
Abstract
PRAS40 is an mTOR binding protein that has complex effects on cell metabolism. Our study tests the hypothesis that PRAS40 knockdown (KD) in C2C12 myocytes will increase protein synthesis via upregulation of the mTOR-S6K1 pathway. PRAS40 KD was achieved using lentiviruses to deliver short hairpin (sh)-RNA targeting PRAS40 or a scrambled control. C2C12 cells were used as either myoblasts or differentiated to myotubes. Knockdown reduced PRAS40 mRNA and protein content by >80% of time-matched control values but did not alter the phosphorylation of mTOR substrates, 4E-BP1 or S6K1, in neither myoblasts nor myotubes. No change in protein synthesis in myotubes was detected, as measured by the incorporation of 35S-methionine. In contrast, protein synthesis was reduced 25% in myoblasts. PRAS40 KD in myoblasts also decreased proliferation rate with an increased percent of cells retained in the G1 phase. PRAS40 KD myoblasts were larger in diameter and had a decreased rate of myotube formation as assessed by myosin heavy chain content. Immunoblotting revealed a 25–30% decrease in total p21 and S807/811 phosphorylated Rb protein considered critical for G1 to S phase progression. Reduction in protein synthesis was not due to increased apoptosis, since cleaved caspase-3 and DNA laddering did not differ between groups. In contrast, the protein content of LC3B-II was decreased by 30% in the PRAS40 KD myoblasts, suggesting a decreased rate of autophagy. Our results suggest that a reduction in PRAS40 specifically impairs myoblast protein synthesis, cell cycle, proliferation and differentiation to myotubes.
Similar content being viewed by others


Introduction
Skeletal muscle is a dynamic and plastic tissue that undergoes both acute and chronic changes in response to various external stimuli. Muscle serves as the largest protein reservoir in the body and can be used as an energy source when the physiological need arises. Protein stores in muscle are maintained by the ingestion of protein/amino acid-containing meals that stimulate protein synthesis and suppress protein breakdown (1,2). Conversely, during periods of starvation or disuse, muscle protein breakdown exceeds protein synthesis (3), providing amino acids to support hepatic gluconeogenesis and acute-phase protein synthesis. Protein synthesis in general is largely regulated at the level of translation initiation, which in turn is regulated by mTOR (mammalian target of rapamycin) (4,5).
Normal cellular function depends on the integration and regulation of cell signaling pathways governing cell cycle, protein synthesis and degradation; cell proliferation; and apoptosis. The serine (S)/threonine (T) protein kinase mTOR plays an important role in these pathways (6–8), and dysregulation of mTOR signaling networks leads to numerous diseases (9). mTOR is contained within two distinct complexes: mTORCl and mTORC2 (5,10,11). The former complex is composed of mTOR, raptor, LST8/G-protein β-subunit-like protein (GβL), proline-rich Akt substrate 40 kDa (PRAS40) and DEP-domain-containing partner of mTOR (DEPTOR) (12).
Exposure of muscle to growth factors and nutrients increases protein translation initiation via the mTOR pathway, thereby stimulating protein synthesis (13). In response to growth factor signaling, the phosphoinositide 3-kinase (PI3K) pathway enhances Akt via phosphoinositide-dependent kinase. Activated Akt then phosphorylates PRAS40 on T246, releasing PRAS40 from the mTOR/raptor complex and enhances its binding to the cellular anchor protein 14-3-3 (14,15). Conversely, in the absence of growth factors, PRAS40 is hypo-phosphorylated and remains bound to mTOR-raptor and thereby inhibits binding of other mTOR substrates, such as the ribosomal protein S6 kinase (S6K1) and the translational repressor eukaryotic initiation factor 4E binding protein (4E-BP1), thereby suppressing cap-dependent protein translation initiation (15).
The necessity of mTOR activation and subsequent phosphorylation of S6K1 and 4E-BP1 has been demonstrated previously; however, the role of PRAS40 in mTORCl is poorly defined. Present data place PRAS40 either at the level of mTOR (as an Akt substrate) or as a direct downstream substrate of mTOR, where it is phosphorylated on S183 (16,17). Several reports have implicated PRAS40 as a negative regulator of mTOR via its inhibition of mTOR substrates, while in contrast, others have shown PRAS40 is required for mTOR signaling (18). These opposing data have given rise to controversies regarding the role of PRAS40 in regulating protein translation initiation via mTORC1. Despite several reports implicating PRAS40 as a regulator of protein translation initiation in a variety of cells, there is a paucity of information related to its role in skeletal muscle. Given the pivotal role mTOR plays in response to environmental cues in regulating protein translation initiation, cell cycle and proliferation, it is reasonable to suspect that one or more of these mTOR functions are altered by PRAS40 in myocytes. Therefore, the purpose of our current investigation was to examine changes in C2C12 myocyte protein synthesis, cell proliferation and cell cycle in response to PRAS40 knockdown using short hairpin (sh)-RNA-based in vitro experimental approaches.
Materials and Methods
Cell Culture
C2C12 myoblasts (American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco’s minimum essential medium (DMEM; Invitrogen; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU/mL) and streptomycin (100 µg/mL) (all from Mediatech, Herndon, VA, USA) under 5% CO2 at 37°C. Myoblasts were subcultured and when 100% confluent, the cells were switched to differentiation medium consisting of DMEM with the above antibiotics-antimycotics and 2% horse serum (Hyclone, Logan, UT, USA) to promote myoblast fusion and differentiation to myotubes. Cells were differentiated for 6 d before experimental manipulation. Myotubes were provided with fresh differentiation medium on day 6 and experiments were performed on day 7. To simulate basal mTOR activity, experiments measuring protein synthesis and the phosphorylation of mTOR substrates were performed with serum-free DMEM without antibiotics-antimycotics. 5-Aminoimidazol-4-carboximide ribonucleoside (AICAR; Toronto Research Chemicals, Ontario, Canada), when present, was added at a final concentration of 2 mmol/L for 8 h. Insulinlike growth factor (IGF)-I, when present, was used at final concentration of 100 ng/mL for the last 20 min of the experiment. These doses maximally suppress and activate protein synthesis in C2C12 cells, respectively (19,20).
