
Abstract
Topoisomerase I is a privileged target for widely used anticancer agents such as irinotecan. Although these drugs are classically considered to be DNA-damaging agents, increasing evidence suggests that they might also influence the tumor environment. This study evaluates in vivo cellular and molecular modifications induced by irinotecan, a topoisomerase I-directed agent, in patient-derived colon tumors subcutaneously implanted in athymic nude mice. Irinotecan was given intraperitoneally at 40 mg/kg five times every 5 d, and expression profiles were evaluated at d 25 in tumors from treated and untreated animals. Unexpectedly, the in vivo antitumor activity of irinotecan was closely linked to a downregulation of hypoxia-inducible factor-1α (HIF1A) target genes along with an inhibition of HIF1A protein accumulation. The consequence was a decrease in tumor angiogenesis leading to tumor size stabilization. These results highlight the molecular basis for the antitumor activity of a widely used anticancer agent, and the method used opens the way for mechanistic studies of the in vivo activity of other anticancer therapies.
Similar content being viewed by others


Introduction
Understanding the molecular mechanisms of anticancer agents is crucial for personalized treatment and for developing more rational drug combinations. However, most mechanistic studies of anticancer agents have been carried out in vitro with isolated tumor cells under conditions that may neither be representative of the tumor environment nor of the actual drug exposure. For example, homogeneous cancer cell lines ignore the clonal heterogeneity of tumors. The interaction of tumor cells with the microenvironment is also absent in cell culture, which precludes a complete analysis of the impact of chemotherapeutic agents such as the influence on tumor angiogenesis. Hypoxia is rarely considered in vitro, nor are the drug-exposure conditions, which usually are much more dynamic in vivo than in vitro.
Considering these caveats, the current study aims to characterize the in vivo tumor response to an anticancer agent by using xenografts of patient-derived tumors. Although orthotropic xenografts models are considered mandatory to evaluate invasion and metastasis in response to therapy, they are challenging to establish, especially from patient-derived tumors. Subcutaneous xenografts of human tumors offer a valuable model to study the mechanisms underlying the effect of experimental therapies in an in vivo context without the bias of using cell lines selected by in vitro culture. In a previous study (1), we showed that xenografting of human tumors alters neither the genetic nor the histological profiles of the original tumors, even after multiple passages, suggesting that clonal selection does not occur in nude mice. Interestingly, as normal cells of human origin do not survive in the xenograft, the tumor compartment is exclusively of human origin, whereas the stromal compartment is principally composed of murine cells, thereby providing a mean to discriminate between the two cell populations.
As a model, we have chosen to study the influence of irinotecan in patient-derived colon cancers. Irinotecan is a topoisomerase I inhibitor widely used for the treatment of colorectal cancer in both first- and second-line treatments (2–6). Clinically used topoisomerase I inhibitors target a covalent DNA-topoisomerase I reaction intermediary (called cleavable complexes), thereby preventing the reannealing of the nicked DNA (7). The cleavable complexes are believed to selectively induce S-phase cytotoxicity when they collide with advancing replication forks (8). If not repaired, the DNA lesions can induce cell-cycle arrest and/or apoptosis, depending on the cellular levels of cleavable complexes and the genetic context of the cells (9,10). Although this process has been clearly described, the subsequent mechanisms underlying the in vivo antitumor effects of irinotecan are less clear.
We define here the molecular signature elicited by irinotecan in human cancer cells as well as in the corresponding murine stromal cells. To the best of our knowledge, this is the first cancer study to apply transcriptome-based masks (11) to distinguish between transcriptomes of human and murine origin. By testing human colon cancer xenografts on human and mouse oligonucleotide microarrays, we were able to identify an in vivo mode of action for irinotecan involving the inhibition of hypoxia-inducible factor-1α (HIF1A) protein expression with a subsequent reduction in the expression levels of HIF1A target genes and inhibition of tumor angiogenesis. These results illustrate how particular cytotoxic antitumor drugs such as topoisomerase I inhibitors can affect in vivo tumor angiogenesis and how the method used can open the way for mechanistic studies of the in vivo activity of anticancer agents.
