
Abstract
Antiinflammatory clinical-grade, plasma-derived human α-1 antitrypsin (hAAT) protects islets from allorejection as well as from autoimmune destruction. hAAT also interferes with disease progression in experimental autoimmune encephalomyelitis (EAE) and in collagen-induced arthritis (CIA) mouse models. hAAT increases IL-1 receptor antagonist expression in human mononuclear cells and T-regulatory (Treg) cell population size in animal models. Clinical-grade hAAT contains plasma impurities, multiple hAAT isoforms and various states of inactive hAAT. We thus wished to establish islet-protective activities and effect on Treg cells of plasmid-derived circulating hAAT in whole animals. Islet function was assessed in mice that received allogeneic islet transplants after mice were given hydrodynamic tail-vein injection with pEF-hAAT, a previously described Epstein-Barr virus (EBV) plasmid construct containing the EBV nuclear antigen 1 (EBNA1) and the family of repeat EBNA1 binding site components (designated “EF”) alongside the hAAT gene. Sera collected from hAAT-expressing mice were added to lipopolysaccharide (LPS)-stimulated macrophages to assess macrophage responsiveness. Also, maturation of peritoneal cells from hAAT-expressing mice was evaluated. hAAT-expressing mice accepted islet allografts (n = 11), whereas phosphate-buffered saline-injected animals (n = 11), as well as mice treated with truncated-hAAT-plasmid (n = 6) and untreated animals (n = 20) rapidly rejected islet allografts. In hAAT-expressing animals, local Treg cells were abundant at graft sites, and the IL-1 receptor antagonist was elevated in grafts and circulation. Sera from hAAT-expressing mice, but not control mice, inhibited macrophage responses. Finally, peritoneal cells from hAAT-expressing mice exhibited a semimature phenotype. We conclude that plasmid-derived circulating hAAT protects islet allografts from acute rejection, and human plasma impurities are unrelated to islet protection. Future studies may use this in vivo approach to examine the structure-function characteristics of the protective activities of AAT by manipulation of the hAAT plasmid.
Similar content being viewed by others


Introduction
Current treatment of type 1 diabetes includes exogenous insulin injections to maintain normal blood glucose levels. However, patients endure uncontrolled glucose spikes, as well as sporadic hyperglycemia, the risk of hypoglycemia and long-term complications associated with diabetes (1). Pancreatic islet transplantation has been evaluated as a procedure that could enable patients to regain physiological glucose control, yet the immunosuppressive protocol that accompanies this procedure excludes diabetogenic corticosteroids, resulting in the exposure of grafted cells to an unopposed inflammatory environment (2). Similar to the process of islet injury during transplantation, the autoimmune response that is directed toward islets in a type 1 diabetic individual appears to overlap with several immune processes that occur during allograft rejection, with recently establish limited responses to T cell-directed clinical therapies (3). The concept by which inflammation serves as the backbone for both alloimmune and autoimmune responses is recently emerging as highly valid (4–10). Thus, there is increasing motivation to identify an islet-protective antiinflammatory immune-modulating agent that is safe for use.
Islets are particularly prone to injury during inflammatory conditions, responding most profoundly to interleukin (IL)-1β (11–13). As can be deduced from transplantation of human and animal islets, injured islets are a source of macrophage chemokines, particularly when inflamed (14–16). These antigen-independent, inflammation-dependent activities precede, as well as determine, the degree of subsequent antigen-specific immune responses.
α-1-Antitrypsin (AAT), the primary protease inhibitor in our circulation, rises during acute-phase responses and possesses antiinflammatory properties (17). For example, AAT increases production of IL-10 and decreases production of IL-6 (18), blocks infiltration of neutrophils and macrophages (19) and reduces nuclear factor (NF)-κB translocation to the nucleus (20). AAT blocks lipopolysaccharide (LPS) responses in human cells (21,22), blocks neutrophil migration and directly binds to IL-8 in lipid rafts (23,24). AAT has been shown to benefit disease parameters and animal models in rheumatoid arthritis (25), multiple sclerosis (26), systemic lupus erythematosus (8,27), ulcerative colitis (6,7,28) and type 1 diabetes (18,19,29–33), and its deficiency appears to be associated with several autoimmune diseases and their complications (6–8,34). Not unexpectedly, nonobese diabetic (NOD) mice have been reported to have 50% less circulating AAT than other strains (35). In addition, cultured islets display superior function in the presence of AAT (36,37). We recently reported that plasma-derived human α-1-antitrypsin (hAAT) facilitates antigen-specific immune tolerance to islet allografts, accompanied by inhibition of dendritic cell maturation and reduction in expression of costimulatory molecules, as well as by graft-site accumulation of T-regulatory (Treg) cells and local expression of the IL-1 receptor antagonist (IL-1Ra) (38). In diabetic individuals and in experimental diabetes, circulating AAT is inactivated by time- and concentration-dependent hyperglycation (39–42).
Irrespective of diabetes research, clinical-grade, plasma-derived affinity-purified hAAT is injected into patients with varying degrees of genetic deficiency in AAT, and prolonged administration has been proven safe (43–46). Yet, the mechanism of action of AAT is unknown, and the involvement of protease inhibition by AAT has not been established as the exclusive means of immune modulation and islet protection. Notably, there are experimental limitations to clinical-grade hAAT; purified from pooled-plasma, hAAT is rigorously processed to remove infectious contaminants (47). Also, depending on the manufacturer, the preparation is reported to contain plasma impurities such as antithrombin III, α1-lipoprotein and immunoglobulin A (47), all carried into the experimental system together with the injected (in vivo) or added (in vitro) material. Finally, the preparation consists of a diverse mixture of >120 human hAAT isoforms.
By using a plasmid construct that contains the genomic sequence of hAAT, rather than a cDNA-based expression segment, Stoll et al. (48) achieved sustained circulating levels of hAAT in mice. In the present study, we use this approach to replace exogenous hAAT with gene-based hAAT therapy and examine the ability of circulating single-isoform gene-delivered hAAT to protect islet allografts from acute rejection. The platform established here allows future research to focus on long-term whole animal studies with mutated hAAT to advance the understanding of the mechanisms that allow the molecule to protect islets from immunemediated destruction.
Materials and Methods
Mice
Islet allograft recipients were 6- to 7-wk-old female mice, heterozygote to the human AAT gene expressed under the control of surfactant promoter (background strain C57BL/6, H-2b) (38,49), achieved by crossbreeding with wild-type C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) or foxp3-green fluorescent protein (GFP) knock-in mice (38) of the same background strain (a gift from TB Strom, Beth Israel Deaconess Medical Center, Boston, MA, USA). Circulating levels of hAAT in heterozygote mice were below detection, as determined by enzyme-linked immunosorbent assay (ELISA) (sensitivity 4 ng/mL; Immunology Consultants Laboratory, Newberg, OR, USA). Islet allograft donors were 6- to 7-wk-old female wild-type BALB/C (H-2d) and DBA/2J (H-2d) mice (The Jackson Laboratory). Experiments were approved by the Ben-Gurion University of the Negev Animal Care and Use Committee (permit number IL-06-02-2010).