shRNA Interference
The lentiviral plasmid used (pLKO.1-mPRAS40) was that described by Vander Haar et al. (21) and targeted the mouse sequence 5′-GAG CCC ACT GAA ACA GAG ACA-3′; the scramble shRNA was used as a negative control as previously reported (15) with a hairpin sequence: CCT AAG GTT AAG TCG CCC TCG CTC TAG CGA GGG CGA CTT AAC CTT AGG (Addgene, Cambridge, MA, USA). The plasmids were transformed in DH5α cells and isolated. The actual DNA sequence was confirmed at the Pennsylvania State University College of Medicine DNA sequence core facility. Packaging plasmids psPAX2 and envelope protein plasmid pMD2.G were obtained from Torono Lab (Addgene). HEK293FT cells (Invitrogen) were grown in DMEM; 80–85% confluent plates were rinsed once with Opti-MEM (Invitrogen) and then incubated with Opti-MEM for 4 h before transfections. psPAX2 and pMD2.G along with either scramble or pLKO.1-mPRAS40 were added after mixing with Lipofectamine 2000 as per the manufacturer’s instructions (Invitrogen). Opti-MEM was changed after overnight incubation with DMEM containing 10% FBS without antibiotics to allow cells to take up the plasmids and recover. Culture media were collected at 36 and 72 h posttransfection for viral particles. Viral particles present in the supernatant were harvested after a 15-min spin at 1,500g to remove cellular debris. The supernatant was further filtered using a 0.45-µm syringe filter. Supernatant-containing virus was either stored at −80°C for long-term storage or at 4°C for immediate use. C2C12 cells at 60% confluence were infected twice overnight with 3 mL of viral supernatant containing 8 µg/mL polybrene in serum-free-antibiotic-free DMEM. Fresh DMEM media containing 10% FBS, antibiotics and 2 µg/mL puromycin (Sigma, St. Louis, MO, USA) were added the next day. Cells that survived under puromycin selection were either harvested (as stable cells) and stored or used immediately.
35S-methionine Labeling
C2C12 myocytes were grown in six-well plates and treated as above. Protein synthesis was measured on day 3 in myoblasts after seeding or on day 7 after addition of differentiation medium in myotubes. For metabolic labeling, 10 µCi of radio-labeled 35S-methionine (MP Biomedicals, Solon, OH, USA) was added to each well of a six-well plate, and radiolabel incorporation into trichloroacetic acid (TCA) precepitable proteins was measured via liquid scintillation as previously described (22).
Multiprobe Template Production for RNase Protection Assay
Primer selection for mouse genes of interest was determined using GeneFisher software (23). The lengths of amplified regions were chosen to allow distinct resolution during electrophoretic separation. Primers were synthesized (IDT, Coralville, IA, USA) with restriction sites for EcoRI or KpnI at the 5′ end and with three extra bases at the extreme 5′ end as follows: for PRAS40 forward (5′-GCA GAA TTC GCC CGA TCG TCA GAT GAG GAG A-3′) and reverse (5′-CCT GGT ACC TCA GCT TCT GGA AGT CGC TGG TA-3′); mTOR forward (5′-GCA GAA TTC GGC CAG TGG ACC AGT TGA GAC A-3′) and reverse (5′-CCT GGT ACC CAG CTC AGA CCA GCA GGA CAC A-3′); raptor forward (5′ -GCA GAA TTC CAT GCA TAG CTG TCG CCG ACA-3′) and reverse (5′-CCT GGT ACC ACA ATG AGC GAA CGG TGC GAA-3′); S6K1 forward (5′-GCA GAA TTC GAC CAT GGG GGA GTT GGA CCA T-3′) and reverse (5′-CCT GGT ACC CTC CAG AAT GTT CCG CTC TGC TT-3′); 4E-BP1 forward (5′-GCA GAA TTC CGG GGA CTA CAG CAC CAC TC-3′) and reverse (5′-CCT GGT ACC GGG CAG TTG GCT CTG GTT GG-3′); and L32 forward (5′-GCA GAA TTC CGG CCT CTG GTG AAG CCC AA-3′) and reverse (5′-GCAGGT ACC CCT TCT CCG CAC CCT GTT GTC A-3′).
PCR was conducted with HotStarTaq DNA polymerase (Qiagen, Valencia, CA, USA), and mouse total RNA was reverse-transcribed with Superscript first-strand synthesis system for RT-PCR (Invitrogen). PCR products were phenol-chloroform extracted, ethanol precipitated and sequentially digested with KpnI and EcoRI (Promega, Madison, WI, USA). Digested products were gel-purified, reextracted and cloned into KpnI/Eco RI-digested pBluescript II SK+ (Stratagene, La Jolla, CA, USA). Plasmid DNA was isolated with both QIAprep spin miniprep and plasmid maxi kits (Qiagen). Final constructs were linearized with EcoRI, gelpurified and quantitated spectrophoto-metrically. The template was prepared so that a 2-µL aliquot contained 10 ng each of PRAS40, S6K1, mTOR, 4E-BP1 and raptor and 20 ng of L32.
RNA Extraction and RNase Protection Assay
Total RNA was extracted from cells using Tri reagent (Molecular Research Center, Cincinnati, OH, USA), exactly as previously described (24). Concentration, purity and integrity of the isolated RNA were assessed using an ultraviolet/visible (VIS) spectrophotometer (Beck-man, Fullerton, CA, USA). mRNA expression was determined by RNase protection assay. A aliquot of template was prepared with T7 polymerase with buffer (Fermentas, Hanover, MD, USA), nucleotide triphosphates (NTPs) and tRNA (Sigma-Aldrich), RNasin and DNase (Promega), and [32P]-UTP (Amersham Biosciences, Piscataway, NJ, USA). Unless otherwise noted, the entire RNase protection assay procedure, including labeling conditions, component concentrations, sample preparation and gel electrophoresis, was as published (24). Gels were exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA, USA), and data were visualized and analyzed by ImageQuant software (version 5.2; Molecular Dynamics). Signal densities within the linear range for mRNAs were normalized to densities for mouse ribosomal protein L32 mRNA.