Materials and Methods
Human Tumor Xenografts and Treatment with Irinotecan
Male athymic nude mice (nu/nu), 6–8 wks old (Charles River, L’Arbresle, France), were maintained under specific pathogen-free conditions. Human tumor tissue fragments were obtained after gaining informed consent from patients undergoing colectomy and/or synchronous liver metastasis resection. Cancer tissues were minced on ice and injected subcutaneously into the right and left flanks of nude mice as previously described (1). Xenografts were further propagated by serial transplantation. For treatment, mice were observed daily for tumor appearance, and irinotecan treatment was initiated when the tumors reached a mean volume of 150–300 mm3. Irinotecan (Campto®, irinotecan chlorhydrate) was given intraperitoneally at 40 mg/kg five times every 5 d (q5dx5). Each group consisted of four to five tumor-bearing mice, corresponding to eight to ten individual tumors. Tumor volume (V) was evaluated once weekly and was calculated as V = (a2 × b)/2, where a is the width of the tumor in millimeters and b is the length. The individual relative tumor volume (RTV) was defined as Vx/V0, where Vx is the volume in mm3 at a given time and V0 is the volume at the start of treatment. Mean RTV and standard error of the mean (SEM) were calculated for each group. Drug efficacy at the end of treatment (d 25) was expressed as the percentage of tumor growth inhibition, which was calculated using the formula 100 — (mean RTVtreated/mean RTVcontrol) × 100. For the analyses, control samples (C) correspond to xenografts of untreated animals, treated samples (T) correspond to xenografts from mice treated with 40 mg/kg irinotecan q5dx5 and collected at the end of treatment period, and regrowth samples (R) correspond to xenografts that have started to grow again after the end of the treatment period.
Tumor Clonogenic Assay Using Human Tumor Xenografts
Freshly xenografted tumors were placed in Liforlab® solution (Oncoscience AG, Wedel, Germany) until preparation of single-cell suspensions for the clonogenic assay. Tumor clonogenic assay was performed as previously described (12). Half-maximal inhibitory concentration (IC50) values were determined using concentrations of SN-38 (Abatra, Xi’an, China), the active metabolite of irinotecan, ranging from 0.001 to 20 µg/mL.
RNA Isolation from Human Tumor Xenografts and Gene Expression Profiling
Xenografted tumors were snap frozen in liquid nitrogen immediately after resection and stored at −80°C. Total RNA was prepared using Trizol (Invitrogen, Cergy Pontoise, France) according to the manufacturer’s instructions. RNA was further purified using spin columns from the RNeasy Mini Kit (Qiagen, Courtaboeuf, France). RNA integrity was analyzed on the Agilent 2100 bioanalyzer. Samples were transcribed in vitro, biotinylated and hybridized to both Affymetrix Human Genome U133 Plus 2.0 GeneChip® arrays and Mouse Genome 430 2.0 GeneChip arrays (Affymetrix, Santa Clara, CA, USA).
Transcriptome Data Analysis
An initial quality control of the raw data from the Affymetrix microarray was performed using RReportGenerator (13), and all arrays fulfilled our quality criteria. Raw data from the Affymetrix expression results were analyzed using a method described by Naef and Huelsken (11) to distinguish mouse and human specific probes within each probe set. Briefly, for transcriptional data analysis of the human tumor component, the probe masks defined by Naef and Huelsken (11), which allow for the removal of individual probes from the U133 Plus 2.0 GeneChip array that may cross-hybridize with mouse mRNA, were applied to the dataset. Settings that allowed a maximum of three mismatches were selected, in agreement with Naef and Huelsken (11), and 16.9% of the individual probes were masked. This choice represents a good compromise between specificity and sensitivity (only 6.3% of the probe sets were discarded because of the lack of sufficient remaining probes), since more stringent conditions, with fewer mismatches tolerated, would significantly reduce the final number of exploitable probe sets.
The newly defined probe sets were then summarized with Robust Multiarray Average (RMA) (14,15), using implementation in Bioconductor (16) under R (www.r-project.org). The summarized and normalized data were then filtered to remove all probe sets that were not expressed in all samples tested (that is, probe sets with log2 expression values of <4.9 for all arrays). Statistical testing was performed based on a two-factor model that allows for separation of (a) tumor specimen origin (that is, patient) and (b) sample treatment type (that is, control, irinotecan-treated or regrowth sample type). The corresponding analysis of variance P values for both factors and for each probe set were then transformed into local fdr (false discovery rate) values (17,18) and were compared against each other. A total of 130 human probe sets, which fulfilled the criteria of fdr-treatment type <0.02 and fdr-treatment type <10,000 × fdr-tumor-specimen origin, were retained for further analysis. The retained probe sets were analyzed for enrichment of Gene Ontology (GO) ontologies using DAVID (Database for Annotation, Visualization, and Integrated Discovery) (19) and for enrichment of pathway interaction maps using the Ingenuity Pathway Analysis tool (www.inge-nuity.com, data not shown). In addition, hierarchical clustering was used to further organize the resulting list of 130 probe sets. Approximate unbiased bootstrap P values were obtained using the R package “pvclust” (20).
To further select human genes to be validated by real-time quantitative reverse transcriptase-polymerase chain reaction (RT-PCR), values for the mean fold-change were calculated using untreated control xenografts as a reference. A 2.0-fold difference was used as a cutoff value. The significance of the changes in gene expression levels was assessed by two-sided Wilcoxon signed-rank tests.
