
Abstract
Effective manipulation of human disease processes may be achieved by understanding transcriptional, posttranscriptional and epigenetic events that orchestrate cellular events. The levels of activation of specific molecules, spatial distribution and concentrations of relevant networks of signaling molecules along with the receptiveness of the chromatin to these signals are some of the parameters which dictate context. Effects elicited by the transcription factor signal transducers and activator of transcription 3 (Stat3) are discussed with respect to the context within which Stat3-mediated effects are elicited within the developing and adult mammalian nervous system. Stat3 signals are pivotal to the proliferation and differentiation of neural stem cells. They also participate in neuronal regeneration and cancers of the nervous system. An analysis of the context in which Stat3 activation occurs in these processes provides a potential predictive paradigm with which novel methods for intervention may be designed.
Similar content being viewed by others


Introduction
Stat3 was discovered as a transcription factor present in complexes which activated the promoters of interferon responsive genes (1), and as a transcription factor which was activated as a result of the acute phase response of liver cells (2,3). It has since been found to subserve diverse roles through evolution, and is implicated ubiquitously in several mammalian physiological systems, and in some pathological events. The mechanisms of Stat3 activation and its implications in mammalian development, its functions in complex physiological systems including the immune and nervous systems, and its role in pathological states including cancer are covered in several reviews, a few of which are referenced here (4–8).
The discussion in this review is focused on possible mechanisms by which Stat3 regulates biological processes in cells within the nervous system. Stat3 plays integral roles during development of the mammalian nervous system, and remains expressed and active in selected regions in the adult brain. It is activated in response to interferons, neuropoietic cytokines, neurotrophic factors, interleukins and notch to name a few. The context in which the Stat3 activating signal is received by cells dictates the outcome of Stat3 activation within the nervous system, that is, the genetic, epigenetic status of the cell along with the levels and activation states of cellular molecules at the time the activating signal is received.
Stat3 also has been implicated in some conditions where the nervous system is compromised, including cancer and neuronal regeneration. Stat3 is involved in complex interactions of proliferation, survival, epithelial-mesenchymal transformation and in the regulation of immune cell functions and inflammatory interactions. While these effects might be relevant to cancer in the nervous system, Stat3 also is implicated in neuronal regeneration. A detailed understanding of the signals which control these physiological and pathological events, including the involvement of Stat3, could lead to the configuration of novel approaches of intervention.
The Activation States of STAT3
The most widely studied function of Stat3 is one of transcriptional transactivation of promoters which include the cognate binding site for Stat3. To be transcriptionally active, Stat3 forms several qualitatively different dimer complexes: Stat3 homodimers and Stat3-Stat1 heterodimers. Each of Stat1 and Stat3 themselves have two isoforms designated a and b; the b isoform is truncated and lacks 38 and 55 C-terminal amino acids respectively. Stat3b has 7 new amino acids at the truncated C-terminus. In addition to transcriptional transactivation, Stat3 also has been implicated in the stabilization of microtubules (9).
Stat3a is a 770 amino acid protein which is posttranslationally modified by phosphorylation and acetylation resulting in a protein “activated” to function as a transcriptional transactivator. The best described modification is phosphorylation of the tyrosine at position 705 by the Janus kinases (JAKs) (1). This phosphorylation event is required for dimerization of Stat3, and the resulting transcriptional transactivation function where it binds to its cognate DNA binding site on Stat-responsive promoters. Phosphorylation of serine 727 also is thought to modulate transcriptional function, and is brought about by several kinases including p42/44 extracellular signal regulated kinases (ERKs) and mammalian target of rapamycin (mTOR) (10,11). Acetylation on lysine 685 by p300 is thought to be relevant to transactivation function (12), while the two N-terminal lysines (lys49, lys87) are reported to be acetylated in some cancer cells (13). In a cervix carcinoma line, leukemia inhibitory factor (LIF) and interleukin-6 (IL-6) have been shown to cause acetylation of Stat3 on lysine 685 via the activation of AKT (14). Methylation has been reported on arginine 31 (15), but has since been questioned (16). The activity of Stat3 is known to be modulated by several ligands. The ones relevant to this discussion include interferon γ (IFNγ), IL-6 and the cytokines which signal through the gp130 receptor subunit which include ciliary neurotrophic factor (CNTF), LIF, leptin, erythropoietin (EPO) and oncostatin M (OSM); neurotrophic factors including nerve growth factor (NGF) and brain derived neurotrophic factor (BDNF); epidermal growth factor (EGF), bone morphogenetic protein 4 (BMP4) and notch.