Plasmid Constructs
The 28-kb pEF-hAAT plasmid (a gift from MP Calos, Stanford University School of Medicine, Stanford, CA, USA) was constructed as described (48). A plasmid construct containing a truncated nonexpressing form of the human α-1-antitrypsin gene (pEF-Δ-hAAT) was generated to serve as a control for the native human α-1-antitrypsin gene-expressing plasmid. Briefly, a 6.5-kb EcoRV-BstBI fragment was removed from the pEF-hAAT plasmid. The remaining 21.5-kb fragment 5’-overhangs were converted into blunt ends using a DNA polymerase I large klenow fragment (New England BioLabs, Hitchin, UK) and then ligated using T4 DNA ligase (New England BioLabs). Negative hAAT expression was verified by specific ELISA (sensitivity 4 ng/mL; Immunology Consultants Laboratory).
In Vivo hAAT Gene Delivery
Delivery of the 28-kb pEF-hAAT construct was carried out as described (48). Briefly, 100 µg pEF-hAAT plasmid DNA was introduced by hydrodynamic (HD) tail-vein injection over a period of 6–9 s in a volume of 1.8 mL PBS (Biological Industries, Beit Haemek, Israel). Circulating levels of hAAT from periodically obtained tail serum samples were determined by ELISA (sensitivity 4 ng/mL; Immunology Consultants Laboratory). Control animals were either noninjected or introduced by HD tail-vein injection with 1.8 mL PBS alone or PBS containing 100 µg pEF-Δ-hAAT. Mice were allowed 12–17 d to recover before further experimentation.
Tissue Lysate Analysis
Liver tissue was homogenized in a radioimmunoprecipitation assay (RIPA) lysis buffer containing 10 mmol/L Tris, pH 8.0, 100 mmol/L NaCl, 5 mmol/L EGTA, 0.1% sodium dodecyl sulfate, 1% NP-40, 45 mmol/L β-mercaptoethanol and 50 mmol/L NaF (all from Sigma-Aldrich, Rehovot, Israel). Protease inhibitor cocktail set III (Calbiochem, Beeston, UK) was added immediately before tissue lysis. Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Aliquots of 100 µg protein were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto a polyvinylidene fluoride membrane (Bio-Rad). Human AAT was detected using anti-human AAT antibody (Fitzgerald Industries International, Acton, MA, USA). For detection of primary antibodies, blots were incubated with horseradish peroxidase conjugated to antirabbit antibodies (Amersham Pharmacia, London, UK). Immobilized antibodies were detected by an enhanced chemiluminescence (ECL) reagent (Amersham Pharmacia). Protein size and specificity were compared with hAAT from affinity-purified pooled human plasma (Aralast™; Baxter, Westlake Village, CA, USA).
Islet Allograft Transplantation
Islet transplantation was performed as described (19). Briefly, heterozygote hAAT transgenic (Tg) mice were rendered diabetic by a single injection of streptozotocin (225 mg/kg i.p.; SigmaAldrich). Donor islets were isolated and grafted under the renal capsule as described (19). Islet allograft rejection was defined as the day blood glucose levels exceeded 300 mg/dL after a period of at least 3 d of normoglycemia.
Immunohistochemistry
Tissues were fixed in 10% neutralbuffered formalin. Histological sections of paraffin-embedded sections were stained with di aminido phenyl indol (DAPI) (Sigma-Aldrich) and hematoxylin and eosin as well as with specific antibodies: hAAT was stained using polyclonal rabbit anti-human AAT (Fitzgerald) and CY-3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA), mouse insulin was stained with guinea pig anti-swine insulin antibody (Dako, Glostrup, Denmark), and CY-3-conjugated donkey anti-guinea pig IgG (Jackson ImmunoResearch) and Foxp3 were stained with Alexa Fluor® 488 anti-mouse foxp3 antibody (BioLegend, San Diego, CA, USA).
Assessment of Explanted Renal Allografts
Grafts were harvested by left nephrectomy as described (38). Upon graft explantation, tissue was maintained on ice and islet graft sites were identified macroscopically on the surface of the kidneys. For histology, samples were fixed in 10% formalin (Sigma-Aldrich). For reverse transcriptase-polymerase chain reaction (RT-PCR), the region containing the graft was removed with a number 11 blade and immediately transferred to liquid nitrogen. An equivalent size of tissue was removed from the opposite renal pole to control for background gene expression.
RT-PCR
Total RNA was extracted using a PerfectPure RNA Tissue Kit (5 PRIME, Gaithersburg, MD, USA), and reverse transcription followed using a Verso cDNA kit (Thermo Scientific, Horsham, West Sussext, UK). Primer sequences: mouse β-actin: forward 5’-GGTCTCAAA-CATGATCTGGG-3’, reverse 5’ GGGTC AGAAGGATTCCTATG-3’; IL-1β: forward 5’-CTCCATGAGCTTTGTACAAGG-3’, reverse 5’-TGCTGATGTACCAGTTGGGG-3’; IL-1 receptor antagonist: forward 5’-GACCCTGCAAGATGCAAGCC-3’, reverse 5’-GAGCGGATGAAGGTA AAGCG-3’; IL-18 binding protein: forward 5’-CCCACCCTACGAAGTACCAA-3’, reverse 5’-CTGGTCAAGGTCATGGTGTG-3’; inducible nitric oxide synthase (iNOS): forward 5’-CCAACCGGAGAAGGGGACGAACT-3’, reverse 5’-GGAGGGTGGTGCGGCTGGAC 3’; monocyte chemoattractant protein (MCP)-1: forward 5’-CTCACCTGCTGCTACTCATTC-3’, reverse 5’-GCTTGAGGTGGTTGTGGAAAA-3’; and tumor necrosis factor (TNF)-α: forward 5’-CCTGAGTTCTGCAAAGGGAG-3’, reverse 5’-AGCAAAAGAGGAGGCAACAA-3’.
Every sample was amplified by at least two different numbers of cycles to ensure that amplification was in the exponential phase of the PCR. Gene expression profile was analyzed by densitometry using National Institutes of Health ImageJ software (https://doi.org/rsbweb.nih.gov/ij) and normalized to β-actin. The results are presented as means ± standard error of the mean (SEM).
Skin-Tissue Allografts and Spleen Treg Analysis
hAAT-Tg × foxp3-GFP knock-in heterozygote mice were implanted intraperitoneally with a single sterile piece of 0.5 cm2 freshly prepared skin tissue, derived from vessel-poor abdominal midline sections of allogeneic donor mice and cleaned from the hypodermis. On day 7 after transplantation, spleens were removed. Erythrocytes were removed from total splenocytes using red blood cell (RBC) lysis buffer (PharM Lyse™; BD Biosciences Pharmingen, San Diego, CA, USA). Total splenocytes from all treatment groups were counted to determine population size and viability (Countess® automated cell counter; Invitrogen, Carlsbad, CA, USA). After washing with a fluorescence-activated cell sorter (FACS) buffer (PBS containing 2% bovine serum albumin, 0.1% sodium azide and 0.1% EDTA, pH 7.4), splenocytes (1 × 106 cells per sample) were either stained with allophycocyanin (APC)-conjugated anti-mouse CD4 (BioLegend) or underwent magnetic bead enrichment for CD4-positive T cells (EasySep® Stem-Cell Technologies, BC, Canada). GFP-positive cells from CD4-positive splenocytes were analyzed by FACS (3 × 105 cells per sample, Cytomics FC 500; Beckman Coulter, Brea, CA, USA). Data were analyzed by FlowJo software (Tree Star, Ashland, Oregon, USA).