Real-Time Relative Quantitative RT-PCR
mRNA expression of selected human genes was evaluated by real-time relative quantitative RT-PCR using the LightCycler® 480 System (Roche Applied Science, Meylan, France) and SYBR Green I technology. Reverse transcription of 2 µg RNA was performed using AMV reverse transcriptase (Finnzymes, Ozyme, Saint Quentin, France) and oligo(dT) primers. Validated QuantiTect® Primer Assays (Qiagen, Courtaboeuf, France) were used for amplification as previously described (21). All quantifications were performed in duplicate and normalized to two independent endogenous reference genes, hydroxymethylbilane synthase (HMBS) and keratin 19 (KRT19). Target cDNA expression was quantified using the comparative Ct method and expressed as the fold-change versus control untreated samples. Two-sided Wilcoxon signed rank tests were used to assess the significance of the fold-change values in irinotecan-treated samples.
Histology and Immunohistochemistry
Resected xenografts were fixed in formalin, embedded in paraffin and processed for histology by hematoxylin and eosin (H&E) and Masson trichrome stains. For Masson trichrome, sections were sequentially stained with Weigert iron hematoxylin and Biebrich Scarlet acid fuchsin solutions. After treatment with phosphotungstic and phosphomolybdic acids, collagen was stained in blue with aniline blue. Ki-67 immunohis-tochemical staining (rabbit anti-human Ki-67 monoclonal antibody, clone SP6, dilution 1:200; Microm Microtech, Francheville, France) was performed using the Ventana automated system (Benchmark®, Ventana Medical Systems, Illkirch, France). For anti-CD31 (anti-platelet endothelial cell adhesion molecule [PECAM-1]) immunohistochemistry, frozen sections were incubated with fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD31 monoclonal antibody (BD Biosciences, Le Pont-De-Claix, France) and then with horseradish peroxidase-conjugated antifluorescein Fab fragments (Roche Molecular Biochemicals, Meylan, France) as previously described (22). Immunostaining was developed with the liquid DAB substrate kit (Roche Molecular Biochemicals, Meylan, France), and the tissue sections were counterstained with hematoxylin. Ki-67-positive cells were quantified in clusters of tumor cells by calculating the ratio of Ki-67-positive cells over total cells in four distinct areas of 0.2 mm2 each, at 400× magnification.
Detection of Tissue Hypoxia by Immunostaining for Pimonidazole Adducts
Pimonidazole (Hypoxyprobe-1™ Plus Kit; Chemicon Upstate, Millipore, Molsheim, France) was administered intraperitoneally at a dose of 100 mg/kg in phosphate-buffered saline for 60 min before euthanizing the mice. Resected xenografts were fixed in formalin, embedded in paraffin and then processed for immunochemistry using the FITC-conjugated hypoxyprobe-1 monoclonal antibody and the horseradish peroxidase-conjugated anti-FITC secondary monoclonal antibody from the Hypoxyprobe-1 Plus Kit following the manufacturer’s instructions.
Immunoblotting
Tumor tissues were crushed on ice in lysis buffer (10 mmol/L Tris, 5 mmol/L MgCl2, 10 mmol/L NaCl, 0.5% NP-40, protease and phosphatase inhibitors) and briefly sonicated. Aliquots (20 µg) of total protein were resolved on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, transferred onto a nitrocellulose membrane and incubated with primary antibodies: mouse anti-human-HIF-1a monoclonal antibody (BD Biosciences, Le Pont-De-Claix, France) that does not cross-react with mouse HIF-1α protein and rabbit polyclonal anti-α/ β-tubulin (Cell Signaling, Ozyme, Saint Quentin, France). Protein expression was detected using horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibodies with enhanced chemiluminescence reagents (Amersham, GE Healthcare, Saclay, France).
Results
Comparison of the In Vitro and In Vivo Efficacy of Irinotecan
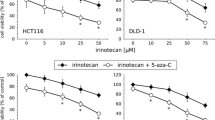
Patient-derived colon tumors and their corresponding liver metastases (Table 1) were used for xenografting to identify a molecular signature associated with the response to irinotecan. First, the in vitro sensitivity to SN-38 (the active metabolite of irinotecan) was determined by clonogenic assays with cells directly dissociated from the tumor xenografts. The tumors showed a wide range of sensitivities with three resistant tumors (xenografts 17, 22 and 36M1) having IC50 values (dose inhibiting 50% tumor cell growth compared with untreated controls) between 17.0 and 25.2 µg/mL and four sensitive tumors with IC50 values between 0.001 and 0.06 µg/mL (Table 2). Next, the in vivo response of the same xenografts was evaluated after irinotecan administration (40 mg/kg intraperitoneally q5dx5). At this dose, irinotecan inhibited tumor growth or induced tumor shrinkage in all xenografts (Figure 1 for two representative examples), including the three xenografts showing in vitro resistance, suggesting that irinotecan may exert additional effects in vivo that are not detectable under in vitro conditions. This observation prompted us to study the in vivo impact of irinotecan at the transcriptome level, with the aim of deciphering novel cellular and molecular mechanisms that could be responsible for the in vivo antitumor effects of irinotecan.

