The asterisk indicates statistical significance (*P < 0.05, **P < 0.01). (D) Cell migration assays were performed in modified Boyden transwell chambers assay. 5 × 104 AGS cells/well were plated and incubated for 24 h. The migratory cells on the bottom of the membrane were fixed and nucleus stained with DAPI (upper figures). The mean number of visible cells was counted by fluorescence microscope in five random high power fields (lower bar diagram). (E) QC 24-Well Collagen-Based Cell Invasion Assay (Millipore, USA) was used to assess cell invasion. Invaded cells were stained with Cell Stain Solution, then detected on a standard microplate reader (560 nm). The asterisk indicates statistical significance (*P < 0.05, **P < 0.01). B, E: ■, Vector; □, HoxD10; C: —?—, vector; —■—, HoxD10.
HoxD10 Inhibits Gastric Tumor Growth In Vivo
To further investigate the effects of HoxD10 on gastric tumor growth, pcDNA3.1-HoxD10 and pcDNA3.1 vector stably transfected MKN28 cells were injected subcutaneously into the flank of immunodeficient mice. The mean volume and wet weight of tumors in HoxD10 transfectants (n = 6) was significantly lower than that of the empty vector transfectants (n = 6) at the end of the experiment (P < 0.001) (Figure 3A, 3B). In addition, tumor growth measured over a 3-wk period showed that tumor volume was decreased significantly in mice with ectopic expression of HoxD10 relative to the empty vector group (Figure 3C). Thus, these results suggest that HoxD10 inhibits tumor growth in gastric cancer in vivo.

HoxD10 inhibits gastric tumor growth in vivo. MKN28 cells (7 × 106 cells) transfected with pcDNA3.1 vector or pcDNA3.1-HoxD10 were injected subcutaneously into the flanks of athymic nude mice (3–4 wk old) mice (n = 6 each group). (A) Representative pictures of tumor growth in nude mice inoculated subcutaneously with empty vector or HoxD10transfected cells at the end of the observation. (B) The tumor from each group was weighed immediately after removal. The average tumor weight is indicated as mean ± SD. (C) After 6 d of implantation of tumor cells, tumor volume (mean ± SD) was assessed every 3 d. The asterisk indicates statistical significance (*P< 0.05, ** P< 0.01). —●—, MKN28/vector; —▲—MKN28/HoxD10. classified into different clusters according
Ectopic Expression of HoxD10 Induces Apoptosis and Cell-Cycle Alteration in Gastric Cancer Cell Lines
To explore the mechanisms underlying the inhibition of cell proliferation by ectopic expression of HoxD10, we assessed cell apoptosis and cell-cycle by flow cytometry. Reexpression of HoxD10 significantly induced apoptosis of gastric cancer cells when compared to empty vector transfected cells in AGS and MKN28 cell lines (Figure 4A). Additionally, we observed that HoxD10-transfected cells (AGS and MKN28) showed lower S phase populations in comparison to the empty vector transfectants (Figure 4B).

HoxD10 induces cell apoptosis and cell-cycle inhibition. (A) The rate of cell apoptosis after stably transfected with HoxD10 was determined by flow cytometry in AGS and MKN28 cell lines. Region A1 indicates early apoptotic cells, A2 shows late apoptotic cells. Experiments were performed in duplicate. (B) Cell-cycle distribution was analyzed by FACS flow cytometry in AGS and MKN28 cells stably transfected with pcDNA3.1-HoxD10 or pcDNA3.1 vector. The mean percentages of cell-cycle phases from representative experiments are shown in each panel.