Immunoblot Analysis and Immunoprecipitation
After treatment, cells were rinsed two times with cold Dulbecco’s Phosphate Buffered Saline (DPBS) and collected on ice in lysis buffer (20 mmol/L HEPES [pH 7.4], 2 mmol/L EGTA, 50 mmol/L β-glycerophosphate, 0.3% 3[(3-Cholamidopropyl) dimethylammonio]-propanesulfonic acid (CHAPS), 100 mmol/L KCl, 2 mmol/L EDTA, 50 mmol/L NaF, 0.5 mmol/L PMSF, 1 mmol/L benzamidine, 1 mmol/L sodium orthovanadate, and 2 µg/mL leupeptin). Lysates were sonicated for 10 min and then kept on a rocker for 30 min in the cold before being clarified (14,000g for 10 min at 4°C). A portion of the resulting cell supernatant was used to determine protein concentration via a bicinchoninic acid assay (Pierce, Rockford, IL, USA). Sample buffer (5×) was added, and samples were loaded according to total protein content (20 µg) on polyacrylamide gels for separation by SDS-PAGE. Proteins were transferred to polyvinylidene fluoride membrane (Biotrace; PALL, Pensacola, FL, USA), blocked in 5% nonfat dry milk and incubated overnight at 4°C with phospho-specific and total antibodies for raptor (S792), mTOR (S2448), S6K1 (T389), AMPK (T172), PRAS40 (T246), 4E-BP1 (T37/46), cleaved caspase-3, poly (ADP-ribose) polymerase (PARP), LC3B, beclin, Atg 7 and p53 (all from Cell Signaling Technology, Boston, MA, USA). Antibodies for total pRb, p21, p27, cyclin-dependent ki-nase (cdk)-4, cdk6, MyoD and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and for myosin heavy chain (MHC) (MF-20, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) were also used. Excess primary antibody was removed by washing in 1× Tris-buffered saline + 0.1% Tween 20, and membranes were incubated with horseradish peroxidase-conjugated goat an-tirabbit or goat antimouse secondary antibody (Sigma) at room temperature for 1 h. Blots were rinsed with 1× Tris-buffered saline + 0.1% Tween 20 to remove excess secondary antibody and were treated with enhanced chemiluminescence (ECL plus) Western blotting reagents as per the manufacturer’s instructions (Amersham, Piscataway, NJ, USA) and then were developed using the Gnome (SynGene, Cambridge, UK). Uncompressed tiff images were analyzed using National Institutes of Health (NIH) ImageJ 1.6 software. After development, blots were stripped and incubated with antibodies for total proteins on blots that were probed earlier with phospho-specific antibodies. Antibodies against β-actin or β-tubulin (Santa Cruz Biotechnology) served as an additional control for equal protein loading of samples.
The PRAS40•raptor, PRAS40•eIF3f and PRAS40•PRAS40 complexes were quantified as described (25). Briefly, to maintain potential protein-protein interactions, cells were homogenized in CHAPS buffer as described above. The homogenate was mixed on a platform rocker and clarified by centrifugation. An aliquot (normalized to equal total protein) of the resulting supernatant was combined with anti-PRAS40 antibody and immune complexes isolated with goat antirabbit BioMag IgG (PerSeptive Diagnostics, Boston, MA, USA) beads. The beads were collected, washed with CHAPS buffer, precipitated by centrifugation and subjected to SDS-PAGE as described above. Blots were then probed with appropriate antibodies to study protein-protein interaction and developed with ECL and analyzed as described above.
Cell Cycle
Myoblasts were transfected with either a scramble (control) shRNA or a shRNA targeting PRAS40. Cells were seeded in 10-cm dishes and used at 60% confluence (∼24 h postseeding). Cells were trypsinized, washed with DPBS and fixed in cold 70% ethanol overnight at −20°C. Cells were then stained with 100 µg/mL solution of propidium iodide buffer containing 0.1% Triton-X100 and 0.001% DNAse free RNAse at 37°C for 30 min immediately before fluorescence-activated cell sorter (FACS) analysis. A total of 10,000 cells per sample were counted, and cell cycle phase was measured by propidium iodide staining intensity using a BD FACS-Calibur flow cytometer (Becton Dickinson, Bedford, MA, USA) and ModFit software LT Version 3.2 (Verity Software, Topsham, ME, USA).
Cell Size and Proliferation
To determine cell size, transfected myoblasts were seeded in 10-cm dishes and used at 60% confluence. For cell number, cells were seeded at similar densities and counted at different time points. Myoblasts were trypsinized and suspended in DMEM with 10% FBS. Cells were then diluted in Isoton II solution (Beckman Coulter, Fullerton, CA, USA) and assayed using the Beckman Coulter counter and particle size analyzer as per the manufacturer’s recommendation (Beckman Coulter). Cell size analysis was performed using the AccuComp® Z2 Coulter counter software (Beckman Coulter).
MTT Assay
C2C12 myoblasts (∼10,000/well) were grown in a 96-well plate for 24 h and then rinsed with phosphate-buffered saline (PBS) to remove the interfering phenol red from the DMEM. This was followed by the addition of methylthiazoletetrazolium (MTT; 50 µg/100 µL) to cells in each well for 4 h at 37°C (26). MTT containing PBS was aspirated, 100 µL DMSO was added to each well to dissolve the resulting formazan and absorbance at 570 nm was read in a plate reader Spectramax (Molecular Devices, Sunnyvale, CA, USA).
DNA Isolation and Analysis
To analyze nucleosomal DNA fragmentation, 2.5 × 106 C2C12 cells were processed as described by Zhivotovsky et al. (27) with minor modifications. A total of 80% confluent cells were trypsinized and centrifuged for 5 min at 2,000g to remove media. The cell pellet was resuspended in total DNA extraction buffer (400 mmol/L NaCl; 10 mmol/L Tris-HCl, pH 7.5; 10 mmol/L EDTA with 50 µg/mL RNAse A and 0.2% SDS) and incubated at 37°C overnight. Proteinase K was added the following day to a final concentration of 50 µg/mL for 4 h at 37°C. NaCl was then added to the DNA extraction mix to obtain a final concentration of 1.23 mol/L, and the extraction mix was left on ice overnight. The following day, the mixture was centrifuged at 23,000g for 1 h at 4°C. The supernatant containing the low-molecular weight DNA was removed and extracted two times with phenol/chloroform followed by a final rinse in chloroform. The extract was then subjected to ethanol precipitation in 0.1 volume of 3 mol/L sodium acetate and 2 volumes of 100% ethanol at −20°C for 1 h. The samples were centrifuged at 2,000g to pellet the DNA, and the pellet was washed with 70% ethanol and air-dried. The pellet was resuspended in Tris-EDTA buffer (pH 8.0), and the amount of DNA was measured spectrophotometrically. Positive control apoptotic DNA from U937 cells was from a Roche DNA-ladder kit (Roche Diagnostics, Indianapolis, IN, USA). DNA (8 µg per sample/lane, and 3 or 5 µg/lane for positive control apoptotic DNA) was loaded onto a 1.5% agarose gel, subjected to electrophoresis (55 V for 2.5 h) in 1× Tris-Borate-EDTA buffer and scanned using the Typhoon fluorescent imager (Molecular Dynamics, Piscataway, NJ, USA).