Growth inhibition of human colon tumor xenografts by irinotecan. Subcutaneous xenograft-bearing mice were treated with irinotecan intraperitoneally at 40 mg/kg q5dx5 on the days indicated by arrows. Tumor growth was evaluated by plotting the mean relative tumor volume ± SEM (n = 8–l10) over time. Two representative examples of xenografts that exhibited different levels of in vitro sensitivity to SN-38 (the active metabolite of irinotecan) are shown (see Table 2). The control (C), treated (T) and regrowth (R) samples used for transcriptome analysis are indicated in the corresponding curves. Four to five mice, each bearing two individual tumor xenografts, were included in each group. Results were confirmed in two independent experiments.
Transcriptional Profile of the Human Tumor Component in Irinotecan-Treated Xenografts
For each of the seven xenografts, three samples were selected for transcriptome analysis using Affymetrix Human Genome U133 Plus 2.0 GeneChip arrays. As illustrated in Figure 1, the three samples correspond to control samples (C) from xenografts of untreated animals, treated samples (T) corresponding to xenografts from mice treated with 40 mg/kg q5dx5 irinotecan and regrowth samples (R) corresponding to xenografts that have started to grow again after the end of the treatment period and that were collected when the relative tumor volumes were comparable with the tumor volumes of the C samples. The R control was included to allow us to identify potential drug-modulated genes that were not associated with the tumor-growth inhibitory activity.
Xenografted human tumors contain an important stromal component of murine cells. To specifically identify the molecular signature elicited by irinotecan treatment in the human cancer cells, raw data from Affymetrix expression results were analyzed using a species-selective approach that removes measurements from oligonucleotides that may cross-hybridize with both human and mouse transcripts (11). After filtering probe sets with marginal expression levels, the remaining 19,818 human probe sets were further analyzed using unsupervised hierarchical clustering on the basis of Euclidean distance and complete linkage. Cluster analysis revealed that C, T and R samples from a given xenograft are more similar to each other than the different C, T or R samples are to each other (Figure 2A). This shows that the tumor-specific component is a major factor directing the outcome of the cluster analysis. Accordingly, all xenografts derived from the same patient clustered together in a separate arm. A second finding was that T samples were always separated from the corresponding C and R samples, indicating that irinotecan treatment elicits a specific signature that is readily discernible in spite of the tumor heterogeneity.

Human gene expression profiles of irinotecan-treated colon tumor xenografts. The results of unsupervised hierarchical clustering of 19,818 human probe sets showing significant expression in xenograft samples are summarized in a dendogram in panel A. Numbers in red above the clusters in the tree indicate the percentage of times these clusters were observed in 3,999 iterations during bootstrap analysis. Panel B shows the results of unsupervised hierarchical clustering of 130 human probe sets, for which variation of expression was more specifically influenced by irinotecan than intertumor variability. Numbers in red above the clusters in the tree indicate the percentage of times these clusters were observed in 3,999 iterations during bootstrap analysis. Red indicates high relative expression; green indicates low relative expression.
To evaluate the transcriptional effects that were specific to irinotecan, a two-factor model was applied to separate the impact of irinotecan treatment from the dominant tumor-specific influence on gene expression. Evaluation of local false discovery rate values defined a highly stringent signature of 130 human probe sets (list available upon request) with a profile that was primarily influenced by irinotecan treatment. Subsequent unsupervised hierarchical clustering of the 130 probe sets (Euclidean distance and complete linkage) showed that the profiles of the irinotecan-treated xenografts can be clearly separated from their corresponding C and R counterparts (Figure 2B). The transcriptome profiles of the T samples revealed most downregulated genes (116 downregulated probe sets out of 130). Functional analysis of the identified genes on the basis of gene ontology classification highlighted genes involved in glucose metabolic processes, such as glucose transporters or glycolytic enzymes (data not shown). In contrast, the analysis did not reveal any modulation of genes classically associated with cell cycle arrest, apoptosis or senescence.