RAW 264.7 Stimulation Assay
RAW 264.7 cells (ATCC) were seeded in a 24-well plate (2.5 × 105 cells per well) in RPMI 1640 medium (Biological Industries) containing 2% fetal calf serum and supplemented with 50 U/mL penicillin and 50 µg/mL streptomycin (Biological Industries). Cells were stimulated with LPS (1 ng/mL; Sigma-Aldrich), with or without one-third volume of serum collected from mice, introduced by HD tailvein injection with either PBS or pEF-hAAT 48 h earlier. For negative control, wild-type mouse serum was used (control [CT]), and for positive control, hAAT was added (Aralast™, 160 µg/mL, corresponding to plasmid-derived hAAT levels in the positive sera). Cell culture supernatants were collected 24 h later and analyzed for IL-6, MCP-1, TNFα and the C-X-C motif ligand 1 keratinocyte-derived chemokine (KC) levels using Q-Plex mouse cytokines chemiluminescence-based ELISA (Quansys Biosciences, Logan, UT, USA) and Q-View imager (Quansys Biosciences). Each cytokine was quantified by densitometry using Quansys Q-View software (Quansys Biosciences). Supernatant IL-1Ra levels were evaluated by specific DouSet ELISA (R&D Systems, Minneapolis, MN, USA).
Peritoneal Cell Population Analysis
Mice were anesthetized by isoflurane inhalation and injected intraperitoneally with cold PBS (8 mL). Peritoneal fluid was recovered, and erythrocytes were lysed by RBC lysis buffer for 10 min at room temperature. After being washed with FACS buffer, peritoneal cells (1 × 106 cells per sample) were stained with R-phycoerythrin (PE)-conjugated anti-mouse CD4O antigen, APC anti-mouse major histocompatibility complex class II (MHCII) and FITC anti-mouse CD11b (all from BioLegend). Isotype control antibodies were used according to the manufacturer’s recommendations. CD40 and MHCII-positive peritoneal cells (3 × 105 cells per sample) were analyzed using an FACS Canto flow cytometer (BD Biosciences Pharmingen). Data were analyzed by FlowJo software. Fluorescent intensity below 103 was defined negative as obtained from unstained samples and isotype controls (not shown). Fluorescent intensity in the range of 104 to 105 was defined as high. The cell size of CD11b-positive cells was determined by forward scatter analysis with gating on CD11b cells.
Statistical Analysis
In the bar graphs, results are expressed as the mean ± SEM. The significance of differences between groups was determined by a two-tailed Wilcoxon t test. Means were considered statistically different at P < 0.05.
All supplementary materials are available online at www.molmed.org .
Results
Production of Circulating hAAT in Mice Protects Islet Allografts from Acute Rejection and Promotes Treg Recruitment to Grafts
As we previously described (38), transgenic mice that carry a single copy of foxp3 promoter-driven GFP and a single copy of surfactant promoter-driven hAAT exhibit green fluorescent-positive foxp3-expressing Treg cells and circulating hAAT that is below detection (38). These strains were necessary to avoid mouse-to-human antibody production. Here, the animals were introduced pEF-hAAT or PBS by a single HD tail-vein injection. Expression of plasmid-derived hAAT in liver cells was confirmed by RT-PCR, Western blot analysis and immunohistochemistry (Figure 1). As shown, hepatocytes from mice that received pEF-hAAT by HD tail-vein injection expressed hAAT mRNA transcripts and produced hAAT, whereas mice that received PBS by HD tail-vein injection lacked hAAT expression. The anti-hAAT antibody detected a product of two sizes, one equivalent to Aralast™ and the other depicted a lower molecular size, reported elsewhere to be the result of posttranslational modifications (50–52).

Production of hAAT in mice by HD injection of pEF-hAAT. Expression of hAAT in mouse liver: Heterozygote hAAT transgenic mice were administered either PBS (HD-PBS, n = 3) or pEF-hAAT (HD-pEF-hAAT, 100 µg, n = 3) via HD tail-vein injection (1.8 mL/6 s). (A) Twenty-four hours later, animals were sacrificed and liver samples were examined for hAAT expression by RT-PCR of extracted liver RNA using primers specific for hAAT (top). Western blot analysis of extracted liver protein using an anti-hAAT antibody is shown (bottom). Protein size and specificity are compared to clinical-grade hAAT (Aralast™). (B) Immunohistochemistry (blue, nuclear DAPI stain; red, hAAT antibody).
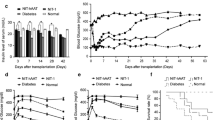
As soon as 1 d after HD tail-vein injection of pEF-hAAT, circulating levels of hAAT reached approximately 450 µg/mL. In 5 of 11 mice, these levels persisted for the >40 d of follow-up, according to periodic serum sample analysis using ELISA specific for hAAT. In 6 of 11 mice, hAAT levels were in the range of 171–350 µg/mL and persisted for the >110 d of follow-up (Figure 2A, representative follow-up, black dashed line, representing hAAT levels). These pEF- hAAT-derived circulating hAAT levels are comparable with those obtained by injections of clinical-grade hAAT at 1 d after injection; however, clinical-grade hAAT-derived circulating levels are short lived and drop to 7.8 ± 2 µg/mL on day 4 after injection, requiring multiple injections (not shown). To assess islet graft survival in hAAT-expressing animals, mice were allowed to recover for 12 d after HD tail-vein injection and were then rendered diabetic by a single streptozotocin injection. Five days later, hyperglycemic injected animals were transplanted with 450 allogeneic islets and glucose follow-up was preformed. As shown in Figures 2A and B, mice that were introduced with either PBS (PBS, n = 11) or a truncated nonexpressing construct of the human α-1-antitrypsin gene (pEF-Δ-hAAT, n = 6) by HD tail-vein injection rejected islet allografts at a comparable rate to untreated transplanted mice (CT, n = 20). In contrast, normo-glycemia persisted for over 100 d in mice that displayed circulating hAAT (Figure 2A, representative mouse; Figure 2B, n = 11, P > 0.0001 between pEF-hAAT-injected animals and the CT group and P > 0.001 between pEF-hAAT and PBS or pEF-Δ-hAAT-injected animals). One mouse out of the pEF-hAAT-treated group exhibited graft failure on day 28 after transplantation (Figure 2B).

Circulating pEF-hAAT-derived hAAT protects islet allografts from acute rejection. Healthy heterozygote hAAT transgenic mice were either non-HD tail-vein-injected animals (CT, n = 20) or administered PBS (n = 11), pEF-hAAT (n = 11) or a truncated modified plasmid pEF-Δ-hAAT (n = 6) via HD tail-vein injection, rendered diabetic by a single streptozotocin (STZ) injection and then grafted with allogeneic islets. (A) Representative follow-up of blood glucose and circulating hAAT levels. pEF-hAAT, pEF-Δ-hAAT and PBS administration (indicated by HD injection arrowhead (▼)) was followed by STZ injection (indicated by STZ arrowhead (▼)) before transplantation of allogeneic islets (day 0, indicated by an up arrow (↑)). hAAT levels are indicated by the black dashed line. Blood glucose levels were assessed periodically in pEF-hAAT (black solid line), pEF-Δ-hAAT (gray dashed line) and PBS (gray solid line) groups. Nephrectomy, removal of the graft-containing kidney. (B) Graft survival curve. The day of islet graft failure is defined as the time after transplantation in which blood glucose levels exceed 300 mg/dL. CT, islet graft recipient mice that were not HD tail-vein injected. (C) Graft histology. Accepted allografts were removed from mice for staining (30–100 d; representative stain of a 72-d graft explant). Graft, islets, noninvasive “cuff” and Treg cells are indicated. Red, insulin staining; blue, DAPI nuclear staining; green, foxp3 Treg cells inside the noninvasive mononuclear “cuff.”