In addition to posttranslational modifications, nucleocytoplasmic shuttling is another level at which control of the effects of activated Stat3 is exerted. Shuttling in and out of the nucleus is mediated by the importins (17). Phosphorylation appears to increase the rate of shuttling into, and retention within, the nucleus. Phosphorylation by v-src causes constitutive activation of Stat3, while regulation by feedback mechanisms is due to cytokines such as IL-6, thus the former is thought to be oncogenic (18). Suppressor of cytokine signaling (SOCS) which regulates Stat3 activation also may translocate to the nucleus, thus adding another level of regulation of Stat3 (19). Unphosphorylated Stat3 also shuttles at a slow pace, and there is a constant presence of Stat3 within the nucleus (20). Unphosphorylated Stat3 (where Y705 is rendered incapable of phosphorylation) when overexpressed causes the upregulation of several genes, and is thought to act by complexing with NFkB (21). However, it is possible that “unphosphorylated” Stat3 is modified in some manner that remains to be defined.
Cellular Context
Activation of Stat3 elicits distinct responses based on the state of the cell that is receiving the signal. This specific state could be termed as the “context” within which the signal is received. Context could be synthesized as the backdrop of all the other signals transpiring in the cell at that particular time, and also the manner in which cumulative signals impact on Stat3.
Context could include three facets, first, the nuclear and cytoplasmic status of the cells as it receives the Stat3 activating signal since this will dictate protein/lipid/carbohydrate activation and complexing events which coincide with the Stat3 signal. The second parameter is the receptiveness of the genome to these cues, and, thirdly, the manner in which Stat3 itself is posttranscriptionally modified. At a global level, context will be decided by several factors including the spatial position and temporal order of activation of signals within the cell and the thresholds of activation of pertinent signaling molecules. This could include all molecules which mediate transcriptional, posttranslational and genomic modifications and facilitating structures such as lipid rafts, specialized membranes and cellular scaffolding. Thresholds will be set by the net effect of intersecting and independent signals (activating and inhibitory) impinging on an effector molecule, and, in turn, the response elicited by the effector will depend on the thresholds of activation achieved. The strength of the signal may be defined by the amplitude of activation and/or the sustained length of activation. Finally, successful transcriptional transactivation requires a genome which is receptive to interaction with proteins and unwinding. A snapshot of the cell capturing all these facets may represent the context within which the signal is interpreted.
Systems biology approaches can envision possible biological networks of interactions between proteins, RNA and DNA and, potentially, lipids and carbohydrates. These technologies may be used to designate hubs of high activity which connect to other signaling molecules and to each other. They may also predict and/or analyze activation events in relation to others, and may provide temporal delineation of these events. Predictions of interactions, and perhaps outcomes, of complex signaling cascades have been modeled in response to various stimuli (22), and the use of microarray-based technologies have yielded astonishing amounts of data describing transcription factor complexes and genes which are induced as a result of Stat3 activation in specific cellular systems, including stem cells (23,24). The perturbation of levels of Stat3 protein in one study related to the placenta permits the description of pathways which are potentially activated in the presence and absence of Stat3, and indicates that several transcription factors coordinately regulate groups of genes which potentially determine functional output (25). On a smaller scale, protein microarrays created using a sandwich method with known antibodies were used to determine relative and parallel levels of activation by phosphorylation of multiple signaling in erythroid cells in response to EPO, providing quantitative and temporal resolution of these events (26). The continuous development of newer technologies and analysis tools will allow these systemwide studies to be performed in several tissues. The delineation of context of Stat3 activation within cells of the nervous system is developing continually with systems and experimental biology paradigms, and may be combined with the current literature to generate testable hypotheses.
STAT3 Biology in the Developing and Adult Mammalian Nervous System
Neurogenesis is dominant during early cortical development, and is followed by gliogenesis which continues into postnatal development. Prior to embryonic d 10.5 (E10.5, approximately E7.5-E10.5) Stat3 mediates specification of neural stem cells (NSCs) by regulating expression of Sox2, which regulates gene expression characteristic of NSCs (27). To maintain survival and proliferation of NSCs, Stat3 appears to be regulated by the cell surface receptor notch and its associated signaling pathways, resulting in the activation of mammalian target of rapamycin (mTOR) (28,29). mTOR activates Stat3 by phosphorylation of ser727 which appears to be a signal for NSC survival and proliferation in this context (Figure 1A). Extracellular signal regulated kinase (ERK2) also has been shown to be required for proliferation of NSCs (30).

Contextual Stat3 signaling in proliferating and differentiation NSCs. (A) Proliferation and survival of NSCs. A context in which Stat3 regulates the proliferation and survival of NSCs appears to include the cell surface receptor notch and its associated signaling pathways resulting in the activation of mTOR and activated ERK within the cell. Both mediate phosphorylation of Stat3 at serine 727. (B)Glial differentiation. Stat3 is activated by phosphorylation on both serine 727 and tyrosine 705. Stat3 signal are activated in a context which includes the notch-mediated signals hes and NF1, retinoic acid signaling and inhibition of neurogenin (ngn). Epigenetic modifications include retinoic acid-mediated histone acetylation, and NFI-mediated demethylation. Other known signals include cytokine and growth factor-mediated activation of JAK and ERK kinases, and change in methylation status CpG on DNA, and on histone H3 to allow transcription. Notch activation may be achieved by release of notch ligands by neighboring neuronal precursors which confers a glial fate on recipient cells. (C) Neuronal differentiation. Signals from NeuroD and ngn are central to neuronal differentiation. This occurs in a context where activities of ERK, disabled (dab) and MeCP2 are high. In addition to induction of neuron-specific genes by NeuroD and ngn, there is active repression of glial-specific promoters in neurons and neuronal precursors. Notch/Stat3-mediated signals are largely silent in this context, in contrast to glial differentiation.