HoxD10 Modulates Multiple Downstream Genes including IGFBP3 and Apoptotic-Related Caspases in Gastric Cancer Cells
To identify potential downstream targets of HoxD10 in gastric cancer cells, we performed cDNA microarray in MNK28 cells. Results demonstrated alterations of multiple gene expression by ectopic overexpression of HoxD10. According to gene functional analysis (http://www.genecards.org), these genes have been reported to participate in cell proliferation, invasion, apoptosis, cell cycle and angiogenesis (representative genes shown in Figure 5A and Table 1). qPCR analysis was further performed to validate the expression pattern in nine selected genes. Our results showed that HCLS1, ANP32A, PDGFRL, IGFBP3 and CXCL9 were upregulated (> 2-fold change), while RAC2, NTS, KRT5 and TUSC3 were downregulated (< -2-fold change) by overexpression of HoxD10 in MKN28 cells (Figure 5B, Supplementary Table 4). These results suggest that HoxD10 potentially regulates downstream genes involved in tumorigenesis.

Gene expression profile changes by ectopic expression of HoxD10. (A) The gene expression profiles in HoxD10 or control vector stable transfectants was visualized in MKN28 cell lines. Representative genes with over a two-fold change are indicated on the right side of this heatmap image. (B) The expression of nine genes was measured by qPCR and calculated using the value of 2−ΔΔCT. Relative fold changes of HoxD10 transfectants were compared with empty vector values normalized to 1. □, Result of cDNA microarray; ■, qRT-PCR data in MKN28 cells. (C) Western blot analysis of IGFBP3 and caspases. The expression of IGFBP3, caspase 3 (pro and cleaved) and caspase 8 (pro and cleaved) were detected after being transected with HoxD10or empty vector in gastric cancer cell lines (AGS and MKN28). Band densities were normalized to β-actin. Relative fold change was expressed when compared with that of empty vector.
We further determined whether HoxD10 modulates IGFBP3 and related apoptotic caspases in gastric cancer cells (AGS and MKN28). Consistent with microarray and qPCR analysis data, the expression of IGFBP3 protein was upregulated significantly by ectopic expression of HoxD10 (Figure 5C, Supplementary Figure 3). It has been reported that IGFBP3 suppresses the growth of tumor cells (prostate and breast cancers) by activation caspases during cell apoptosis (28). Indeed, our results showed that reexpression of HoxD10 leads to activating cleaved caspase 8 and caspase 3 (see Figure 5C). These results indicate that the induction of cell apoptosis by HoxD10 may be mediated through IGFBP3 and subsequent activation of apoptotic caspases.
HoxD10 Transcriptionally Regulates the Expression of IGFBP3 in Gastric Cancer Cells
To determine whether HoxD10 directly binds to the promoter of IGFBP3 gene, ChIP assay was adopted. We used the PROMO program to search for HoxD10 transcription factor binding site in the putative IGFBP3 promoter. Five potential DNA sequences (H1-H5) were revealed as potential binding sites in the upstream 2.3-kb of IGFBP3 (Figure 6A). The ChIP assays were considered positive if the predicted HoxD10-binding sequence was enriched three-fold greater than the IgG antibody control. Quantitative PCR using the HoxD10 antibody-precipitated chromatin showed that four predicted binding sites (A1, A2, A3 and A4) were enriched 2.5, 4.4, 12.7 and 7.1 times, respectively (Figure 6B). Results demonstrated that as a transcription factor, HoxD10 could bind to multiple binding sites (A2, A3 and A4) of upstream IGFBP3.