Grafts were removed for inspection after graft acceptance was established (Figure 2C). Insulin staining confirmed the presence of viable islets, and hematoxylin and eosin staining revealed the presence of a noninvasive mononuclear cell “cuff,” similar to the observation described using exogenous hAAT injections (38) and other tolerogenic approaches (53). To identify foxp3-positive Treg cells, anti-mouse foxp3 staining was performed. As shown in Figure 2C (bottom), a scattered appearance of Treg cells is detected within the noninvading mononuclear cell population, comparable to the findings observed using exogenous hAAT during islet allograft transplantation in mice (38). By contrast, mice that were introduced with PBS by HD tail-vein injection, depicted islet allograft rejection as evidenced by the low residual insulin staining, and foxp3-positive Treg cells were infrequent or absent (Supplementary Figure 1, PBS). To facilitate comparison, the original Figure 2C pEF-hAAT immunofluorescent panel is presented adjacent to PBS.
Cytokine Expression Profile in Islet Allografts
RT-PCR was performed on explanted tissue to study the inflammatory and antiinflammatory-related gene expression profile in the allografts (Figure 3). Renal tissue from the pole opposite to the transplantation site was used for background tissue gene expression. Grafts from day 2 after transplantation in mice that were introduced PBS by HD tail-vein injection were used to represent an inflammatory graft environment (PBS); these were compared with grafts from day 60 to 110 after transplantation in mice that were introduced pEF-hAAT by HD tail-vein injection (pEF-hAAT), representing the end-point inflammatory profile of islet grafts in the presence of hAAT. Importantly, graft-derived MCP-1 was evaluated in light of its reported correlation with poor allograft prognosis in human islet transplantation (14–16,54). As shown, expression of MCP-1 reached as low as background values in mice that had positive circulating levels of pEF-hAAT-derived hAAT and was 2.7 ± 0.43-fold lower compared with PBS-treated mice (P = 0.0156). A similar trend was found in the levels of TNFα and iNOS (P = 0.0488 and P = 0.0313, respectively). IL-1β expression responded to circulating hAAT by a significant reduction of 1.8 ± 0.35-fold compared with PBS-treated mice (P = 0.0391), whereas the expression of the antiinflammatory IL-1Ra was elevated by 1.52 ± 0.6 compared with PBS-treated mice (P = 0.0313). IL-18BP expression levels were increased 1.45 ± 0.21-fold compared with PBS-treated animals (P = 0.0313).

Cytokine expression profile in explanted islet allografts. Mice were grafted with allogeneic islets and graft mRNA expression levels were assessed. Renal tissue (CT, n = 8) was obtained from the opposite pole to islet grafts of non-HD tail-vein-injected animals, to represent background gene expression levels. Graft tissue: PBS (n = 8, PBS administered by HD tail-vein injection, islets grafted 17 d later and grafts explanted at 48 h); pEF-hAAT (n = 8, pEF-hAAT administered by HD tail-vein injection, islets grafted 17 d later and grafts explanted 30–100 d later). The results are presented as arbitrary units (AU) of mean ± SEM normalized to β-actin mRNA expression; *P> 0.05.
Circulating pEF-hAAT-Derived hAAT Modifies the Posttransplantation foxp3 Treg Cell Population
We sought to examine the effect of hAAT on the Treg cells under inflammatory conditions in vivo. Skin-tissue grafts were used to evoke a strong immune response. Foxp3-GFP knock-in transgenic mice were used as recipients, since they allow the detection of graft-evoked GFP-positive Treg cells within the CD4-positive lymphocytic spleen population by FACS analysis (Figure 4). Specifically, mice were introduced pEF-hAAT or PBS by HD tail-vein injection (n = 6 per treatment) 4 d before intraperitoneal skin-tissue transplantation (skin-tissue graft). Mice that were not grafted with skin-tissue (CT and pEF-hAAT HD tail-vein-injected mice, n = 6 per group) were used to determine background Treg cell population size in each group. Circulating hAAT level at the time of transplantation was 0.5 ± 0.1 mg/mL (mean ± SEM). On day 7 after transplantation, total splenocyte count from control and treated mice depicted comparable population size and viability. CD4-positive cell population and foxp3-GFP-positive cell population were assessed out of total spleen (Supplementary Figures 2Aa and B). The expression of CD4 was in the range of 19.1–19.4% for all treatment groups (Supplementary Figure 2A), whereas the expression of foxp3-GFP out of total splenocytes varied in the low range of 1.4–2.27% between treatment groups (Supplementary Figure 2B). To better depict changes in foxp3-GFP expression, splenocytes were enriched for CD4-positive cells. Subsequently, GFP-positive cells out of CD4-enriched cells were identified by FACS analysis. As shown, skin-tissue grafted mice that were introduced with PBS exhibited 4.5 ± 0.7% foxp3-positive cells, whereas mice that were introduced pEF-hAAT exhibited 7.5 ± 0.6% cells, a 1.67-fold increase in Treg cell population size (P = 0.0469) (Figure 4A). Representative scatter images are shown (Figure 4B).

Circulating pEF-hAAT-derived hAAT modifies posttransplantation foxp3 Treg cell population. (A) Mice were grafted intraperitoneally with allogeneic skin tissue, and splenic Treg cell population size was assessed 7 d after transplantation. Control (nontransplanted mice): CT, background Treg cell population size in mice that were not injected by HD tailvein injection (n = 6); pEF-hAAT, Treg cell population size from mice that were HD tail-vein injected with pEF-hAAT (n = 6). Transplantation (skin-tissue graft): PBS, allogeneic skin-tissue transplantation into mice that were HD tail-vein injected with PBS (n = 6). pEF-hAAT, allogeneic skin-tissue transplantation into mice that were HD tail-vein injected with pEF-hAAT (n = 6). Treg cell population size was assessed by FACS after CD4-positive T-cell enrichment. Mean ± SEM; *P> 0.05. (B) Representative FACS analysis images (y axis: forward scatter (FSC); x axis: Foxp3-GFP).