Activation of Stat3 appears to be a dominant signal for glial differentiation (Figure 1B) (31–34). Stat3 activation may be achieved by several ligands including CNTF, LIF, BMP and notch. CNTF and LIF cause optimal activation of Stat3 by activating the JAK and ERK pathways, which phosphorylate the tyr705 and ser727 residues respectively; BMP4 phosphorylates ser727 via mTOR; and notch activates Stat3 by direct complexing with its downstream activator hes (31,33–35). Gain of function experiments with notch suggest that neuronal differentiation signals, presumably including neurogenin (ngn), are inhibited while signals regulating glial differentiation are promoted (36). Canonical activation of Stat3 involves tyrosine phosphorylation at tyr705 and serine phosphorylation at ser727. Although all studies agree that phosphorylation on tyr705 by JAK kinase is required for glial differentiation, activation of ERKs and, by extension, phosphorylation of ser727 seems to confer effects which are interpreted depending on the assay used. While one study concluded that ERK activation was required for optimal glial differentiation (33), another indicated that ERK activation inhibits glial differentiation (31). BMP-mediated phosphorylation of ser727 by way of mTOR promoted glial differentiation (34). In contrast, phosphorylation of ser727 by notch, also mediated by mTOR, promoted survival, proliferation and maintenance of NSCs rather than differentiation (28,29). It is possible that none of these conclusions are mutually exclusive and other facets of the context require examination. In contrast to these contexts where Stat3 activation leads to glial differentiation, activation of Stat3 by IFNγ in NSCs results in the upregulation of p27 and neuronal maturation along with neurite outgrowth (37). The context in which this IFNγ-mediated Stat3 activation occurs is unclear.
Neuronal differentiation in NSCs and retinal stem cells occurs in the context of the neurogenin, notch and reelin pathways along with a repression of Stat3 activity (Figure 1C). During directed differentiation of NSCs into neurons and glia, the activation of the basic helix-loop-helix transcription factors NeuroD and ngn initiate neuronal differentiation. Conversely, suppression of Stat3 activation also results in neuronal differentiation (38,39). Differentiation of late retinal stem cells may be regulated by a gradient of CNTF where fate choice is dictated by concentration of the ligand (40). This study showed that, at high concentration of CNTF, Stat3 is activated by JAK and ERK which causes suppression of the neuronal pathway and leads to glial differentiation to Muller cells, while low concentrations of CNTF result in bipolar neuron differentiation, probably due to an ERK-mediated expression of transcription factors which promote neuronal differentiation. Optimal Muller cell differentiation requires modulation of the activation of Stat3 by notch-mediated effects (40). Along with notch, reelin is another molecule which plays a central role in the development of the mammalian nervous system, and it signals through a cytoplasmic adapter protein called disabled (dab). Gliogenesis was increased at the expense of neurogenesis in dab-deficient NSCs due to decreased expression of NeuroD and enhanced expression of JAK-Stat (41). Interestingly, ratios of differentiation in wild-type NSCs were not affected by reelin, suggesting that dab could signal independently of reelin.
Epigenetic factors contribute extensively to the context in which the activation signal is interpreted (see Figures 1B, C). NSCs derived from embryos prior to E12 differentiate into glia less efficiently than late NPCs (42). Subsequent studies showed that this is probably due to CpG methylation of the Stat3 site on the glial fibrillary acidic protein (GFAP) promoter (43), and histone methylation at Stat3 site at the GFAP promoter (44). Gliogenesis is more efficient in cells lacking the DNA methyltransferase 1 causing the genome and chromatin to be hypomethylated, resulting in increased expression of Stat1 and GFAP (45). Another series of experiments suggest that notch ligands are expressed by neuronal progenitors, which then “unblocks” the methylation of the GFAP promoter of the neighboring stem cells so that they may become astrocytes. Notch causes activation of NF1, which, upon binding to astrocyte-specific promoters including GFAP promoter, promotes demethylation (46). It thus appears that glial genes are suppressed by cell-autonomous epigenetic means in neuronal cells. Along similar lines, MeCP2 is highly expressed in neurons, and the overexpression of MeCP2 leads to the suppression of astrocytic gene expression (47). Retinoic acid is another important morphogen which is active in the postnatal d 1 (P1) brain at the time when astrocytic differentiation occurs. Retinoic aci induces histone H3 acetylation in the proximity of the Stat3 site at the GFAP promoter, allowing increased transcriptional activity by Stat3 (48).