Cell Differentiation
Approximately 0.5 × 106 myoblasts transfected with scramble or shPRAS40 were seeded in 10-cm plates and photographed daily using a Nikon digital camera (Nikon, Tokyo, Japan) mounted on a binocular microscope using a 10× objective lens. Images were composed and edited in Photoshop 7.0 (Adobe Systems, San Jose, CA, USA). Background was reduced using contrast and brightness adjustments to enhance reprint, and all modifications were applied to the whole image. Similarly treated plates were collected at day 3, 5, 7 and 9 for Western blotting to measure MHC expression as a functional end point to measure differentiation biochemically, since myoblasts were allowed to fuse and form differentiated postmitotic myotubes.
Cell Apoptosis and Autophagy Controls
PRAS40 KD and scramble control myoblasts of the same passage were treated with 2 µmol/L staurosporine in culture media for 4 h to induce apoptosis and were collected in the CHAPS media. To induce autophagy, C2C12 myoblasts were treated with Hanks’ buffered salt solution (HBSS) for 6 h, and cells were collected in CHAPS buffer.
Statistics
Results for individual cell experiments (n ≥ 6) were replicated in at least three independent experiments and when applicable are presented as means ± SE calculated from the pooled data. Data were analyzed by unpaired Student t test in two-group comparisons or with analysis of variance (ANOVA) and Tukey posttest in multigroup comparisons to determine treatment effect when ANOVA indicated a difference among the means. GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA, USA) was used for analysis. Differences between groups were considered significant at P < 0.05.
Results
Effect of PRAS40 Knockdown in C2C12 Myotubes
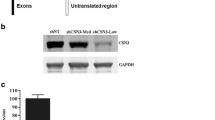
C2C12 stable cell lines deficient in PRAS40 or scramble controls were created using shRNA. shRNA were retrovirally delivered to myoblasts, and some of these myoblasts were allowed to differentiate and form myotubes after puromycin selection. shRNA directed toward PRAS40 in myotubes reduced PRAS40 protein levels by >80%, compared with scramble control values (Figure 1A, B). As anticipated, PRAS40 knockdown also reduced the PRAS40 mRNA content by ∼65% in infected myotubes (Figure 1C). In contrast, PRAS40 knockdown did not alter the mRNA content for 4E-BP1, mTOR, S6K1 or raptor, proteins central to the functioning of the mTOR signaling pathway.

Effect of PRAS40 knockdown in C2C12 myotubes. (A) Representative Western blot of PRAS40 protein in control (shScramble) and PRAS40 knockdown (shPRAS40) myotubes. (B) Quantification of Western blot data. Values are means ± SE; n = 10 per group. *P< 0.05 compared with time-matched scramble control values. (C) Representative autoradiographs from RNase protection assays for PRAS40 and other proteins important in controlling protein synthesis. Except for PRAS40, there are no changes in the mRNA expression. L32 serves as a loading control.
Knockdown of PRAS40 in differentiated myotubes did not alter global protein synthesis compared with scramble controls, as measured by 35S-methionine incorporation into protein (Figure 2A). To determine whether the responsiveness of the PRAS40 knockdown cells to external stimuli was altered, cells were incubated with either an anabolic (IGF-I) or catabolic (AICAR) agent. Addition of IGF-I to the myotubes increased protein synthesis, whereas AICAR inhibited protein synthesis (Figure 2A). Contrary to expectations, the magnitude of the changes produced by these agents in myotubes was the same in both control and PRAS40 knockdown cells. To confirm protein synthesis data, we performed Western blotting for mTOR and its substrates and binding partners. PRAS40 knockdown cells remained responsive to both types of stimuli and their response was similar and comparable to the scramble controls (Figure 2B). For example, IGF-I increased phosphorylation of S6K1 (T389) and PRAS40 (T246), while AICAR increased raptor phosphorylation (S792).

Effect of IGF-I and AICAR on shScramble and shPRAS40 knockdown myotubes. Myotubes were transfected with shScramble and shRPAS40 and incubated with vehicle (control), AICAR (2 mmol/L; 8 h) or IGF-I (100 ng/mL; 20 min) and labeled with 35S-methionine. (A) Protein synthesis in myotubes. Values are means ± SE for n = 8–10 for each condition. Means not sharing the same superscript (a, b and c) are significantly different (P < 0.05). For quantification, data were normalized to scramble control values. (B) Representative Western blots of various total and phosphorylated proteins where cells were treated as described above except that the isotope was omitted. Representative blot of three independent experiments with four replicates per experiment is shown.
PRAS40 Knockdown Decreases Protein Synthesis in C2C12 Myoblasts
While the preceding data were obtained from postmitotic differentiated myotubes (>95%), we also determined whether myoblasts would yield comparable results. In myoblasts, the knockdown of PRAS40 decreased global protein synthesis by ∼25% under basal conditions (Figure 3A). Despite the decrease in basal protein synthesis in the PRAS40 knockdown cells, the ability of these cells to respond positively or negatively to IGF-I or AICAR, respectively, was unaltered. Contrary to expectations, the decreased protein synthesis observed in PRAS40 knockdown cells under basal conditions was not associated with any difference in phosphorylation state of the mTOR substrates S6K1 and 4E-BP1, compared with the scramble control values (Figure 3B) or changes in protein-protein interaction of PRAS40-raptor-eIF3 between the two groups (Figure 3C). In myoblasts, the ability of IGF-I to stimulate T389 phosphorylation of S6K1 and AICAR to increase S792 phosphorylation of raptor did not differ between scrambled and PRAS40 knockdown cells (Figure 3D).