Validation of Microarray Data by Quantitative RT-PCR
To focus on the most prominent transcriptional changes induced by the in vivo irinotecan exposure, 37 probe sets, with a mean fold-change of at least 2.0 between T and C samples (P < 0.05), were selected, with 34 downregulated and 3 upregulated probe sets that corresponded to 27 downregulated and 3 up-regulated genes (Table 3). Five of the genes were poorly documented in the literature and were therefore excluded from further analysis. Real-time quantitative RT-PCR (qRT-PCR) was used to validate the remaining 24 downregulated and 1 upregulated genes.
The expression levels of these 25 genes were measured by qRT-PCR in the same xenograft samples as those that were originally used for the microarray experiments and were normalized to two different housekeeping genes (Table 3). A total of 24 out of the 25 genes that had been shown to be down- or upregulated by irinotecan in the microarray experiments were successfully confirmed by qRT-PCR with a similar fold-change (P < 0.05). In agreement with the array data, no significant changes were observed between the C and R samples (Table 3). Only the downregulation of PTPN12 gene expression could not be confirmed by qRT-PCR. For further control, a set of five genes, for which the expression levels did not exceed 2.0-fold in the microarray experiment, were included as negative controls. Their expression levels, as measured by qRT-PCR, remained below the 2.0-fold cutoff, with a P value that did not reach the significance level (Table 3).
The In Vivo Signature of Irinotecan Treatment Is Characterized by the Downregulation of HIF1A Target Gene Expression
A total of 18 of the 23 genes that were significantly downregulated by irinotecan represented HIF1A- or hypoxiainduced target genes (23–25). These genes include VEGFA, SLC2A1, SLC2A3, SLC6A8, PFKFB3, PFKP, ALDOC, PGK1, ENO2, ADM, ANKRD37, BHLHB2, BNIP3L, DDIT4, ERO1L, NDRG1, P4HA1 and PLOD2 (Table 3). The relevance of this signature was evaluated in an independent xenograft (xenograft 40) that was quite insensitive to irinotecan, with only 19% tumor growth inhibition on d 25 of treatment. This tumor model is derived from a right-sided human stage IV colon tumor (pT4N1M1 with liver metastasis) that was surgically removed from a patient first treated with FOLFOX and then with the FOLFIRI regimen and who subsequently developed new liver metastatic lesions during FOLFIRI treatment. Irinotecan had only a marginal effect on the tumor xenografted in mice, which is consistent with the absence of clinical response observed in the patient. When comparing T and C samples for this insensitive tumor, no obvious down-regulation of the 23 genes associated with tumor growth inhibition in irinotecan-sensitive tumors had occurred (data not shown). These results strongly suggest that downregulation of HIF1A target genes are specifically associated with the in vivo response of irinotecan-sensitive human tumor xenografts.
Irinotecan Inhibits HIF1A Protein Accumulation and Angiogenesis in Human Colon Tumor Xenografts
To characterize the molecular mechanisms underlying the irinotecan-induced downregulation of HIF1A target genes, the expression of HIF1A mRNA was evaluated. Microarray and qRT-PCR analyses revealed that RNA transcript levels for HIF1A did not change in response to irinotecan (Table 3). In contrast, a complete inhibition of human HIF1A protein accumulation was observed, which was partially reversed in R samples after treatment arrest (Figure 3A). Interestingly, the inhibition of HIF1A protein accumulation occurred independently of significant changes in intratumoral hypoxia (Figure 3B). In addition, tumor sections from T samples showed a decrease in the number of tumor cells, as evaluated by H&E staining, and an increase in extracellular matrix deposition, as evaluated by trichrome staining, in comparison with C samples (Figure 3C). These alterations were completely reversed in R samples. Within clusters of tumor cells, the number of proliferating cells, as detected by Ki-67 staining, is identical in C (93% of positive cells), T (90% of positive cells) and R (94% of positive cells) tumors (Figures 3C, D). After irinotecan treatment, all tumors showed a dramatic disorganization of their vascular network and exhibited only isolated CD31-positive endothelial cells (Figure 3C). Altogether, these observations indicate that irinotecan inhibits HIF1A protein accumulation and angiogenesis in human colon cancer xenografts in vivo.