HoxD10 transcriptionally regulates the expression of IGFBP3 in gastric cancer cells. (A). Schematic representation of IGFBP3 putative promoter with HoxD10 binding sites (BS). Upstream 2.3-kb sequence from the transcription start site of IGFBP3 were input to PROMO software to predict the possible HoxD10 binding sites (H1-H5). IGFBP3 DNA fragments containing of HoxD10 BS (−2,251 to −2,073 for BS1, and −1,727 to −943 for BS2) were cloned into pGL3-promoter vector and generated luciferase reporter gene. (B). Chromatin immunoprecipitation assays. The chromatin was immunoprecipitated with normal rabbit IgG and HoxD10 antibody in BGC823 cells. Quantitative real-time PCR was performed to detect DNA fragments in the promoter regions of IGFBP3 (A1 for H1 and H2, A2 for H3, A3 for H4, A4 for H5 respectively). Results were expressed as fold change compared to rabbit IgG normalized to 1. The experiments were performed in triplicate. (C). Luciferase reporter gene assay. Gastric cancer cell lines (BGC823 and SGC7901) were transfected with pcDNA3.1-HoxD10 or empty vector pcDNA3.1, pGL3-promoter vector or pGL3-BS, and pRL-TK vector. The relative fold of firefly activity was expressed normalized to renilla activity in pGL3-promoter vector. The experiments were performed in triplicate. **P < 0.01 indicates of HoxD10 versus empty vector. B: □, Rabbit IgG; ■, HoxD10 antibody; C: □, BGC-vector;  , BGC-HoxD10;
, BGC-HoxD10;  , SGC-vector; ■, SGC-HoxD10.
, SGC-vector; ■, SGC-HoxD10.
To delve further into the HoxD10 regulation of the transcription of IGFBP3, we have cloned two different DNA fragments (BS1 and BS2) in the promoter regions into a pGL3-promoter vector to generate luciferase reporter gene (see Figure 6A). The luciferase reporter assays showed that when co-transfected with pcDNA3.1-HoxD10 expression plasmid, luciferase activity of pGL3-BS2 promoter activity was enhanced by 4.0- and 4.4fold respectively, when compared to gastric cancer cells (BGC823 and SGC7901) transfected with pcDNA3.1 vector (P < 0.01). However, pGL3-BS1 promoter activity showed no significant change after being transfected with pcDNA3.1-HoxD10 (Figure 6C). It should be emphasized that DNA fragments in HoxD10 (BS2) have predicted IGFBP3 binding sites (A2, A3 and A4), and BS1 fragments with A1 binding sites. These results indicate that HoxD10 could directly bind to promoter sites (A2, A3 and A4), thus enhancing the expression of IGFBP3. Taken together, our results suggested that HoxD10 transcriptionally regulates the expression of IGFBP3 in gastric cancer cells.
Promoter Hypermethylation Contributes to HoxD10 Gene Silencing in Gastric Cancer Cells
To address whether HoxD10 is silenced though promoter hypermethylation in gastric cancer, we examined the expression of HoxD10 before and after treatment with 5-aza-2′-deoxycytidine (5-Aza) for 72 h. As shown in Figure 7A, the expression of HoxD10 mRNA and protein was reactivated dramatically after 5-Aza treatment in gastric cancer cell lines (AGS, MKN28 and MKN45). While return of silencing expression of HoxD10 was observed in MKN28 and MKN45 cells after withdrawing 5-Aza for another 48 h (Figure 7B). These suggested that HoxD10 may be reactivated transiently after 5-aza treatment. Next, the methylation status of HoxD10 in gastric cancer cells was determined by methylation-specific PCR. All tested gastric cancer cell lines showed full (MKN28, MKN45 and NCI-N87) or partial (AGS) methylation of HoxD10 promoter DNA (Figure 7C). These results suggest that DNA methylation may be associated with the transcriptional silencing of HoxD10 in gastric cancer cells.

DNA promoter of HoxD10is hypermethylated in gastric cancer. (A) HoxD10 expression was examined in gastric cancer cells (AGS, MKN28, MKN45 and NCI-N87) after treatment with 5-Aza for 3 d (d3) by RT-PCR (left diagram) and Western blot (right diagram). (B) HoxD10expression was examined in gastric cancer cells (AGS, MKN28 and MKN45) after withdrawn of 5-Aza for another 2 d (d5). Aza: 5-aza-2′-deoxycytidine. (C) The methylation status of HoxD10promoter is detected by methylation-specific PCR (MSP) in the above gastric cancer cell lines. (D) The methylation status of HoxD10promoter in primary gastric cancer, intestinal metaplasia and normal stomach tissues was detected by MSP. Representative images are shown. T: Gastric tumor tissues; A: Adjacent nontumor tissues; IM: Intestinal metaplasia with atrophic gastritis tissues; N: Normal stomach tissue; M: Methylated; U: Unmethylated. (E) Survival curves were plotted based on Kaplan-Meier survival analysis. Methylation status of HoxD10promoter was used as the variate to separate two lines (n = 58, P = 0.028, log-rank test).  , Unmethylated;
, Unmethylated;  , methylated.