Effect of IGF-I and AICAR on protein synthesis in control and PRAS40 knockdown C2C12 myoblasts. Myoblasts were incubated with vehicle (control), AICAR or IGF-I as described in Figure 2. (A) Protein synthesis in myoblasts were measured on day 3, and values are means ± SE for n = 8 for each condition. Means not sharing the same superscript (a) are significantly different (P< 0.05). (B) Effect of PRAS40 knockdown on total and phosphorylated 4E-BP1, S6K1 and PRAS40 in C2C12 myoblasts. Representative Western blots for proteins involved in the mTORC1 complex mediated translation initiation. Except for the reduction in total and phosphorylated PRAS40, there were no significant differences between the two groups. (C) Effect of PRAS40 KD on protein-protein interaction. Equal amount of total protein from shScramble (four left lanes) and shPRAS40 KD (four right lanes) myoblasts were immunoprecipitated using an excess amount of anti-PRAS40 antibody to pull down proteins interacting with PRAS40. The antigen-antibody immune complex was then probed with antibodies against PRAS40, raptor and eIF3f. The lower three blots represent the whole cell lysate (WCL), which was probed with total antibody against raptor and phospho-specific and total antibody against PRAS40. The amount of raptor and eIF3f bound to PRAS40 did not differ between control and PRAS40 KD cells. (D) Representative Western blots of various total and phosphorylated proteins treated as described in Figure 2. Representative blot of three independent experiments with four replicates per experiment is shown.
PRAS40 Knockdown Alters Myoblast Cell Size and Proliferation
Vander Haar et al. (21) reported that overexpression of wild-type PRAS40 decreased cell size in HEK293 cells, whereas knockdown of Lobe (a PRAS40 ortholog in Drosophila) increased cell size (15). Hence, we hypothesized that knocking down PRAS40 would also increase cell size in myocytes. PRAS40 knockdown increased the diameter (16.8 ± 0.1 µm) of low passage proliferating (∼60% confluent) myoblasts compared with scramble control cells (14.0 ± 0.1 µm) as measured using either the Coulter counter particle size analyzer (Figure 4A) or FACS flow cytometry analysis (data not shown). Mean cell volume was also increased in PRAS40 knockdown cells (Figure 4B). However, unexpectedly we found that PRAS40 knockdown cells grew slower than time-matched scramble controls (Figure 5A), although both cell types were seeded at the same initial density. To exclude anchorage dependence and altered capacity to attach, cells were seeded and counted 4–8 h after seeding to allow for attachment. An equal number of cells were harvested after trypsinization in both the control and PRAS40 knockdown cells, suggesting no significant difference in the ability of PRAS40 knockdown cells to attach to the culture plates (data not shown). To confirm that the proliferation rate of PRAS40 knockdown cells was slower, we used an independent colorimetric assay based on the conversion of the MTT tetrazolium salt to its formazan product. Consistent with the above presented data, the MTT assay revealed that PRAS40 knockdown cells had a 25% lower rate of proliferation (Figure 5B).

Effect of PRAS40 knockdown on cell size in C2C12 myoblasts. (A) Cell size was measured using the Coulter counter particle size analyzer and shown in parentheses. n = 8 for each condition. (B) Mean cell volume of myoblasts as described in (A); mean cell volume in cells with PRAS40 knockdown is increased. Bar graph is mean ± SE; n = 7–9 for each condition. *P < 0.0001. Where absent, standard error bars are too small to be visualized.

Effect of PRAS40 knockdown on C2C12 myoblasts proliferation. (A) Proliferation rate was determined in stably transfected myoblasts with shScramble and shPRAS40. Myoblasts were seeded at the same density and counted using the Coulter counter as described in Materials and Methods. Time intervals are indicated; n = 5 for each treatment time point; experiments were repeated at least three times. (B) To measure proliferative rate acutely (24 h), an independent alternative approach using MTT was used. Values are means ± SE for n = 32 for each condition (*P< 0.0001).
PRAS40 and Apoptosis
To determine whether increased apoptosis in PRAS40 knockdown cells was responsible for the slower proliferation rate, we isolated low-molecular weight DNA and performed an apoptosis DNA laddering assay. Figure 6A illustrates there is no difference between the scramble control and the PRAS40 knockdown cells and that neither group of cells were undergoing active apoptosis within the detectable limits of the assay. These findings were confirmed by Western blotting for caspase-3/PARP cleavage, which failed to detect a significant difference between the groups. Myoblasts incubated with staurosporine were used as a positive control and demonstrated increased caspase-3 and PARP cleavage (Figure 6B). Collectively, these data suggest that the decreased protein synthesis and reduced proliferation in PRAS40 knockdown myoblasts cannot be attributed to increased apoptosis.

Effect of PRAS40 knockdown on apoptosis in C2C12 myoblasts. (A) DNA laddering assay. Low-molecular weight DNA was extracted from stably transfected myoblasts with shScramble and shPRAS40. No regular laddering pattern was observed in either the shScramble or shPRAS40 transfected myoblasts. For sample DNA, 8 µg/lane was loaded, and for positive apoptotic control DNA, 3 or 5 µg/lane was loaded. (B) Myoblasts were transfected with either control (shScramble) shRNA or shRNA targeting PRAS40, and cell lysates were collected for Western blotting. Representative Western blots of whole cell lysates probed using antibodies against cleaved caspase-3, PARP and phosphorylated and total PRAS40 are shown. Staurosporine-treated myoblasts served as the positive control.
PRAS40 Knockdown Inhibits Cell Cycle Progression
To determine the mechanism for the lower proliferation rate in PRAS40 knockdown cells, we stained myoblasts with propidium iodide to study cell cycle events. PRAS40 knockdown myoblasts had a greater proportion of cells in G1/G0 of the cell cycle and fewer cells in the active S phase compared with control values (Figure 7A and B, respectively; Table 1).

Effect of PRAS40 knockdown on C2C12 myoblast cell cycle. Myoblasts were transfected with either control (shScramble) shRNA or shRNA targeting PRAS40. Myoblasts were grown in DMEM supplemented with 10% FBS for 24 h and stained with propidium iodide stain to study cell cycle using FACS. Representative forward scatter histogram highlighting G1 and G2 phases of cell cycle for control (shScramble; A) and PRAS40 knockdown (shPRAS40kd; B) are shown. (C) Cell lysates from myoblasts as described in (A) and (B) were collected for Western blotting analysis. Representative Western blots of whole cell lysates probed using antibodies against total and p-PRAS40 (T246), total p21 and pRb (S807/811) and actin are shown. (D) Cell lysates from myoblasts as described in (A) and (B) were collected for Western blotting analysis. Representative Western blots of whole cell lysates probed using antibodies against Rb, p53, Cyclin D1, cdk4 and cdk6 are shown.