Irinotecan inhibits HIF1A protein accumulation and angiogenesis in colon tumor xenografts. Western blot analysis of HIF1A protein accumulation in human colon tumor xenografts is shown in panel A. Tumors from mice treated with irinotecan (T) show a complete inhibition of HIF1A protein accumulation compared with control tumors from untreated mice (C). The inhibition of HIF1A protein accumulation is reversed in xenografts that have started to grow again (R) after the end of the treatment period. Results were confirmed in two independent experiments. Panel B shows the detection of intratumor hypoxia in human colon tumor xenografts of mice treated with irinotecan. Mice were injected intraperitoneally with 100 mg/kg pimonidazole 1 h before euthanasia. Pimonidazole adducts in hypoxic areas of the tumors were identified using hypoxyprobe monoclonal antibodies on paraffin-embedded sections. Bars represent 250 µm. Results were confirmed in two independent experiments. Panel C shows histological examinations of human colon tumor xenografts in mice treated with irinotecan. H&E and trichrome staining show a decrease in tumor cellularity and an increase in extracellular matrix deposition, respectively, in tumors from irinotecan-treated mice (bars represent 2.5 mm). Cellular proliferation was evaluated with Ki-67 labeling within clusters of tumor cells (bars represent 500 µm). CD31 staining shows well-formed vessels (arrows) in the control tumors of untreated mice, whereas the vascular network is completely disorganized in the tumors of irinotecan-treated mice. This effect is reversed in tumors that have started to grow again after the end of the treatment period (bars represent 1 mm). Results were confirmed in two independent experiments. Panel D shows the quantification of the impact of irinotecan treatment on cell proliferation as assessed by Ki-67 staining. The mean values for the average percentage of Ki-67-positive cells were calculated along with SEM. No significant difference was observed in tumor cells from control (C), irinotecan-treated (T) and regrowth (R) samples.
Discussion
We used human tumor xenografts to identify a molecular signature for tumor growth inhibition by irinotecan, a topoisomerase I inhibitor, which principally relies on the inhibition of HIF1A protein accumulation accompanied by transcriptional downregulation of HIF1A target genes.
HIF1A is a key transcriptional activator of genes needed for crucial aspects of cancer biology, including angiogenesis, glycolytic metabolism, cell survival and invasion (24–28). In nondiseased tissue, HIF1A is generally expressed at very low levels. However, in cancer tissue, and colon cancers in particular, HIF1A is frequently upregulated (29–31; GeneAtlas and NCI-60 transcription profiles, http://biogps.gnf.org), in agreement with the results for our untreated control xenografts. Several of the HIF1A target genes, which became downregulated after irinotecan treatment, are crucial for overcoming the hypoxic status of tumors and may thereby interfere with the capacity of tumor cells to adapt to a hypoxic environment. In particular, irinotecan reduced the expression of several genes encoding glucose transporters (SLC2A1, SLC2A3) as well as glycolytic enzymes (PFKFB3, PFKP, ALDOC, PGK1, ENO2). Reduced expression of these genes may result in decreased ATP production by the glycolytic pathway, which is needed for tumor growth during hypoxia (32,33). This in vivo signature is in agreement with our previous observations showing that downregulation of HIF1A by siRNA in colon cancer cells induces antiproliferative effects in hypoxia conditions, validating HIF1A as a functionally important target (21).
A notable feature of irinotecan-treated xenografts was the inhibition of tumor angiogenesis that correlated well with the observed tumor size stabilization, indicating that in vivo irinotecan had a cytostatic rather than cytotoxic effect. This activity may result from the downregulation of genes encoding VEGFA and ADM, both of which are potent angiogenic factors, and of ERO1L, an endoplasmic reticulum oxidoreductase that activates VEGF secretion under hypoxic conditions (34,35). Irinotecan treatment was also accompanied by intense reactive fibrosis within the tumor microenvironment. Mouse tissue-specific analysis of the same initial RNA samples revealed a broad profile of differential gene expression, although the induction of some mouse extracellular matrix components such as lumican or laminin α2 and the downregulation of angiogenesis-related genes such as endothelial cell-specific molecule 1 or placental growth factor were still evident (data not shown). The expression of the mouse hif1a gene was not modulated by irinotecan. Altered expression of genes linked to protein kinases was also observed, suggesting that their involvement in the regulation of the stromal compartment during irinotecan treatment might implicate protein networks.
Whereas irinotecan downregulated a large number of genes in the human tumor cells in vivo, only a few genes were upregulated. The only upregulated human gene that could be robustly confirmed by qRT-PCR was poly (ADP-ribose) polymerase family, member 12 (PARP12), a member of the PARP super-family belonging to the subgroup of CCCH-type zinc-finger PARPs (36). Like other recently discovered PARP proteins, the structure of PARP12 is characterized by a PARP catalytic domain that lacks the critical glutamic acid residue important for poly-ADP-ribosylation. This result suggests that this enzyme, of yet unknown function, may act as a mono-ADP-ribosyltransferase, although the physiological role of this modification remains to be elucidated (37).
Importantly, our in vivo study did not identify genes associated with cell cycle regulation or apoptosis, in clear contrast to previous cDNA microarray studies that were performed on colon or other cancer cell lines in vitro under normoxic conditions (38–43). The discrepancies in expression profiles underline the important influence of the microenvironment and/or pharmacological parameters on the in vivo tumor response. This result supports the view that definitively in vivo mechanisms have to be analyzed to address the whole complexity of different pathways involved in response to chemotherapeutic agents.