, methylated.
Promoter Hypermethylation of HoxD10 Is Involved in Gastric Carcinogenesis
To further illustrate the relationship between HoxD10 expression and promoter methylation, we investigated HoxD10 methylation status in different endoscopic biopsy specimens from primary gastric cancers, atrophic gastritis with intestinal metaplasia and normal stomach tissues (Supplementary Table 4). The MSP results showed that the DNA promoter of HoxD10 in primary gastric cancer biopsy tissues exhibited frequent methylation (85.7%, 24/28). Interestingly, as a precancerous lesion, atrophic gastritis with intestinal metaplasia tissues showed a relatively high rate of HoxD10 methylation (60%, 18/30), whereas no methylation was observed in normal stomach tissues (0/10) (Figure 7D). To rule out the possibility that endoscopic biopsy specimens may not be representative of the whole tumor due to intratumor heterogeneity, we further evaluated the HoxD10 methylation status in surgically resected samples from primary gastric cancers, as well as samples from adjacent nontumor tissues. Results illustrated that HoxD10 promoter methylation was detected frequently in primary tumor samples (82.6%, 57/69), while most adjacent nontumor tissues did not demonstrate this methylation (see Figure 7D). We further analyzed the effect of HoxD10 promoter methylation on survival of surgical gastric cancer patients using Kaplan-Meier survival analysis. HoxD10 promoter hypermethylation was significantly associated with the lower survival times of gastric cancer patients (P = 0.028, Log-rank test) (Figure 7E). There was no significant correlation between the methylation of HoxD10 promoter and clinicopathological features such as gender and Lauren type. Interestingly, HoxD10 promoter hypermethylation seems to be associated with the TNM stage and pathological differentiation in surgical gastric cancer patients (Supplementary Table 5). These results indicate that HoxD10 promoter methylation contributes to the aberrant expression of HoxD10, and may be involved in gastric carcinogenesis.
Discussion
In this study, we demonstrated that downregulation of HoxD10 is a common event during the gastric tumorigenesis. Ectopic expression of HoxD10 inhibited proliferation, impaired migration and invasion of gastric cancer cells. In particular, reexpression of HoxD10 suppresses tumor growth in a mouse xenograft model. Taken together, our results suggest that HoxD10 functions as a novel tumor suppressor in gastric cancer. HoxD10 expression also was reported to be downregulated commonly other cancers including breast and endometrial tumors relative to normal tissues based on both in situ hybridization and RT-PCR analysis (24,25). Tumor growth suppression also was observed in prolactinoma with delivery of HoxD10 using adenovirus/adeno-associated virus (Ad/AAV) (29). Collectively, the expression of HoxD10 frequently is absent in cancer progression, and restoration of HoxD10 might be an important strategy for gastric cancer therapy.