Other less defined elements of cellular context relating to Stat3 are emerging. The composition of lipid rafts confers a differentiation signal in the developing mouse cortex. Rafts enriched in phosphatidyl glucoside direct astrocytic differentiation by regulating the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) which results in the activation of JAK/Stat3 pathway (49). One of the results of the activation of Stat3 is the expression of molecules which regulate not only Stat3 itself, but also other transcription factors and cellular proteins. This provides a continuity of signals which either maintain the cellular environment or initiate a unique one. Suppressor of cytokine signaling 3 (SOCS3), and protein inhibitor of activated Stat3 (PIAS3) are the most common regulators of Stat3 activity. SOCS3 suppresses AP1 activity by inhibiting the transcription factor JNK-1 (50), downregulating IFNβ levels and also by affecting signaling from chemokine receptors by binding directly to them (51). Such dynamic interactions with other intracellular pathways could modify the context in which a signal is being propagated. As one might envisage, the expression levels of receptors on the cell surface, concentration/gradients of soluble and membrane-bound ligands, availability of signaling molecules and modifying enzymes, will continuously contribute to the elicited result.
The leptin-responsive neurons in the hypothalamus are the second distinct population where Stat3 is activated in the adult brain, the first being the “niche” area of the subventricular zone where NSCs remain in the adult brain (5). The regulation of leptin-related responses in the hypothalamic-pituitary axis regulates appetite. Leptin induces the JAK/Stat pathway in neurons of the hypothalamic area (52). Leptin also induces protein tyrosine phosphatase1b (PTP1b) which, in turn, regulates leptin signaling (53). More details regarding the context of Stat3 signaling in these neurons are awaited.
STAT3 Biology in Pathologies of the Nervous System
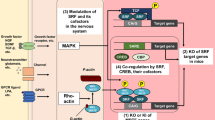
Stat3 appears to have a prominent role in the biology of several cancers, including gliomas (8,54). The activation of Stat3 by LIF or IL-6 causes increased proliferation of glioma stem cells (55,56). Inhibition of Stat3 in neurosphere cultures of cancer stem cells derived from glioblastoma multiforme (GBM) causes an irreversible inhibition of proliferation, and a downregulation of genes associated with the stem cell phenotype (57). mTOR also activates Stat3 in GBM cells downstream of the serine kinase Akt (58). This is in line with the BMP-activated, mTOR-mediated, activation of Stat3 described previously in NSCs (34), and also aligns with the observation that BMP causes differentiation of GBM-derived stem cells (Figure 2) (59). While no epigenetic regulation of Stat3 itself has been uncovered in gliomas, there is documented epigenetic regulation of genes which might control Stat3 activity. Hypermethylation of SOCS1 and SOCS3 correlates with poor patient outcome (60,61). Release of hypermethylation of SOCS1 genes in gliomas leads to an increased sensitivity to radiation, presumably through the ERK pathway. Methylation of the BMP receptor 1a gene promoter in GBMs causes silencing of this gene (62), reminiscent of the fact that BMP promotes differentiation of GBM-derived stem cells (59). Epigenetic regulation also may be exerted by micro-RNAs, a group of small stable RNAs which regulate gene expression. The micro-RNA miR21 shows the greatest differential expression in GBMs (63). Its known targets include p53, PTEN and others. Although it had not been known to regulate Stat3 activity directly, it does regulate increased survival, a function also mediated by Stat3. Similarly miR7 and miR34a are associated with the regulation of EGFR/Akt and notch pathways respectively (64,65), and, again, may be associated with the regulation of Stat3 activity.

Stat3 interactions in glioma biology. There is evidence for a role for Stat3 in tumor cells, immune cells and endothelial cells within gliomas. Stat3 mediated proliferative/survival signals downstream of LIF or IL-6. It also possibly mediates differentiation downstream of BMP/mTOR activation. Several genetic and epigenetic mutations/interactions which impact the Stat3 pathway have been documented, including mutations in the EGFR, hypermethylation of SOCS and BMPR promoters and misregulation of several miRNAs including miR326 which affects notch signals. Stat3 also appears to be an important regulator of mesenchymal transformation events. These interactions are impacted by levels of ERK and notch signals present within the cells. Inflammation results in the activation of several pathways which impinge directly on levels of activated Stat3. These include upregulation of notch signaling and recruitment of OSM and IFNγ mediated signals. Stat3 plays an important role in the immunotolerance of the tumor by the host. It also is involved in the production of angiogenic factors in the tumor microenvironment, and in the mediation of VEGF signaling in endothelial cells within the tumor.
A systems biology approach was applied to identify a putative transcriptional network which drives the mesenchymal transformation of high-grade gliomas (66). Using a unique algorithm, these authors were able to identify a group of six transcription factors which exert significant control over the expression of a gene cassette designated as mesenchymal in these tumors. Promoter occupancy studies using ChIP assays showed that, along with interactions with each other, there are also feed-forward loops of self-regulation. Stat3 and C/EBP were identified as the two factors which were highest in the hierarchy of events causing mesenchymal phenotype in glioma cells. Presumably these effects are elicited in the context of the network of transcriptional events at the hub of which are six transcription factors: Stat3, C/EBP, bHLH-B2, RUNX1, FOSL2 and ZNF238. By using next-generation sequencing technologies, a comprehensive gene expression analysis of gliomas was performed with the aim of uncovering copy number variations and gene mutations which are correlative with gliomas (67). Parsons et al. identified a prevalence of mutations in the isocitrate dehydrogenase (IDH-1) in 12% of the tumors, in addition to mutations in other expected genes including p53, retinoblastoma, PTEN and EGFR. Again, the perturbations in these genes would contribute to the context within which Stat3 elicits its effects in gliomas.