Macrophages and Peritoneal Cell Population Exhibit an Antiinflammatory Response Profile in the Presence of Circulating pEF-hAAT-Derived hAAT
It was important that we demonstrate the antiinflammatory function of circulating hAAT. The ability of serum collected from mice that were introduced to either pEF-hAAT or PBS to reduce the inflammatory response of macrophages was examined using stimulated RAW 264.7 cells (Figure 5A). The cells were added sera obtained from untreated mice, sera from untreated mice supplemented with clinical-grade hAAT (160 µg/mL) or sera from mice that were introduced PBS/pEF-hAAT by HD tailvein injection, all at a 1-to-3 dilution ratio inside culture media. Cells were then stimulated with LPS for 48 h. Culture supernatants were collected, and cytokine levels were analyzed. As shown, stimulation by LPS evoked the secretion of the proinflammatory cytokines and chemo-kines IL-6, TNFα, KC and MCP-1. To a lesser extent, LPS also evoked IL-1Ra release. Proinflammatory cytokine secretion was significantly reduced by both exogenous hAAT and pEF-hAAT-derived hAAT, but not by sera from mice that were administered PBS. Between PBS-introduced animals and pEF-hAAT-introduced animals (Figure 5A, two far right bars), IL-6 was reduced by 45.7 ± 1.3% (mean ± SEM, P = 0.0156); MCP-1 by 60.1 ± 1.8% (P = 0.0078) and TNFα by 33.3 ± 1.9% (P = 0.0078). In contrast, IL-1Ra was elevated by 74 ± 3.6% in the presence of sera from pEF-hAAT-introduced mice compared with sera from PBS-introduced mice (P = 0.0078). Analysis of sera before introduction to wells revealed minute levels of inflammatory cytokines with insignificant changes between treatments (IL-1β >40 pg/mL, IL-6 >20 pg/mL, IL-10 >2 pg/mL, MCP-1 >1 ng/mL and KC >30 pg/mL). However, hAAT-positive sera contained elevated IL-1Ra levels (137.8 ± 30 ng/mL control sera, 434.4 ± 72 ng/mL hAAT-positive sera, mean ± SEM, P = 0.0234).

Macrophages exhibit an antiinflammatory response and a reduced maturation profile in the presence of circulating pEF-hAAT-derived hAAT. (A) RAW 264.7 cells were stimulated with LPS (1 ng/mL) in the presence of mouse serum (33% volume) from the following sources: nonstimulated and CT (serum from noninjected mice, n = 8); serum + hAAT (serum from noninjected mice plus 160 µg/mL hAAT, n = 8); serum pEF-hAAT (serum from plasmid-injected mice expressing 500 µg/mL hAAT, n = 8); and serum PBS (serum from PBS-injected mice, n = 8). The 48-h supernatant cytokine levels were analyzed. (B) Surface levels of CD40 and MHCII in peritoneal cell populations directly obtained from islet-grafted mice. The peritoneal cell population (1 × 106 cells per sample) from CT nongrafted mice (black dashed line, n = 6) or islet-grafted mice that were administered PBS (gray solid line, n = 6) or pEF-hAAT (thick black solid line, n = 6) via HD tail-vein injection was assessed for the expression of CD40 (upper panels) and MHCII (lower panels) in total cells (left) and CD11bHI peritoneal cells (middle) by FACS (representative images are shown). Bar graphs depict events from CD11bHI peritoneal cells, presented as fold from CT. Mean ± SEM; *P > 0.05, **P > 0.01.
Antiinflammatory function of circulating pEF-hAAT-derived hAAT was assessed in islet-grafted mice in vivo. Islet-grafted mice that were introduced PBS or pEF-hAAT by HD tail-vein injection at day 30 after transplantation were used to assess the expression of CD40 and MHCII in the peritoneal cell population by flow cytometry. These mice were compared with nongrafted mice (CT), representing the steady-state expression profile of CD40 and MHCII in peritoneal cells. At the time of peritoneal lavage on day 30, PBS-treated islet-grafted mice exhibited hyperglycemia, indicating islet graft failure. As shown in Figure 5B, pEF-hAAT-treated islet-grafted mice exhibit an expression profile of total CD40 and MHCII that was similar to nongrafted mice (CT) and significantly reduced compared with PBS-treated islet-grafted mice (Figure 5B, left upper and lower panels, representative images and Supplementary Figure 3, pooled results, P = 0.0313). Similarly, CD11b-positive peritoneal cells from pEF-hAAT-treated mice displayed significantly reduced surface levels of both CD40 and MHCII compared with PBS-treated islet grafted mice (P = 0.0313). However, the expression of MHCII in this cell population was not significantly altered in both PBS-treated islet grafted mice and control nongrafted mice (CT) (Figure 5B, middle upper and right lower panels, representative images, and their corresponding bar graphs).
Discussion
Despite the discouraging 5-year outcomes of human islet transplants, multiinternational collaboration groups have made tremendous progress in various aspects of islet transplantation (reviewed in 2). The ongoing developments include improvements in islet-isolation techniques, advances in pretransplant islet culture conditions and progress toward single-donor transplants.
AAT has been administered to AAT-deficient patients for three decades; the low plasma levels of AAT are augmented by lifelong weekly injections of affinity-purified hAAT, reaching 4- to 6-fold higher circulating levels than normal. Consistent with its role as an antiinflammatory agent, AAT augmentation therapy was reported to reduce the incidences of pneumonia and lung cancer in treated individuals (43–46). Recently, various studies suggested that AAT possesses qualities that are favorable for islet graft survival (30,33,35,55–58). In particular, our group studied the protective attributes of AAT in the context of islet allograft survival using repeated injections of clinical-grade hAAT in mice (18,38).
An increasing amount of evidence points at an association between α-1-antitrypsin deficiency or its impaired activity and a variety of immune disorders with an inflammatory component, such as rheumatoid arthritis (25), multiple sclerosis (26), systemic lupus erythematosus (8,27), ulcerative colitis (6,7,28) and diabetes mellitus (29–33,59,60). The study of hAAT in animal models for prolonged periods of time is essential to elucidate the underlying mechanisms for the protective effect of α-1-antitrypsin in these immune disorders and its possible clinical implication. However, the use of exogenous hAAT in animal models imposes substantial limitation in prolonged experimental setups. For example, adverse effects were reported when administering multiple injections of hAAT to animals, including induction of inflammation at the site of administration (35) and induction of a humoral response against the human protein (35). Human AAT is cleared twice as rapidly in mice and therefore requires more frequent injections (61). Lastly, the presence of other plasma proteins and chemical impurities in clinical-grade preparations of human AAT that are carried into the experimental system together with the injected (in vivo) or added (in vitro) material require revisiting with a gene-derived product to confirm outcomes. In light of the wellrecognized genetic AAT deficiency in humans, AAT is considered a primary candidate for gene therapy (62). Hydrodynamic-based transfection of nonadenoviral naked DNA allows long-term expression of the transgene without evoking host immune responses (48,63). pEF-hAAT is a 28-kb plasmid that holds the entire human α-1-antitrypsin sequence, including exons and introns, and the ability to self-replicate without integrating into the genome (48). The plasmid also holds an Epstein Barr viral backbone that provides affinity for hepatocytes, the cells in which the promoter for AAT is most active. Using pEF-hAAT, expression of hAAT extends for the longest period of time compared with various cDNA-based hAAT plasmids (48). Moreover, recent work highlighted the potential to translate the HD delivery method to large animals and humans through the use of catheters to access liver circulation (64–67).
In the present study, we show that hAAT-expressing mice accept allogeneic islet grafts. Accepted grafts revealed local reduction in proinflammatory cytokine and chemokine expression and an increase in the expression of antiinflammatory molecules, similar to the reported findings using repeated injections of clinical-grade hAAT (38). Of particular importance, the proinflammatory cytokine IL-1β and the chemokine MCP-1 were significantly reduced in intragraft sites of hAAT-expressing mice. These mediators were reported to cause islet graft failure via antigen-independent inflammation and correlate with poor transplant outcome (12,68–70). Thus, the ability of hAAT to diminish inflammatory mediators in the graft tissue may provide the basis for the diminished alloimmune response and prolonged islet graft survival. Furthermore, a noninvasive mononuclear cell “cuff” containing foxp3-positive T-regulatory cells was recruited, agreeing with the results obtained by Lewis et al. (19) and others (53). Within the pEF-hAAT-treated group there was one case of graft failure. We attribute the late failure to the development of ascites, which appears to have progressed gradually. According to liver histology, which was obtained on a different day throughout the first month after HD injection in related experiments, we detected an injured liver in the immediate time after injection, but a regenerated liver from day 14 onward (not shown), consistent with the report by Stoll et al. (48). The proportion of cases with HD injection-induced ascites was thus minimized by allowing a 17-day follow-up before islet transplantation protocol and the exclusion of animals that exhibited signs of intra-abdominal fluid accumulation.