In agreement with previous data suggesting that irinotecan and other topoisomerase inhibitors may inhibit angiogenesis (44–51), our findings obtained in patient-derived colon tumors illustrate the mechanistic basis for this activity, which is linked to decreased accumulation of HIF1A protein. These results are consistent with the downregulation of the HIF1A protein induced by the polyethylene glycol (PEG) conjugate of SN-38 in neuroblastoma and glioblastoma xenograft tumors (50,51). Also interestingly, a systematic in vitro screen of 2,000 chemically diverse compounds for specific inhibitors of the transcriptional activity of HIF1A under hypoxic conditions in a glioblastoma cell line tumor model identified four compounds, among which three were topoisomerase I inhibitors (52,53). Therefore, the link between HIF1A and topoisomerase I inhibitors has now been identified both in vitro and in vivo and by several different approaches. The molecular mechanisms that link TOP1 inhibition to the attenuation of HIF1A protein levels remain to be identified, but are not on the basis of transcriptional downregulation of the HIF1A gene. Similar observations dissociating the inhibition of HIF1A protein accumulation from mRNA modulation have been recently published for PEGy-lated SN-38 and irinotecan in glioblastoma xenografts (51). These results are also consistent with those reported for topotecan, another topoisomerase I inhibitor that did not affect HIF1A mRNA accumulation or protein half-life, but rather acted on the translational level (54). In addition, we have recently shown that inhibition of HIF1A protein accumulation by irinotecan does not rely on inhibition of the mammalian target of rapamycin (mTOR) pathway (21).
Conclusion
In conclusion, this study reveals the activity of a cytotoxic antitumor drug on tumor angiogenesis and highlights its mechanistic basis. Use of transcriptome-based masks for separate analysis of the tumor and the stromal component of xenograft models opens the way for innovative mechanistic studies of the in vivo activity of anticancer agents. Such studies are crucial for the development of more rational drug combinations, as exemplified by our recent work demonstrating a cooperative inhibition of the mTOR/HIF1A axis by the combined use of irinotecan and rapamycine, a novel association that was designed on the basis of the results of the present study (21).
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Guenot D, et al. (2006) Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol. 208:643–52.
Saltz LB, et al. (2000) Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer: Irinotecan study group. N. Engl. J. Med. 343:905–14.
Douillard JY, et al. (2000) Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 355:1041–7.
Cunningham D, et al. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 351:337–45.
Hurwitz H, et al. (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350:2335–42.
Van Cutsem E, et al. (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360:1408–17.
Champoux JJ. (2001) DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369–413.
Liu LF, et al. (2000) Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci. 922:1–10.
Bhonde MR, et al. (2006) Equivalent effect of DNA damage-induced apoptotic cell death or long-term cell cycle arrest on colon carcinoma cell proliferation and tumour growth. Oncogene. 25:165–75.
Pommier Y. (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 6:789–802.
Naef F, Huelsken J. (2005) Cell-type-specific transcriptomics in chimeric models using transcriptome-based masks. Nucleic Acids Res. 33:e111.
Fiebig HH, Maier A, Burger AM. (2004) Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur. J. Cancer. 40:802–20.
Raffelsberger W, et al. (2008) RReportGenerator: automatic reports from routine statistical analysis using R. Bioinformatics. 24:276–8.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 19:185–93.
Irizarry RA, et al. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31:e15.
Gentleman RC, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80.
Reiner A, Yekutieli D, Benjamini Y. (2003) Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 19:368–75.
Strimmer K. (2008) fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 24:1461–2.
Dennis G, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4:P3.
Suzuki R, Shimodaira H. (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 22:1540–2.
Pencreach E, et al. (2009) Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor-1alpha axis. Clin. Cancer Res. 15:1297–307.
Prewett MC, et al. (2002) Enhanced antitumor activity of anti-epidermal growth factor receptor monoclonal antibody IMC-C225 in combination with irinotecan (CPT-11) against human colorectal tumor xenografts. Clin. Cancer Res. 8:994–1003.
Denko NC, et al. (2003) Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 22:5907–14.
Semenza GL. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 3:721–32.
Semenza GL. (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 29:625–34.
Pouysségur J, Dayan F, Mazure NM. (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 441:437–43.
Gordan JD, Simon MC. (2007) Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 17:71–7.
Rankin EB, Giaccia AJ. (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 15:678–85.
Zhong H, et al. (1999) Overexpression of hypoxia-inducible factor 1 alpha in common human cancers and their metastases. Cancer Res. 59:5830–5.
Su AI, et al. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U. S. A. 101:6062–7.
Simiantonaki N, Taxeidis M, Jayasinghe C, Kurzik-Dumke U, Kirkpatrick CJ. (2008) Hypoxia-inducible factor 1 alpha expression increases during colorectal carcinogenesis and tumor progression. BMC Cancer. 8:320.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7:11–20.