Inhibition of Stat3 in GBMs reverses the tolerance in immune cells isolated from the tumor (68). Stat3 promotes proliferation and survival of tumor cells, and the production angiogenic factors in the tumor microenvironment (69). It has been shown in animal models that the responsiveness of tumors to the immune system of the host and to adoptively transferred T-cells may be enhanced by the inhibition of signaling molecules such as Stat3 (70,71). In these models, expression of Stat3 was inhibited selectively in cells of the myeloid lineage by targeting these cells with siRNAs directed against Stat3. The intracellular context in which the Stat3 signal is received by the host T-cells, or by the adoptively transferred T-cells, is an important question. Stat3 signaling also is relevant to the regulation of angiogenesis in gliomas. In gliomas as well as medulloblastomas, Stat3 is activated by vascular endothelial growth factor receptor 2 (VEGFR2), and activated Stat3 increases the expression of VEGF in an autocrine activation loop. In this study, activated Stat3 was localized primarily to the endothelial cells of vascular regions of the tumor (see Figure 2) (72).
Stat3 activity is modulated in pathological conditions which result in inflammation in the nervous system by at least three mechanisms. Inflammation causes the expression of jagged, a ligand of notch, on the cell surface of astrocytes. This results in the activation of notch which mediates the expression of GFAP, inducible nitric oxide synthase (iNOS) expression, and cytokines in astrocytes. In addition, jagged induces the expression of JAK2 and SOCS3, thus modulating Stat3 function (73). Another report has shown that during inflammation, IFNγ is secreted by astrocytes, microglia and macrophages. Under these conditions IFNγ-dependent Stat3 activation is dependent in glycogen synthase kinase 3b which complexes with Stat3, and chaperones it to the IFNγ receptor where it is activated by tyrosine phosphorylation (74). OSM, a ligand which signals through the gp130 family of receptors, is produced during inflammation by activated microglia, reactive astrocytes and infiltrating leukocytes. It is implicated in the inflammatory response, and also during proliferation and differentiation in the brain. OSM induces the expression of SOCS3 in astrocytes causing a complex of Stat3, the AP1 proteins fos and jun, p300 and CBP to activate the SOCS promoter (75). In this context it might provide a feedback mechanism for the regulation of Stat3- and API-mediated responses during inflammation in the nervous system.
As mentioned previously, Stat3 initially was discovered as part of the gene cassette involved in the acute phase response in the liver. It has since been strongly implicated in regeneration in the liver, where it is thought to initiate events related to liver regeneration in response to IL-6 and tumor necrosis factor (76). Stat3 also is involved in regeneration in the nervous system. The potential to regenerate is more apparent in the mammalian peripheral nervous system (PNS) than in the central nervous system (CNS). Axotomy of the superior cervical ganglion (SCG) is a model for neuronal injury, where transection of the postganglionic axons causes a decrease in the neurotransmitter proteins and an increase in proteins related to regeneration which largely include galanin and vasoactive intestinal peptide (VIP). That this “switch” in phenotype is dependent on LIF, which is secreted by the nonneuronal cells in the SCG and is mediated by Stat3, has been demonstrated in vivo (77,78). These interactions are mirrored in in vitro explant cultures of SCG, where it was demonstrated that the neuronally present Stat3 is activated by LIF under conditions of injury, but is inhibited by nerve growth factor (79) which is available to the SCG neurons from the target in an uninjured SCG (80). This NGF-modulated context of LIF-mediated Stat3 activation is mirrored by BDNF, but not NT4, and is mediated by a serine kinase (79). A systems biology approach investigating the differences in gene expression between dorsal root ganglia (DRG) and CNS neurons was used to identify genes, which might promote regeneration and neurite extension in adult CNS neurons (81). A panel of 32 candidate transcription factors were identified by defining interactive networks and frequency of transcription factor binding to mutual promoters, and include Stat3, JUN, ATF3 and KLF6. Although none of these proteins were effective when tested independently, attesting to the fact that several of these networks establish the context for the correct signal, the transfection of constitutively active Stat3 served as a powerful cue for neurite extension from CNS neurons. There is evidence for injury-related activation of Stat3 in the CNS. Stat3 causes axonal growth in axotomized retinal ganglion cells in response to EPO (another member of the gp130 responsive family of neuropoietic cytokines (82), and Stat3 is activated in conditions of seizure and results in the expression of γ amino-butyric acid receptor α1 subunit. Interestingly, in the seizure model, JAK/Stat3 pathway appears to be activated by BDNF by an unknown mechanism (83). Insights from these studies may be included in attempts to promote regeneration in the CNS.