Because PRAS40 knockdown cells were arrested in G1/G0 of the cell cycle, we assessed whether proteins regulating the cell cycle, especially the G1 to S transition, were concomitantly altered. Figure 7C illustrates there was a 25–30% reduction in S807/811 phosphorylation of Rb, consistent with reduced progression from the G1 to S phase. In myoblasts with PRAS40 knockdown, a 20–30% reduced expression of p21 was also detected in these cells. There was no difference in the other proteins analyzed that regulate the cell cycle—p53, cdk 4/6, p27 and cyclin D1 (Figure 7D).
PRAS40 Alters Myogenesis
Our data demonstrate the presence of a concomitant delay in proliferation and altered cell cycle in PRAS40 knockdown myoblasts. Because mTOR also regulates autophagy, which in turn plays an important role in cell differentiation (28,29), we determined the expression of proteins important in regulating autophagy. Although there were no changes in the early markers for autophagy, including Atg 7 and Beclin 1 (Figure 8A), our data indicate that PRAS40 KD decreased the ratio of LC3B-II/LC3B-I (Figure 8A, B).

Effect of PRAS40 knockdown on autophagy. Myoblasts were transfected with either control (shScramble) shRNA or shRNA targeting PRAS40, and cell lysates were collected for Western blotting. (A) Representative Western blots of whole cell lysates probed using antibodies against total Atg 7, Beclin 1, LC3B and PRAS40 are shown. Nutrient-starved myoblasts treated with Hanks’ balanced salt solution served as the positive control. (B) Quantification of Western blot data. Values are means ± SE; n= 10 per group. *P < 0.05 compared with time-matched scramble control values.
Next we determined whether such changes might be of physiological relevance to skeletal muscle development. In this regard, we seeded the same number of myoblasts and tracked their progression to form myotubes (Figure 9). We observed that control cells reached confluent status earlier than the PRAS40 knockdown and began fusion to form a substantial number of myotubes by day 5, whereas PRAS40 knockdown cells only sparsely formed myotubes by day 5. These data suggest that myotube formation and myogenesis is delayed in PRAS40 knockdown cells.

Effect of PRAS40 knockdown on C2C12 differentiation. Myoblasts were transfected with either control (shScramble) shRNA or shRNA targeting PRAS40. Cells were plated at the same density and photographed daily (10 × objective magnification; Nikon digital camera mounted on binocular microscope) to visually record changes in cell proliferation (time to reach confluence) and formation of myotubes. On day 3 when the plates were fully confluent, the media were switched to 2% horse serum (DM, differentiation media) to induce myotube formation.
To quantitate these findings, cell lysates were collected at various stages of development of myoblasts and myotubes to measure the expression of MHC—a protein expressed only in differentiated matured myotubes. Although MHC expression was absent in myoblasts (day 3) and there was an initial delay in MHC expression in PRAS40 knockdown cells (days 5 and 7), by day 9, the expression of MHC in both scramble control and PRAS40 knockdown cells was comparable (Figure 10A, B). The protein content of the muscle transcription factor MyoD did not differ between scramble control and PRAS40 KD cells (Figure 10A).

PRAS40 knockdown in C2C12 myocytes delays MHC protein expression. (A) Representative Western blots for MHC and the muscle-specific transcription factor MyoD in samples treated as in Figure 9. (B) Quantification of MHC Western blots in (A). Values are means ± SE for n = 6 for each condition. *P < 0.05, compared with time-matched control values. ND, not detected.
Discussion
In vitro studies performed in HEK293 and other rapidly dividing cell lines have identified PRAS40 as an mTORC1 binding protein and a regulator for mTOR activity (15,16,18,21). As the reduction of PRAS40 using RNAi leads to increased phosphorylation of mTOR substrates S6K1 and 4E-BP1, it has been posited that PRAS40 functions as a negative regulator of mTOR and translation initiation. To the contrary, our results show PRAS40 is required for protein synthesis in rapidly dividing myoblasts, and the reduction of PRAS40 in these cells decreases protein synthesis. In contradistinction, reduction of PRAS40 did not significantly affect protein synthesis in differentiated myotubes, indicating a developmental-specific effect of PRAS40 in this cell type. The reason for this difference between our findings and earlier reports is unclear but may be related to differences in cell type, experimental conditions, end point measured and/or the extent of PRAS40 knockdown. To this end, we did not detect a change in the protein-protein interaction of PRAS40-raptor-eIF3 complex after PRAS40 KD in myoblasts. Such data suggest that the amount of available PRAS40 is sufficient to allow mTOR signaling toward its known substrates in this cell type, despite a >80% reduction in PRAS40 protein in knockdown cells. Furthermore, knockdown of PRAS40 in both myoblasts and myotubes did not alter the ability of these cells to respond to either an anabolic (IGF-I) or catabolic (AICAR) stimuli. Our data show that addition of IGF-I increased phosphorylation on S6K1 (T389) and PRAS40 (T246) in these myocytes, while treatment with AICAR increased the phosphorylation on raptor (S792) and AMPK (T172). Correspondingly, protein synthesis was altered as anticipated in both myoblasts and myotubes in response to IGF-I and AICAR. Collectively, these data demonstrate the normal responsiveness of PRAS40 knockdown cells—both myoblasts and myotubes.