Graft gene expression profile was considerably antiinflammatory. We compared 48-h untreated graft gene expression profiles with >60-day pEF-hAAT-treated accepted grafts. These time points were found to be optimal to allow the examination of inflammatory events that govern rejection versus the inflammatory environment of a treated graft; untreated islet grafts did not survive until day 60, preventing time-parallel inspection of longstanding accepted treated allografts.
To specifically address the possibility that gene-derived hAAT modifies Treg cells under inflammatory conditions, we examined spleen cells after graft provocation in vivo in animals that were introduced pEF-hAAT. Skin-tissue transplantation represents a more stringent allograft model than islet transplantation, since it contains numerous professional antigen-presenting cells with the capacity to migrate from the graft and efficiently stimulate recipient T cells (71). In addition, a larger number of animal recipients is more easily achievable in a short experimental session, minimizing variance in outcomes. Indeed, expression of hAAT resulted in a greater number of Treg cells in the spleen (Figure 4) and elevated foxp3 transcripts in the explanted long-lasting islet allografts (Figure 3). The rise in Treg cell population size is desired not only in experimental setups but also in the clinical environment (72), where mild changes in Treg cell population size have been shown to partially predict outcomes of transplants (73–75).
Macrophages are the primary cell population that the engrafted islets encounter in current surgical practice and that exert the greatest rapid damage to islets. To examine the implications of circulating plasmid-derived hAAT on macrophage cell activation, we exposed a murine macrophage cell line to animal sera after pEF-hAAT introduction, in the presence of LPS. The effect of sera from hAAT-expressing mice on macrophage cell stimulation revealed a blunted inflammatory profile that is consistent with graft survival. The importance of early macrophage inhibition can also be appreciated by the requirement that hAAT therapy cover the initial 3 days after transplantation, as previously reported by Lewis et al. (18). Here, serum contained changes in cytokines that evolved because of the presence of circulating hAAT, consistent with findings obtained from exogenous hAAT treatments (38). However, quite notably, IL-1Ra levels exhibited a 3.1-fold rise in the hAAT group. The induction of IL-1Ra, the naturally occurring endogenous inhibitor of IL-1, by hAAT was reported by others in other experimental setups (17,76), and the role of IL-1Ra in IL-1 blockade and islet protection has had a recent surge in reports (12,77–79). These changes may have influenced the outcome of macrophage stimulation. If this is indeed the case, it may be advantageous to use prolonged hAAT pretreatment in the context of islet transplantation, thus raising IL-1Ra levels and aiming at preconditioning the recipient toward an improved islet transplant prognosis.
Peritoneal cell population from hAAT-expressing islet-grafted mice depicted marked reduction in surface costimulatory molecules CD40 and MHCII expression. The reduction in surface costimulatory molecule expression was more profound in CD11b-positive peritoneal cells. The expression of costimulatory molecules on the surface of antigenpresenting cells is crucial in determining the nature and extent of the immune response. Elevated surface expression of costimulatory molecules is generally associated with increased immune activation. The expression of the surface markers CD11b, CD40 and MHCII is a well-established phenotype of macrophages. However, recent data suggest that using these markers for identifying macrophages may not exclude other cell types (80). In summary, the reduced expression of costimulatory molecules in peritoneal cells from islet-grafted hAAT-expressing mice suggest an overall diminished maturation profile.
Although AAT appears to modify the immune response to pancreatic islet allografts, AAT does not cause immune incompetence (43–46,81–87). In support of this finding, two independent studies report that AAT does not inhibit T-cell proliferative and cytokine release responses to IL-2, anti-CD3/CD28 or concanavalin A (18,38). In addition, in vivo studies demonstrate an ability for AAT treatment to protect against TNFα- and endotoxin-induced lethality (84), as well as to suppress live bacterial proliferation in two chronic lung infection models (81,83). Clinical studies indicate that augmentation therapy with AAT for α-1-antitrypsin genetically deficient patients effectively elevates AAT levels and function in serum and lung epithelial fluid and attenuates microbial colonization, loss of lung tissue and function, airway inflammation, and the frequency and severity of acute chronic obstructive pulmonary disease (COPD) exacerbation (43–46,82,85–87). In these reports, administration of AAT at doses that exceed normal plasma values is reported to be well tolerated and safe (44,85,86). Taken together, the outcomes of islet grafting in the presence of circulating hAAT, as described in the current study, and the widely reported competence of the immune system under these conditions, suggest that AAT is a nontoxic agent with a tolerogenic function.
Islet protection by AAT was successfully executed using gene therapy approaches in the autoimmune NOD mouse model. For example, in the study by Lu et al. (58), a single intramuscular injection of recombinant adeno-associated virus 1 (rAAV1)-expressing hAAT resulted in prevention of diabetes in 9 of 10 NOD mice. This protective effect of hAAT was regarded specific, since the treatment with rAAV1 vector-expressing human elafin resulted in prevention of diabetes development in only 1 of 10 NOD mice. However, circulating elafin levels were considerably low (10 ng/mL). The animals responded to hAAT with a strong humoral immune response, primarily since the material is from a human source, but perhaps enhanced by the fact that they are prone to a hyperactive immune system (88). The present study is novel in that the mouse model used harbors an intact immune system and does not mount neutralizing antibodies against the human material.
In related experiments, using animals with low circulating pEF-hAAT-derived hAAT (1.0 ± 0.02 pg/mL, n = 5, not shown), we found protection of islet allografts and reduction in inflammation to be similarly achieved. A protective effect by clinical-grade hAAT could not be achieved with this 500-fold lower dose. Thus, this finding implies that clinicalgrade hAAT might contain AAT with impaired islet-protective activity, requiring dose compensation. Because elastase inhibition by the clinical preparation is intact, according to the manufacturer and as verified by us, our findings suggest that the islet-protective attributes of AAT may be protease independent. Using the molecular approach reported here, a mutation of the plasmid at the elastase-binding site would allow direct examination of the protease-independent activities of hAAT in vivo.
In conclusion, plasmid-derived circulating hAAT protects islet allografts from acute rejection, and human plasma impurities are unrelated to islet protection. Consistent with the availability as augmentation therapy for genetic deficiency in AAT and its favorable safety profile (43–46), the use of AAT for recently diagnosed type 1 autoimmune diabetes is under two clinical trials in the U.S. (NCT01319331 and NCT01183455); yet the complete mechanism behind the favorable activities of AAT is still being investigated. Future studies may use the approach described here to examine the structure-function attributes of the islet-protective activities of AAT in vivo by gene manipulation of the hAAT plasmid.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Nathan DM, et al. (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 353:2643–53.
Robertson RP. (2010) Islet transplantation a decade later and strategies for filling a half-full glass. Diabetes. 59:1285–91.
Ludvigsson J. (2010) Immune intervention in children with type 1 diabetes. Curr Diab Rep 10:370–9.