We revealed that demethylating treatment with 5-aza could restore the expression of HoxD10 in gastric cancer cell lines (see Figure 7A). Furthermore, methylation of HoxD10 promoter frequently was detected in gastric cancer cell lines, as well as in primary gastric cancer tissues (see Figure 7C, 7D). These findings strongly suggest that aberrant DNA methylation is one of the key mechanisms underlying HoxD10 downregulation in gastric cancer. DNA hypermethylation has been viewed as a global process but only a few genes are functionally significant in tumorigenesis (5,7). DNA regions enriched with CpG dinucleotides, called CpG islands, once hypermethylated, can result in the silencing of tumor suppressor genes (TSGs) in cancer cells (5,6). Here, we identified HoxD10 as another novel TSG inactivated though promoter hypermethylation in gastric cancer. It has been well reviewed that the paralogous Hox genes, including HoxA5 and HoxA7, also are silenced epigenetically by promoter methylation in cancer progression (20,21,30), but we cannot exclude the appearance of other mechanisms in the silencing of HoxD10 in gastric cancer. Another “epigenetic” modification by microRNAs (miRNAs) are encoded in Hox cluster (19,31). Such miRNAs could regulate Hox gene expression by ways of mRNA cleavage or protein degradation (31,32). HoxD10 is indeed an important target of miR-10b, and constitutive expression of HoxD10 completely abrogated miR-10b-induced cell motility and invasion in breast tumor (32). In addition, an intervening RNA, called hox transcript antisense intergenic RNA (HOTAIR), is emerging as a controlling factor of the expression of HoxD genes (33).
We attempted to search for novel downstream targets of HoxD10 to identify pathways responsible for its tumor suppressing effect. Numerous genes are altered significantly by ectopic expression of HoxD10 with cDNA microarray analysis. These candidate targets have been reported to participate in cellular growth, apoptosis, adhesion and angiogenesis (34–41). We further validated that HCLS1, ANP32A, PDGFRL, IGFBP3 and CXCL9 were upregulated, while RAC2, NTS, KRT5 and TUSC3 downregulated by qPCR analysis. One gene of particular interest is IGFBP-3, which plays key roles on proliferation, migration and apoptosis in many cancers, including gastric cancer (28,34,35). Overexpression of IGFBP-3 could induce cell apoptosis by activation of caspase 8 and caspase 3 (28). Our results revealed that ectopic expression of HoxD10 could regulate IGFBP3 transcriptionally by Chip and promoter luciferase analysis, and subsequent activation of caspase 3 and caspase 8. Furthermore, two other HoxD10 downstream targets, ANP32A and HCLS1, also were involved in cell apoptosis. ANP32A is an important cofactor of caspase activation, which may enhance the activity of caspase 3 and determine sensitivity to apoptosis (36–38). HCLS1 is reported to increase apoptosis by activation of caspase 3 activity (39). Thus, these results support our observation that HoxD10 modulates several targets, at least including IGFBP3, ANP32A and HCLS1, that mediate caspase activation and induce cell apoptosis in gastric cancer. In addition to the regulation of apoptotic pathways, ectopic expression of HoxD10 upregulates CXCL9, an inhibitor of tumor growth and metastasis (40). RAC2, Ras-related Rho GTPases, which was downregulated by HoxD10 in our observation, enhances cell survival and migration (41). The identification of HoxD10 target genes may facilitate our understanding of its functions as tumor suppressor.
To evaluate the potential utility of HoxD10 as a biomarker in gastric oncogenesis in vivo, we examined the promoter methylation of HoxD10 in primary gastric cancer tissues, as well as precancerous tissues. We observed that the HoxD10 promoter DNA is hypermethylated in gastric cancer tissues obtained from endoscopic biopsies. Also, we found that HoxD10 promoter hyperme-thylation was present in 82.6% (57/69) of surgically resected gastric cancer tissues, which removes the possibility of intratumor heterogeneity. Interestingly, precancerous lesions tissues showed a 60% (18/30) rate of HoxD10 promoter hypermethylation relative to no methylation in normal stomach tissues (0/10). These results suggest that HoxD10 methylation is a frequent event in gastric cancer and its early involvement in gastric carcinogenesis. Recent studies have shown that although the abnormal epigenetic silencing of genes can occur at any time during tumor progression, it occurs most frequently during the early stages of the neoplastic process, as well as during the precancerous stage (42,43). We and others have shown previously that many genes were silenced frequently through promoter hypermethylation in precancerous lesions such as intestinal metaplasia (12,14,44). These epigenetic alterations might initiate the expansion of premalignant cells which accumulate during the early stages of tumorigenesis (7,12,44,45). We also demonstrated that the HoxD10 promoter hypermethylation was associated significantly with poor survival in gastric cancer patients. Our results indicate that the HoxD10 promoter hypermethylation may be associated with gastric carcinogenesis and potentially utilized as a biomarker for the progression of gastric cancer.