Projections and Conclusions
The concentrations and spatial distribution of signaling molecules in the cytoplasmic and nuclear milieu, state of chromatin in the nucleus and cross-talk between pathways create a context which dictates outcome. While Stat3 signals are activated during NSC proliferation and differentiation into glial and neuronal fates, the context within which the activation is realized dictates the outcome (see Figures 1A–C). The proliferation of NSCs and their differentiation into neuronal and glial fates is dependent on activation of Stat3, and is controlled by receptors in response to extracellular soluble ligands. Importantly, the response is elicited within the context that activation of the notch receptor confers on the cell. An effective context within which Stat-3 mediated glial differentiation occurs includes notch activation along with reduced MeCP2 or increased demethylase activity, along with adequate histone deacetylation. The “threshold” of notch activation required for glial differentiation would be one which suppresses NeuroD and ngn levels within the cell to levels which do not permit neuronal differentiation. On the other hand, a context for effective neuronal differentiation is sufficient activation of NeuroD and ngn occurring in conjunction with, or leading to, the suppression of Stat3 activation, coupled with high levels of MeCP2 expression. As notch represses ngn, absence of notch activation is possibly a requirement for the neuronal context. Other qualifying events (including activation of dab and ERK2 for neuronal context, and specialized lipid rafts and EGFR in glial differentiation) may be required, and, conversely, a dynamic change in context could alter outcome (such as neuronal maturation resulting from Stat3 activation by IFNγ).
The possibility of subtle qualitative differences in the effects resulting from the phosphorylated products of Stat3 also arises. Notch signals to Stat3 in two conditions: on ser727 during proliferation, in which case it mediates cell survival and proliferation, and secondly, during glial differentiation, where it potentially causes phosphorylation of ser727 along with JAK-mediated tyr705 phosphorylation in collaboration with other factors (see Figures 1A, B). Differential complex formation, subcellular localization, stability and intensity of signals might result. The length of activation of the Stat3 signal also needs to be considered as this will contribute to the overall strength of the signal. While cytokine-mediated Stat3 interaction is quite short-lived in liver cells, it is prolonged in NSCs in response to CNTF and IFNγ, and is evident up to 24 hours after stimulation (33,37).
A detailed study of signals which collaborate and mediate events in stem cell biology will permit a predictive interrogation of these pathways in cancer, as the same cellular signals which mediate stem cell proliferation, differentiation and survival go awry in transformed cells (Figure 2). An example which arises from the analysis presented here is notch. Notch provides an essential context for the proliferation and glial differentiation of NSCs, which is mirrored by the relevance of notch particularly in the proliferation and survival of gliomas (84). Notch is overexpressed in gliomas (85) and promotes cell survival and confers radioresistance on gliomas (86). An inhibitor of notch, γ-secretase, caused a reduction of cell growth in vitro and in vivo (87). The micro RNA miR326 provides a very interesting mutual feedback loop where it downregulates notch expression and activity, and is, in turn, suppressed by notch (88). miR326 expression in decreased in GBM, thus generating another potential point of intervention in GBM with respect to the notch pathway. Some of the more obvious predictions of new points of intervention in glioma therapies generated from this analysis include MeCP2 signaling, miRNAs, other epigenetic modifications and gene mutations for molecules in the JAK/STAT/SOCS, BMP/mTOR and EGFR signaling pathways, dab, ngn and potential roles for acetylated Stat3 in gliomas. The design of novel therapies which account for complexities of several molecules/pathways providing the context for the aberrant transforming signal must be considered.
A potential role exists for Stat3 in manipulating regeneration in the mammalian nervous system. From the current literature, it appears the context defined by inhibitory signals from the neurotrophic factors with respect to Stat3 activation needs to be explored further in light of the gene candidates identified by systems biology approaches. While this review has focused on the involvement of Stat3 in selected cells of the mammalian nervous system and related pathologies, obviously several other cellular events occur simultaneously with the Stat-centric signals discussed. The mechanisms by which these pathways intersect, or function in parallel, with the Stat3 events may define novel insights and help confer specificity in our analysis and interventions.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Shuai K, Schindler C, Prezioso VR, Darnell JE Jr. (1992) Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science. 258:1808–12.
Akira S, et al. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 77:63–71.
Raz R, Durbin JE, Levy DE. (1994) Acute phase response factor and additional members of the interferon-stimulated gene factor 3 family integrate diverse signals from cytokines, interferons, and growth factors. J. Biol. Chem. 269:24391–5.
Dewilde S, Vercelli A, Chiarle R, Poli V. (2008) Of alphas and betas: distinct and overlapping functions of STAT3 isoforms. Front Biosci. 13:6501–14.
Dziennis S, Alkayed NJ. (2008) Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev. Neurosci. 19:341–61.
Lim CP, Cao X. (2006) Structure, function and regulation of STAT proteins. Mol. Biosyst. 2:536–50.
Sehgal PB. (2008) Paradigm shifts in the cell biology of STAT signaling. Semin. Cell Dev. Biol. 19:329–40.