Cell cycle progression is linked to cell size, and typically cells must attain a certain size before they replicate and divide. However, exceptions to this norm may be observed under artificial (for example, pharmaceutical drugs or transformed immortalized cancer cells) or disease (for example, cardiac hypertrophy) conditions. mTOR also plays an important role in regulating cell growth (8,30–32). In this regard, the overexpression of PRAS40 decreases cell size in HEK293 cells. Conversely, knockdown of PRAS40 in the same cells and of Lobe (an ortholog of PRAS40 in drosophila S2 cells) increased cell size (15,21). Consistent with these earlier observations, knockdown of PRAS40 in myoblasts also increased cell size. However, unexpectedly, PRAS40 knockdown cells were fewer in number compared with the scramble controls. Furthermore, we observed that PRAS40 knockdown myoblasts had a slower rate of proliferation. This change could not be attributed to differential cell binding or attachment to the culture plates. Another potential explanation for the decreased cell number is increased apoptosis in myoblasts with PRAS40 knockdown. However, the role of PRAS40 in regulating apoptosis is controversial. Whereas knockdown of PRAS40 inhibited tumor growth and proliferation via induction of apoptosis in melanoma cells (33), PRAS40 knockdown reduced the ability of tumor necrosis factor (TNF)-α and cycloheximide to induce apoptosis in HeLa cells (34). Using two different approaches—caspase 3 and PARP cleavage and DNA laddering—our data suggest that the lower proliferation rate in myoblasts could not be attributed to increased apoptosis. In general, it is believed that mTOR integrates signals to regulate cell size and cell cycle. However, increased cell size due to inhibition of myostatin was shown to be insensitive to rapamycin, suggesting an mTOR-independent regulation of muscle size. Also, maintenance of the hypertrophy during chronic myostatin deficiency does not require altered Akt/mTOR activity (35,36). Previously, Hentges et al. (37) implicated mTOR in regulating cell size and proliferation via different and independent mechanisms. Therefore, although PRAS40 knockdown cells are larger than controls, cells deficient in PRAS40 have lower proliferation, suggesting PRAS40 may potentially be an important modulatory binding partner of mTOR, which uncouples cell size and cell cycle.
mTOR regulates cell cycle as evidenced by the ability of rapamycin to arrest cells in the G1 phase (4,32,37–40). Propidium iodide staining revealed that myoblasts with PRAS40 knockdown had a greater proportion of cells in the G1/G0 phase, compared with scramble controls, and fewer cells in the active S phase. Collectively, these data suggest PRAS40 is required for mTOR activity in regulating cell cycle and that knockdown of PRAS40 in myoblasts retarded cell cycle progression. Alternatively, we cannot exclude the possibility that the reduction in PRAS40 alters cell cycle kinetics by an undetermined mechanism that is mTOR independent. Because PRAS40 knockdown cells were arrested in G1/G0 of the cell cycle, we focused on elucidating potential underlying mechanisms that might produce cell cycle arrest. Regulation of cell cycle progression by the cyclin-dependent kinase (cdk) inhibitor p21 blocks cells from entering into the DNA synthesis (or S) phase in many cell types. The opposite role of p21 in skeletal muscle growth and differentiation, compared with its role in HEK293 cells, has received recent attention. In HEK293 cells, AICAR increased phosphorylation of p53 with an increased expression of p21 (41,42). While p21 null mice develop normally during embryogenesis (43) because of the presence and activation of another redundant cdk inhibitor (p57) (44), myocytes from these mice have difficulty differentiating to myotubes (45). C2C12 myoblasts treated with AICAR were arrested in G1, and H9c2 cardiomyocytes had reduced expression of the p21 protein (42). Because PRAS40 knockdown myoblasts were arrested in G1, we screened for proteins that regulate the cell cycle, especially in the G1 to S transition. Retinoblastoma protein (pRb) regulates G1 exit in the cell cycle. Rb is phosphorylated upon mitogenic activation, which disrupts Rb binding to the E2F transcription factor, thus allowing transcription of proteins that are essential for the G1 to S transition. We found that after PRAS40 knockdown, there was a reduction of pRb S807/811 phosphorylation consistent with reduced progression from the G1 to S phase. In contrast, no change in the total amount of p53, cdk 4/6, cyclin D1 or p27 was detected. It has been reported that protein expression of p21 can be independent of these other regulatory proteins (46,47). However, we observed that similar to AICAR treatment, PRAS40 knockdown myoblasts had reduced expression of p21. Because decreased p21 adversely affects myotube formation and differentiation, we determined whether PRAS40 knockdown myoblasts exhibited delayed differentiation.
These results are consistent with those of Williamson et al. (42) showing that prolonged G1/G0 and reduced p21 expression in C2C12 myocytes produced by AICAR decreased cell cycling and delayed myotube formation. To determine whether PRAS40 knockdown would delay myoblast fusion and thereby myotube formation, we monitored the progression and ability of these cells to form myotubes in culture. Time lapse imaging and Western blotting analysis for myosin heavy chain (a marker for matured myotubes) indicated that knockdown of PRAS40 in C2C12 myoblasts delayed myotube formation. Autophagy is another mTOR regulated cellular event that plays an important role in differentiation of myoblasts to mature myocytes (48–51). Our results indicate that PRAS40 KD decreases autophagy in myoblasts, as inferred from the reduction in the LC3BII/LC3B-I ratio. These changes suggest PRAS40 regulates muscle proliferation and differentiation via regulation of cell cycle and autophagy regulatory proteins.
In summary, PRAS40 knockdown in differentiated myotubes did not alter protein synthesis. In contrast, PRAS40 knockdown in C2C12 myoblasts decreased protein synthesis independent of a change in the phosphorylation of S6K1 and 4E-BP1, suggesting that PRAS40 is not a negative regulator of mTOR-mediated translation initiation in this cell type. Moreover, both myoblasts and myotubes remained responsive to anabolic and catabolic stimuli when PRAS40 was reduced. Knockdown of PRAS40 inhibited G1 to S phase transition of cell cycle and lowered proliferation rate in myoblasts, supporting the contention that PRAS40 is required for this aspect of mTOR signaling. Our data suggest that PRAS40 knockdown in C2C12 myoblasts impairs the ability of mTOR to regulate cell size and proliferation and that PRAS40 is required for these mTOR-associated functions. We confirm that PRAS40 plays an important role in regulation of cell size and show that it also affects cell proliferation and differentiation. Understanding the role of PRAS40 in proliferation and differentiation of myocytes as outlined here may prove important in designing new strategies to manage the muscle wasting associated with catabolic insults such as sepsis, alcohol abuse and aging.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Kimball SR, Jefferson LS. (2006) Signaling pathways and molecular mechanisms through which branched-chain amino acids mediate translational control of protein synthesis. J. Nutr. 136 (Suppl. 1): 227S–31S.
Anthony JC, et al. (2002) Contribution of insulin to the translational control of protein synthesis in skeletal muscle by leucine. Am. J. Physiol. Endocrinol. Metab. 282:E1092–101.
Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. (2005) Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am. J. Physiol. Endocrinol. Metab. 289:E969–80.