Conclusion
In summary, we identified HoxD10 as a candidate tumor suppressor gene which is epigenetically silenced in gastric cancer.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Kamangar F, Dores GM, Anderson WF. (2006) Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 24:2137–50.
Hartgrink HH, et al. (2009) Gastric cancer. Lancet. 374:477–90.
Ushijima T, Sasako M. (2004) Focus on gastric cancer. Cancer Cell. 5:121–5.
de Vries AC, et al. (2008) Gastric cancer risk in patients with premalignant gastric lesions: a nationwide cohort study in the Netherlands. Gastroenterology. 134:945–52.
Jones PA, Baylin SB. (2007) The epigenomics of cancer. Cell. 128:683–92.
Esteller M. (2008) Epigenetics in cancer. N. Engl. J. Med. 358:1148–59.
Baylin SB, Ohm JE. (2006) Epigenetic gene silencing in cancer — a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer. 6:107–16.
Dehan P, et al. (2009) DNA methylation and cancer diagnosis: new methods and applications. Expert Rev. Mol. Diagn. 9:651–7.
Laird PW. (2003) The power and the promise of DNA methylation markers. Nat. Rev. Cancer. 3:253–66.
Wang LJ, et al. (2009) ZIC1 is downregulated through promoter hypermethylation in gastric cancer. Biochem. Biophys. Res. Commun. 379:959–63.
Liu X, et al. (2010) Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. 29:442–50.
Yu J, et al. (2009) Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology. 136:640–51.
Wang X, et al. (2009) CHD5 is down-regulated through promoter hypermethylation in gastric cancer. J. Biomed. Sci. 16:95.
Wang LJ, et al. (2011) Klotho is silenced through promoter hypermethylation in gastric cancer. Am. J. Cancer Res. 1:111–9.
Botas J. (1993) Control of morphogenesis and differentiation by HOM/Hox genes. Curr. Opin. Cell Biol. 5:1015–22.
Samuel S, Naora H. (2005) Homeobox gene expression in cancer: insights from developmental regulation and deregulation. Eur. J. Cancer. 41:2428–37.
Jung C, et al. (2005) HOXB13 is downregulated in colorectal cancer to confer TCF4-mediated transactivation. Br. J. Cancer. 92:2233–9.
Friedmann Y, et al. (1994) Hox genes in normal and neoplastic mouse mammary gland. Cancer Res. 54:5981–5.
Shah N, Sukumar S. (2010) The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer. 10:361–71.
Raman V, et al. (2000) Compromised HOXA5 function can limit p53 expression in human breast tumors. Nature. 405:974–8.
Rauch T, et al. (2007) Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc. Natl. Acad. Sci. U. S. A. 104:5527–32.
Shiraishi M, et al. (2002) HOX gene clusters are hotspots of de novo methylation in CpG islands of human lung adenocarcinomas. Oncogene. 21:3659–62.
Jung C, et al. (2004) HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res. 64:3046–51.
Osborne J, et al. (1998) Expression of HOXD10 gene in normal endometrium and endometrial adenocarcinoma. J. Soc. Gynecol. Investig. 5:277–80.
Carrio M, Arderiu G, Myers C, Boudreau NJ. (2005) Homeobox D10 induces phenotypic reversion of breast tumor cells in a three-dimensional culture model. Cancer Res. 65:7177–85.
McCabe CD, Spyropoulos DD, Martin D, Moreno CS. (2008) Genome-wide analysis of the homeobox C6 transcriptional network in prostate cancer. Cancer Res. 68:1988–96.