Semenza GL. (2009) Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 19:12–6.
May D, et al. (2005) Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 24:1011–20.
Nikitenko LL, Fox SB, Kehoe S, Rees MC, Bicknell R. (2006) Adrenomedullin and tumour angiogenesis. Br. J. Cancer. 94:1–7.
Schreiber V, Dantzer F, Ame JC, de Murcia G. (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 7:517–28.
Kleine H, et al. (2008) Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell. 32:57–69.
Zhou Y, et al. (2002) Transcriptional regulation of mitotic genes by camptothecin-induced DNA damage: microarray analysis of dose- and time-dependent effects. Cancer Res. 62:1688–95.
Daoud SS, et al. (2003) Impact of p53 knockout and topotecan treatment on gene expression profiles in human colon carcinoma cells: a pharmacogenomic study. Cancer Res. 63:2782–93.
Minderman H, et al. (2005) In vitro and in vivo irinotecan-induced changes in expression profiles of cell cycle and apoptosis-associated genes in acute myeloid leukemia cells. Mol. Cancer Ther. 4:885–900.
Souza V, Dong YB, Zhou HS, Zacharias W, McMasters KM. (2005) SW-620 cells treated with topoisomerase I inhibitor SN-38: gene expression profiling. J. Transl. Med. 3:44.
Bhonde MR, et al. (2006). DNA damage-induced expression of p53 suppresses mitotic checkpoint kinase hMps1: the lack of this suppression in p53MUT cells contributes to apoptosis. J. Biol. Chem. 281:8675–85.
Guo X, et al. (2006) Analysis of common gene expression patterns in four human tumor cell lines exposed to camptothecin using cDNA microarray: identification of topoisomerase-mediated DNA damage response pathways. Front. Biosci. 11:1924–31.
Nakashio A, Fujita N, Tsuruo T. (2002) Topotecan inhibits VEGF- and bFGF-induced vascular endothelial cell migration via downregulation of the PI3K-Akt signaling pathway. Int. J. Cancer. 98:36–41.
Petrangolini G, et al. (2003) Antiangiogenic effects of the novel camptothecin ST1481 (gimatecan) in human tumor xenografts. Mol. Cancer Res. 1:863–70.
Kamiyama H, Takano S, Tsuboi K, Matsumura A. (2005) Anti-angiogenic effects of SN38 (active metabolite of irinotecan): inhibition of hypoxia-inducible factor 1 alpha (HIF-1alpha)/vascular endothelial growth factor (VEGF) expression of glioma and growth of endothelial cells. J. Cancer Res. Clin. Oncol. 131:205–13.
Ji Y, et al. (2007) The camptothecin derivative CPT-11 inhibits angiogenesis in a dual-color imageable orthotopic metastatic nude mouse model of human colon cancer. Anticancer Res. 27:713–8.
Bocci G, et al. (2008) Antiangiogenic and anticolorectal cancer effects of metronomic irinotecan chemotherapy alone and in combination with semaxinib. Br. J. Cancer. 98:1619–29.
Takano S, et al. (2010) Metronomic treatment of malignant glioma xenografts with irinotecan (CPT-11) inhibits angiogenesis and tumor growth. J. Neurooncol. 99:177–85.
Pastorino F, et al. (2010) Tumor regression and curability of preclinical neuroblastoma models by PEGylated SN38 (EZN-2208), a novel topoisomerase I inhibitor. Clin. Cancer Res. 16:4809–21.
Sapra P, et al. (2011) Potent and sustained inhibition of HIF-1α and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis. 14:245–53.
Rapisarda A, et al. (2002) Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 62:4316–24.
Rapisarda A, et al. (2004) Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 64:6845–8.
Rapisarda A, et al. (2004) Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 64:1475–82.
Acknowledgments
The authors wish to acknowledge the biocomputing, bioinformatics and microarray facilities at the Institut de Génétique et de Biologie Moléculaire et Cellulaire. They also gratefully acknowledge Christiane Arnold, Mathilde Arrivé, Nathalie Hamelin and Laetitia Ruck for excellent technical assistance.
This work was supported by the Ligue Régionale contre le Cancer (Haut-Rhin, Bas-Rhin), the Institut National du Cancer (CETIRICOL, PL06.008), the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique, the Université de Strasbourg and the Hôpitaux Universitaires de Strasbourg.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Guérin, E., Raffelsberger, W., Pencreach, E. et al. In Vivo Topoisomerase I Inhibition Attenuates the Expression of Hypoxia-Inducible Factor 1α Target Genes and Decreases Tumor Angiogenesis. Mol Med 18, 83–94 (2012). https://doi.org/10.2119/molmed.2011.00120
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00120