Leavy O. (2011) Autoimmunity: joint damage without antigen. Nat. Rev. Immunol. 11:678.
Stadelmann C, Wegner C, Bruck W. (2011) Inflammation, demyelination, and degeneration: recent insights from MS pathology. Biochim. Biophys. Acta. 2011:275–82.
Gambichler T, et al. (2006) Complex extraintestinal complications of ulcerative colitis in a patient with alpha1-antitrypsin deficiency. Eur. J. Med. Res. 11:135–8.
Yang P, et al. (2000) Alpha1-antitrypsin deficiency and inflammatory bowel diseases. Mayo Clin. Proc. 75:450–5.
Zhang XH, et al. (1989) Changes of neutrophil elastase and alpha 1-antitrypsin in systemic lupus erythematosus. Proc. Chin. Acad. Med. Sci. Peking Union Med. Coll. 4:26–9.
Goldstein DR. (2011) Inflammation and transplantation tolerance. Semin. Immunopathol. 33:111–5.
Hanidziar D, Koulmanda M. (2010) Inflammation and the balance of Treg and Th17 cells in transplant rejection and tolerance. Curr. Opin. Organ Transplant. 15:411–15.
Mandrup-Poulsen T, et al. (1986) Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia. 29:63–7.
Schwarznau A, et al. (2009) IL-1beta receptor blockade protects islets against pro-inflammatory cytokine induced necrosis and apoptosis. J. Cell Physiol. 220:341–7.
Mandrup-Poulsen T, et al. (1993) Involvement of interleukin 1 and interleukin 1 antagonist in pancreatic beta-cell destruction in insulin-dependent diabetes mellitus. Cytokine. 5:185–91.
Solomon MF, Kuziel WA, Simeonovic CJ. (2004) The contribution of chemokines and chemokine receptors to the rejection of fetal proislet allografts. Cell Transplant. 13:503–14.
Piemonti L, et al. (2002) Human pancreatic islets produce and secrete MCP-1/CCL2: relevance in human islet transplantation. Diabetes. 51:55–65.
Chen MC, et al. (2001) Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 betaexposed human and rat islet cells. Diabetologia. 44:325–32.
Tilg H, et al. (1993) Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1 beta synthesis by human peripheral blood mononuclear cells. J. Exp. Med. 178:1629–36.
Lewis EC, et al. (2005) Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc. Natl. Acad. Sci. U. S. A. 102:12153–8.
Lewis EC, et al. (2008) Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc. Natl. Acad. Sci. U. S. A. 105:16236–41.
Churg A, et al. (2001) Alpha-1-antitrypsin and a broad spectrum metalloprotease inhibitor, RS113456, have similar acute anti-inflammatory effects. Lab. Invest. 81:1119–31.
Subramaniyam D, et al. (2010) Effects of alpha 1-antitrypsin on endotoxin-induced lung inflammation in vivo. Inflamm. Res. 59:571–8.
Nita IM, Serapinas D, Janciauskiene SM. (2007) Alpha1-antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int. J. Biochem. Cell Biol. 39:1165–76.
Subramaniyam D, et al. (2010) Cholesterol rich lipid raft microdomains are gateway for acute phase protein, SERPINA1. Int. J. Biochem. Cell Biol. 42:1562–70.
Bergin DA, et al. (2010) Alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Invest. 120:4236–50.
Grimstein C, et al. (2011) Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J. Transi. Med. 9:21.
Subramanian S, Shahaf G, Ozeri E, et al. (2011) Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab. Brain Dis. 26:107–13.
Lacki JK, et al. (1995) IgA-alpha-1-antitrypsin complex in systemic lupus erythematosus: preliminary report. Lupus. 4:221–1.
Elzouki AN, et al. (1999) The prevalence and clinical significance of alpha 1-antitrypsin deficiency (PiZ) and ANCA specificities (proteinase 3, BPI) in patients with ulcerative colitis. Inflamm. Bowel Dis. 5:246–52.
Koulmanda M, Strom TB. (2010) T-cell-directed treatment strategies for type 1 diabetes and the confounding role of inflammation. Immunotherapy. 2:431–6.
Pileggi A, et al. (2008) Alpha-1 antitrypsin treatment of spontaneously diabetic nonobese diabetic mice receiving islet allografts prolonged islet allograft survival by alpha-1 antitrypsin: the role of humoral immunity. Transplant Proc. 40:457–8.
Ma H, et al. (2010) Intradermal alpha1-antitrypsin therapy avoids fatal anaphylaxis, prevents type 1 diabetes and reverses hyperglycaemia in the NOD mouse model of the disease. Diabetologia. 53:2198–204.
Koulmanda M, et al. (2008) Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc. Natl. Acad. Sci. U. S. A. 105:16242–7.
Weir GC, Koulamnda M. (2009) Control of inflammation with alpha1-antitrypsin: a potential treatment for islet transplantation and new-onset type 1 diabetes. Curr. Diab. Rep. 9:100–2.
Lolin YI, Ward AM. (1995) Alpha-1-antitrypsin phenotypes and associated disease patterns in neurological patients. Acta. Neurol. Scand. 91:394–8.
Song S, et al. (2004) Recombinant adeno-associated virus-mediated alpha-1 antitrypsin gene therapy prevents type I diabetes in NOD mice. Gene Ther. 11:181–6.
Kalis M, et al. (2010) Alpha 1-antitrypsin enhances insulin secretion and prevents cytokinemediated apoptosis in pancreatic beta-cells. Islets. 2:185–9.
Loganathan G, et al. (2010) Culture of impure human islet fractions in the presence of alpha-1 antitrypsin prevents insulin cleavage and improves islet recovery. Transplant. Proc. 42:2055–7.
Lewis EC, et al. (2008) Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc. Natl. Acad. Sci. U. S. A. 105:16236–41.
Hernandez-Espinosa D, et al. (2009) Hyperglycaemia impairs antithrombin secretion: possible contribution to the thrombotic risk of diabetes. Thromb. Res. 124:483–89.
Lisowska-Myjak B, et al. (2006) Serum protease inhibitor concentrations and total antitrypsin activity in diabetic and non-diabetic children during adolescence. Acta Diabetol. 43:88–92.
Yaghmaei M, et al. (2009) Serum trypsin inhibitory capacity in normal pregnancy and gestational diabetes mellitus. Diabetes Res. Clin. Pract. 84:201–4.
Hashemi M, et al. (2007) Impaired activity of serum alpha-1-antitrypsin in diabetes mellitus. Diabetes Res. Clin. Pract. 75:246–8.
Blanco I, et al. (2008) Long-term augmentation therapy with alpha-1 antitrypsin in an MZ-AAT severe persistent asthma. Monaldi Arch. Chest Dis. 69:178–82.
Petrache I, Hajjar J, Campos M. (2009) Safety and efficacy of alpha-1-antitrypsin augmentation therapy in the treatment of patients with alpha-1-antitrypsin deficiency. Biologics. 3:193–204.
Hubbard RC, Crystal RG. (1988) Alpha-1-antitrypsin augmentation therapy for alpha-1-antitrypsin deficiency. Am. J. Med. 84:52–62.
Mordwinkin NM, Louie SG. (2007) Aralast: an alpha 1-protease inhibitor for the treatment of alpha-antitrypsin deficiency. Expert Opin. Pharmacother. 8:2609–614.