Lei H, Juan AH, Kim MS, Ruddle FH. (2006) Identification of a Hoxc8-regulated transcriptional network in mouse embryo fibroblast cells. Proc. Natl. Acad. Sci. U. S. A. 103 27:10305–9.
Ingermann AR, et al. (2010) Identification of a novel cell death receptor mediating IGFBP-3-induced antitumor effects in breast and prostate cancer. J. Biol. Chem. 285:30233–46.
Cho MA, et al. (2008) HoxD10 gene delivery using adenovirus/adeno-associate hybrid virus inhibits the proliferation and tumorigenicity of GH4 pituitary lactotrope tumor cells. Biochem. Biophys. Res. Commun. 371:371–4.
Chen H, Chung S, Sukumar S. (2004) HOXA5-induced apoptosis in breast cancer cells is mediated by caspase 2 and 8. Mol. Cell. Biol. 24:924–35.
Yekta S, Tabin CJ, Bartel DP. (2008) MicroRNAs in the Hox network: an apparent link to posterior prevalence. Nat. Rev. Genet. 9:789–9.
Ma L, Teruya-Feldstein J, Weinberg RA. (2007) Tumor invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 449:682–8.
Gupta RA, et al. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 464:1071–6.
Oy GF, et al. (2010) Biological effects induced by insulin-like growth factor binding protein 3 (IGFBP-3) in malignant melanoma. Int. J. Cancer. 126:350–61.
Natsuizaka M, et al. (2010) Insulin-like growth factor-binding protein-3 promotes transforming growth factor-{beta}1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 31:1344–53.
Pan W, et al. (2009) PHAPI/pp32 suppresses tumorigenesis by stimulating apoptosis. J. Biol. Chem. 284:6946–54.
Hill MM, et al. (2004) Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 23:2134–45.
Hoffarth S, et al. (2008) pp32/PHAPI determines the apoptosis response of non-small-cell lung cancer. Cell Death Differ. 15:161–70.
Lee AY, et al. (2008) HS1-associated protein X-1 is cleaved by caspase-3 during apoptosis. Mol. Cells. 25:86–90.
Walser TC, et al. (2007) Immune-mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J. Immunother. 30:490–8.
Gu Y, et al. (2003) Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 302:445–9.
Feinberg AP, Tycko B. (2004) The history of cancer epigenetics. Nature Rev. Cancer. 4:143–53.
Ibáñez de Cáceres I, Cairns P. (2007) Methylated DNA sequences for early cancer detection, molecular classification and chemotherapy response prediction. Clin. Transl. Oncol. 9:429–37.
Kang GH, Lee S, Kim JS, Jung HY. (2003) Profile of aberrant CpG island methylation along the multistep pathway of gastric carcinogenesis. Lab. Invest. 83:635–41.
Kanai Y, Hirohashi S. (2007) Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis. 28:2434–42.
Acknowledgments
The project was supported by National Natural Science Foundation of China (30900676,81071961), National Basic Research Program of China (973 Program) (2012CB945004) and Science and Technology Key project of Zhejiang Province in China (2009C03012-3). The authors are grateful to Robert Weinberg of the Massachusetts Institute of Technology for kindly providing HoxD10 cDNA. We thank Lei Guo for helpful suggestions and discussion; Yan Shan, Xiaotong Hu and Fubiao Zhang for excellent technical assistance; and Manish Gala for critical review and discussion of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
10020_2012_1803389_MOESM1_ESM.pdf
Homeobox D10 Gene, a Candidate Tumor Suppressor, Is Downregulated through Promoter Hypermethylation and Associated with Gastric Carcinogenesis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Wang, L., Chen, S., Xue, M. et al. Homeobox D10 Gene, a Candidate Tumor Suppressor, Is Downregulated through Promoter Hypermethylation and Associated with Gastric Carcinogenesis. Mol Med 18, 389–400 (2012). https://doi.org/10.2119/molmed.2011.00172
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00172