Yu H, Jove R. (2004) The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer. 4:97–105.
Ng DC, et al. (2006) Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 172:245–57.
Yokogami K, Wakisaka S, Avruch J, Reeves SA. (2000) Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 10:47–50.
Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. (1995) Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 267:1990–4.
Yuan ZL, Guan YJ, Chatterjee D, Chin YE. (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 307:269–73.
Ray S, Boldogh I, Brasier AR. (2005) STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. 129:1616–32.
Ohbayashi N, et al. (2007) LIF- and IL-6-induced acetylation of STAT3 at Lys-685 through PI3K/Akt activation. Biol. Pharm. Bull. 30:1860–4.
Mowen KA, et al. (2001) Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 104:731–41.
Komyod W, Bauer UM, Heinrich PC, Haan S, Behrmann I. (2005) Are STATS arginine-methylated? J. Biol. Chem. 280:21700–5.
Reich NC, Liu L. (2006) Tracking STAT nuclear traffic. Nat. Rev. Immunol. 6:602–12.
Herrmann A, et al. (2007) Nucleocytoplasmic shuttling of persistently activated STAT3. J. Cell Sci. 120:3249–61.
Lee KH, et al. (2008) Increased cytoplasmic levels of CIS, SOCS1, SOCS2, or SOCS3 are required for nuclear translocation. FEBS Lett. 582:2319–24.
Pranada AL, Metz S, Herrmann A, Heinrich PC, Muller-Newen G. (2004) Real time analysis of STAT3 nucleocytoplasmic shuttling. J. Biol. Chem. 279:15114–23.
Yang J, Stark GR. (2008) Roles of unphosphorylated STATs in signaling. Cell Res. 18:443–51.
Ma’ayan A, et al. (2009) SNAVI: Desktop application for analysis and visualization of large-scale signaling networks. BMC Syst. Biol. 3:10.
Kidder BL, Yang J, Palmer S. (2008) Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One. 3:e3932.
Sekkai D, et al. (2005) Microarray analysis of LIF/Stat3 transcriptional targets in embryonic stem cells. Stem Cells. 23:1634–42.
Jiang K, et al. (2009) Ablation of Stat3 by siRNA alters gene expression profiles in JEG-3 cells: a systems biology approach. Placenta. 30:806–15.
Korf U, et al. (2008) Quantitative protein microarrays for time-resolved measurements of protein phosphorylation. Proteomics. 8:4603–12.
Foshay KM, Gallicano GI. (2008) Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev. 17:269–78.
Androutsellis-Theotokis A, et al. (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 442:823–6.
Nagao M, Sugimori M, Nakafuku M. (2007) Cross talk between notch and growth factor/cytokine signaling pathways in neural stem cells. Mol. Cell. Biol. 27:3982–94.
Imamura O, Satoh Y, Endo S, Takishima K. (2008) Analysis of extracellular signal-regulated kinase 2 function in neural stem/progenitor cells via nervous system-specific gene disruption. Stem Cells. 26:3247–56.
Bonni A, et al. (1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 278:477–83.
Nakashima K, et al. (1999) Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science. 284:479–82.
Rajan P, McKay RD. (1998) Multiple routes to astrocytic differentiation in the CNS. J. Neurosci. 18:3620–9.
Rajan P, Panchision DM, Newell LF, McKay RD. (2003) BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J. Cell Biol. 161:911–21.
Kamakura S, et al. (2004) Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6:547–54.
Gaiano N, Nye JS, Fishell G. (2000) Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 26:395–404.
Lum M, et al. (2009) Inhibition of neurosphere proliferation by IFNgamma but not IFNbeta is coupled to neuronal differentiation. J. Neuroimmunol. 206:32–8.
Gu F, et al. (2005) Suppression of Stat3 promotes neurogenesis in cultured neural stem cells. J. Neurosci. Res 81:163–71.
Sun Y, et al. (2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 104:365–76.
Bhattacharya S, Das AV, Mallya KB, Ahmad I. (2008) Ciliary neurotrophic factor-mediated signaling regulates neuronal versus glial differentiation of retinal stem cells/progenitors by concentration-dependent recruitment of mitogenactivated protein kinase and Janus kinase-signal transducer and activator of transcription pathways in conjunction with Notch signaling. Stem Cells. 26:2611–24.
Kwon IS, et al. (2009) Expression of Disabled 1 suppresses astroglial differentiation in neural stem cells. Mol. Cell. Neurosci. 40:50–61.
Molne M, et al. (2000) Early cortical precursors do not undergo LIF-mediated astrocytic differentiation. J. Neurosci. Res 59:301–11.
Takizawa T, et al. (2001) DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell. 1:749–58.
Song MR, Ghosh A. (2004) FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat. Neurosci. 7:229–35.
Fan G, et al. (2005) DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 132:3345–56.
Namihira M, et al. (2009) Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev. Cell. 16:245–55.
Kohyama J, et al. (2008) Epigenetic regulation of neural cell differentiation plasticity in the adult mammalian brain. Proc. Natl. Acad. Sci. U. S. A. 105:18012–7.