Louie SG, Sclar DA, Gill MA. (2005) Aralast: a new alpha1-protease inhibitor for treatment of alpha-antitrypsin deficiency. Ann. Pharmacother. 39:1861–9.
Stoll SM, et al. (2001) Epstein-Barr virus/human vector provides high-level, long-term expression of alpha1-antitrypsin in mice. Mol. Ther. 4:122–9.
Dhami R, et al. (1999) Pulmonary epithelial expression of human alpha1-antitrypsin in transgenic mice results in delivery of alpha1-antitrypsin protein to the interstitium. J. Mol. Med. 77:377–85.
Kuiperij HB, et al. (2009) Serpina1 (alpha1-AT) is synthesized in the osteoblastic stem cell niche. Exp. Hematol. 37:641–7.
Seta N, et al. (1996) Diagnostic value of Western blotting in carbohydrate-deficient glycoprotein syndrome. Clin. Chim. Acta. 254:131–40.
Lomas DA, Elliott PR, Carrell RW. (1997) Commercial plasma alpha1-antitrypsin (Prolastin) contains a conformationally inactive, latent component. Eur. Respir. J. 10:672–5.
Walters S, et al. (2009) Increased CD4+Foxp3+ T cells in BAFF-transgenic mice suppress T cell effector responses. J. Immunol. 182:793–801.
Lee I, et al. (2003) Blocking the monocyte chemoattractant protein-1/CCR2 chemokine pathway induces permanent survival of islet allografts through a programmed death-1 ligand-1-dependent mechanism. J. Immunol. 171:6929–35.
Koulmanda M, et al. (2008) Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc. Natl. Acad. Sci. U. S. A. 105:16242–7.
Molano RD, et al. (2008) Prolonged islet allograft survival by alpha-1 antitrypsin: the role of humoral immunity. Transplant. Proc. 40:455–6.
Zhang B, et al. (2007) Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes. 56:1316–23.
Lu Y, et al. (2006) Alpha1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum. Gene Ther. 17:625–34.
Lewis EC, et al. (2008) Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc. Natl. Acad. Sci. U. S. A. 105:16236–41.
Lewis EC, et al. (2005) Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc. Natl. Acad. Sci. U. S. A. 102:12153–8.
Kalsheker N, Morley S, Morgan K. (2002) Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem. Soc. Trans. 30:93–8.
Cruz PE, Mueller C, Flotte TR. (2007) The promise of gene therapy for the treatment of alpha-1 antitrypsin deficiency. Pharmacogenomics. 8:1191–8.
Zhang G, Song YK, Liu D. (2000) Long-term expression of human alpha1-antitrypsin gene in mouse liver achieved by intravenous administration of plasmid DNA using a hydrodynamicsbased procedure. Gene Ther. 7:1344–9.
Fabre JW, et al. (2008) Hydrodynamic gene delivery to the pig liver via an isolated segment of the inferior vena cava. Gene Ther. 15:452–62.
Yoshino H, Hashizume K, Kobayashi E. (2006) Naked plasmid DNA transfer to the porcine liver using rapid injection with large volume. Gene Ther. 13:1696–702.
Alino SF, et al. (2007) Pig liver gene therapy by noninvasive interventionist catheterism. Gene Ther. 14:334–43.
Khorsandi SE, et al. (2008) Minimally invasive and selective hydrodynamic gene therapy of liver segments in the pig and human. Cancer Gene Ther. 15:225–30.
Bertuzzi F, et al. (2004) Tissue factor and CCL2/monocyte chemoattractant protein-1 released by human islets affect islet engraftment in type 1 diabetic recipients. J. Clin. Endocrinol. Metab. 89:5724–8.
Abdi R, et al. (2004) Differential role of CCR2 in islet and heart allograft rejection: tissue specificity of chemokine/chemokine receptor function in vivo. J. Immunol. 172:767–75.
Boni-Schnetzler M, et al. (2008) Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 93:4065–74.
Jones ND, et al. (2001) Differential susceptibility of heart, skin, and islet allografts to T cell-mediated rejection. J. Immunol. 166:2824–30.
Allan SE, et al. (2008) Generation of potent and stable human CD4+ T regulatory cells by activationindependent expression of FOXP3. Mol. Ther. 16:194–202.
Joffre O, et al. (2008) Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat. Med. 14:88–92.
Ashton-Chess J, et al. (2009) Using biomarkers of tolerance and rejection to identify high- and low-risk patients following kidney transplantation. Transplantation. 87:S95–9.
Bestard O, et al. (2008) Presence of FoxP3+ regulatory T cells predicts outcome of subclinical rejection of renal allografts. J. Am. Soc. Nephrol. 19:2020–6.
Pott GB, et al. (2009) Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J. Leukoc. Biol. 85:886–95.
Panakanti R, Mahato RI. (2009) Bipartite vector encoding hVEGF and hIL-1Ra for ex vivo transduction into human islets. Mol. Pharm. 6:274–84.
Glas R, et al. (2009) Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia. 52:1579–88.
Hsu BR, et al. (2009) Interleukin-1 receptor antagonist enhances islet engraftment without impacting serum levels of nitrite or osteopontin. Transplant Proc. 41:1781–5.
Ghosn EE, Cassado AA, Govoni GR, et al. (2010) Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc. Natl. Acad. Sci. U. S. A. 107:2568–73.
Cantin AM, Woods DE. (1999) Aerosolized prolastin suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Am. J. Respir. Crit. Care Med. 160:1130–5.
Chan ED, et al. (2007) Alpha-1-antitrypsin (AAT) anomalies are associated with lung disease due to rapidly growing mycobacteria and AAT inhibits Mycobacterium abscessus infection of macrophages. Scand. J. Infect. Dis. 39:690–6.
Dhami R, et al. (2000) Acute cigarette smokeinduced connective tissue breakdown is mediated by neutrophils and prevented by alpha1-antitrypsin. Am. J. Respir. Cell Mol. Biol. 22:244–52.
Libert C, et al. (1996) Alpha1-antitrypsin inhibits the lethal response to TNF in mice. J. Immunol. 157:5126–9.
Stocks JM, et al. (2006) Multi-center study: the biochemical efficacy, safety and tolerability of a new alpha1-proteinase inhibitor, Zemaira. COPD. 3:17–23.
Wencker M, et al. (2001) Longitudinal follow-up of patients with alpha(1)-protease inhibitor deficiency before and during therapy with IV alpha(1)-protease inhibitor. Chest. 119:737–44.
Lieberman J. (2000) Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 118:1480–5.
von Herrath M, Nepom GT, von Herrath MG. (2009) Remodeling rodent models to mimic human type 1 diabetes. Eur. J. Immunol. 39:2049–54.
Acknowledgments
The authors wish to thank Valeria Frishman for her excellent technical assistance. The study was funded by the Juvenile Diabetes Research Foundation (2-2007-103) and Israel Science Foundation (1027/07).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
10020_2011_1791000_MOESM1_ESM.pdf
α-1-Antitrypsin Gene Delivery Reduces Inflammation, Increases T-Regulatory Cell Population Size and Prevents Islet Allograft Rejection
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shahaf, G., Moser, H., Ozeri, E. et al. α-1-Antitrypsin Gene Delivery Reduces Inflammation, Increases T-Regulatory Cell Population Size and Prevents Islet Allograft Rejection. Mol Med 17, 1000–1011 (2011). https://doi.org/10.2119/molmed.2011.00145
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00145