Asano H, et al. (2009) Astrocyte differentiation of neural precursor cells is enhanced by retinoic acid through a change in epigenetic modification. Stem Cells. 27:2744–52.
Kinoshita MO, et al. (2009) Lipid rafts enriched in phosphatidylglucoside direct astroglial differentiation by regulating tyrosine kinase activity of epidermal growth factor receptors. Biochem. J. 419:565–75.
Miao T, et al. (2008) SOCS3 suppresses AP-1 transcriptional activity in neuroblastoma cells through inhibition of c-Jun N-terminal kinase. Mol. Cell. Neurosci. 37:367–75.
Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. (2008) Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J. Immunol. 181:3167–76.
Gong L, et al. (2008) Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology. 149:3346–54.
Benomar Y, et al. (2009) Leptin but not ciliary neurotrophic factor (CNTF) induces phosphotyrosine phosphatase-1B expression in human neuronal cells (SH-SY5Y): putative explanation of CNTF efficacy in leptin-resistant state. Endocrinology. 150:1182–91.
Turkson J, Jove R. (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 19:6613–6626.
Penuelas S, et al. (2009) TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 15:315–27.
Wang H, et al. (2009) Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 27:2393–404.
Sherry MM, Reeves A, Wu JK, Cochran BH. (2009) STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 27:2383–92.
Aeder SE, Martin PM, Soh JW, Hussaini IM. (2004) PKC-eta mediates glioblastoma cell proliferation through the Akt and mTOR signaling pathways. Oncogene. 23:9062–9.
Piccirillo SG, et al. (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 444:761–5.
Martini M, et al. (2008) Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer. 123:2955–60.
Zhou H, et al. (2007) Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 13:2344–53.
Lee J, et al. (2008) Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 13:69–80.
Martinez R, Esteller M. (2010) The DNA methylome of glioblastoma multiforme. Neurobiol. Dis. 39:40–6.
Kefas B, et al. (2008) microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 68:3566–72.
Li Y, et al. (2009) MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 69:7569–76.
Carro MS, et al. (2010) The transcriptional network for mesenchymal transformation of brain tumours. Nature. 463:318–25.
Parsons DW, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science. 321:1807–12.
Hussain SF, et al. (2007) A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 67:9630–6.
Yu H, Kortylewski M, Pardoll D. (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7:41–51.
Herrmann A, et al. (2010) Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 70:7455–64.
Kortylewski M, et al. (2009) In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 27:925–32.
Schaefer LK, Ren Z, Fuller GN, Schaefer TS. (2002) Constitutive activation of Stat3alpha in brain tumors: localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2). Oncogene. 21:2058–65.
Morga E, et al. (2009) Jagged1 regulates the activation of astrocytes via modulation of NFkappaB and JAK/STAT/SOCS pathways. Glia. 57:1741–53.
Beurel E, Jope RS. (2008) Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol. Chem. 283:21934–44.
Baker BJ, Qin H, Benveniste EN. (2008) Molecular basis of oncostatin M-induced SOCS-3 expression in astrocytes. Glia. 56:1250–62.
Fausto N. (2000) Liver regeneration. J. Hepatol. 32:19–31.
Rajan P, Stewart CL, Fink JS. (1995) LIF-mediated activation of STAT proteins after neuronal injury in vivo. Neuroreport. 6:2240–4.
Rao MS, et al. (1993) Leukemia inhibitory factor mediates an injury response but not a target-directed developmental transmitter switch in sympathetic neurons. Neuron. 11:1175–85.
Rajan P, Gearan T, Fink JS. (1998) Leukemia inhibitory factor and NGF regulate signal transducers and activators of transcription activation in sympathetic ganglia: convergence of cytokine-and neurotrophin-signaling pathways. Brain Res. 802:198–204.
Shadiack AM, Vaccariello SA, Sun Y, Zigmond RE. (1998) Nerve growth factor inhibits sympathetic neurons’ response to an injury cytokine. Proc. Natl. Acad. Sci. U. S. A. 95:7727–30.
Smith RP, et al. (2010) Transcriptional profiling of intrinsic PNS factors in the postnatal mouse. Mol. Cell. Neurosci. 46:32–44.
Kretz A, Happold CJ, Marticke JK, Isenmann S. (2005) Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol. Cell. Neurosci. 29:569–79.
Lund IV, et al. (2008) BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci. Signal. 1:ra9.
Purow BW, et al. (2005) Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 65:2353–63.
Kanamori M, et al. (2007) Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 106:417–27.
Wang J, et al. (2010) Notch promotes radioresistance of glioma stem cells. Stem Cells. 28:17–28.
Chen J, et al. (2010) Inhibition of Notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer. 1:822–35.
Kefas B, et al. (2009) The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 29:15161–8.
Acknowledgments
P Rajan would like to thank R Srinivasan for helpful suggestions on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rajan, P. STATus and Context within the Mammalian Nervous System. Mol Med 17, 965–973 (2011). https://doi.org/10.2119/molmed.2010.00259
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2010.00259