Dunlop EA, Tee AT. (2009) Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell Signal 21:827–35.
Gingras AC, Raught B, Sonenberg N. (2004) mTOR signaling to translation. Curr. Top. Microbiol. Immunol. 279:169–97.
Hay N, Sonenberg N. (2004) Upstream and downstream of mTOR. Genes Dev. 18:1926–45.
Hall MN. (2008) mTOR: what does it do? Transplant. Proc. 40 (Suppl. 10):S5–8.
Schmelzle T, Hall MN. (2000) TOR, a central controller of cell growth. Cell 103:253–62.
Balasubramanian S, et al. (2009) mTOR in growth and protection of hypertrophying myocardium. Cardiovasc. Hematol. Agents Med. Chem. 7:52–63.
Holz MK, Ballif BA, Gygi SP, Blenis J. (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123:569–80.
Kim DH, et al. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110:163–75.
Peterson TR, et al. (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137:873–86.
Lang CH, et al. (2003) Alcohol impairs leucinemediated phosphorylation of 4E-BP1, S6K1, eIF4G, and mTOR in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 285:E1205–15.
Kovacina KS, et al. (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J. Biol. Chem. 278:10189–94.
Sancak Y, et al. (2007) PRAS40 is an insulinregulated inhibitor of the mTORC1 protein kinase. Mol. Cell. 25:903–15.
Wang L, Harris TE, Lawrence JC Jr. (2008) Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 283:15619–27.
Oshiro N, et al. (2007) The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 282:20329–39.
Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. (2007) PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 282:24514–24.
Williamson DL, Bolster DR, Kimball SR, Jefferson LS. (2006) Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am. J. Physiol. Endocrinol. Metab. 291:E80–9.
Frost RA, Lang CH, Gelato MC. (1997) Transient exposure of human myoblasts to tumor necrosis factor-alpha inhibits serum and insulin-like growth factor-I stimulated protein synthesis. Endocrinology 138:4153–9.
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell. Biol. 9:316–23.
Hong-Brown LQ, Brown CR, Huber DS, Lang CH. (2007) Alcohol regulates eukaryotic elongation factor 2 phosphorylation via an AMP-activated protein kinase-dependent mechanism in C2C12 skeletal myocytes. J. Biol. Chem. 282:3702–12.
Giegerich R, Meyer F, Schleiermacher C. (1996) GeneFisher—software support for the detection of postulated genes. Proc. Int. Conf. Intell. Syst. Mol. Biol. 4:68–77.
Krawiec BJ, Nystrom GJ, Frost RA, Jefferson LS, Lang CH. (2007) AMP-activated protein kinase agonists increase mRNA content of the muscle-specific ubiquitin ligases MAFbx and MuRF1 in C2C12 cells. Am. J. Physiol. Endocrinol. Metab. 292:E1555–67.
Lang CH, Frost RA, Vary TC. (2008) Acute alcohol intoxication increases REDD1 in skeletal muscle. Alcohol Clin. Exp. Res. 32:796–805.
Das A, Desai D, Pittman B, Amin S, El-Bayoumy K. (2003) Comparison of the chemopreventive efficacies of 1,4-phenylenebis(methylene)selenocyanate and selenium-enriched yeast on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in A/J mouse. Nutr. Cancer 46:179–85.
Zhivotovsky B, Nicotera P, Bellomo G, Hanson K, Orrenius S. (1993) Ca2+ and endonuclease activation in radiation-induced lymphoid cell death. Exp. Cell. Res. 207:163–70.
Srinivas V, Bohensky J, Shapiro IM. (2009) Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs 189:88–92.
Zeng M, Zhou JN. (2008) Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal 20:659–65.
Shaw RJ, Cantley LC. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441:424–30.
Fingar DC, et al. (2004) mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 24:200–16.
Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. (2002) Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes. Dev. 16:1472–87.
Madhunapantula SV, Sharma A, Robertson GP. (2007) PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 67:3626–36.
Thedieck K, et al. (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2:e1217.
Sartori R, et al. (2009) Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell. Physiol. 296:C1248–57.
Welle S, Burgess K, Thornton CA, Tawil R. (2009) Relation between extent of myostatin depletion and muscle growth in mature mice. Am. J. Physiol. Endocrinol. Metab. 297:935–40.
Hentges KE, etal. (2001) FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. U. S. A. 98:13796–801.
Zhang LH, Lin FR. (2009) Effect of rapamycin on leukemia cell lines [in Chinese]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 17:870–3.
Shafer A, Zhou C, Gehrig PA, Boggess JF, Bae-Jump VL. (2010) Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int. J. Cancer 126:1144–54.
Rosner M, Fuchs C, Siegel N, Valli A, Hengstschlager M. (2009) Functional interaction of mammalian target of rapamycin complexes in regulating mammalian cell size and cell cycle. Hum. Mol. Genet. 18:3298–310.
Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. (2001) Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287:562–7.
Williamson DL, Butler DC, Alway SE. (2009) AMPK inhibits myoblast differentiation through a PGC-1alpha-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 297:E304–14.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82:675–84.
Zhang P, et al. (1999) p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes. Dev. 13:213–24.
Hawke TJ, et al. (2003) p21 is essential for normal myogenic progenitor cell function in regenerating skeletal muscle. Am. J. Physiol. Cell. Physiol. 285:C1019–27.
Lee YJ, et al. (2009) Involvement of a p53-independent and post-transcriptional upregulation for p21WAF/CIP1 following destabilization of the actin cytoskeleton. Int. J. Oncol. 34:581–9.
Parker SB, et al. (1995) p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science 267:1024–7.
Mammucari C, Schiaffino S, Sandri M. (2008) Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 4:524–6.
Annovazzi L, et al. (2009) mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 29:3087–94.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH. (2010) mTOR regulation of autophagy. FEBS Lett. 584:1287–95.
Ravikumar B, et al. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–95.
Acknowledgments
We thank Drs. Ly Hong-Brown and Robert Frost for discussions and critical readings of the manuscript. We thank Danuta Huber and Anne Pruznak for technical support, Dr. David Spector for technical help with viral purification and transfection, Dr. Arun Das with MTT assay and reagents, and Dr. Samina Alam for help with the DNA laddering assay and reagents. We also thank David Stanford of the Penn State Flow Cytometry Core facility for help with cell cycle analysis imaging. This work was supported in part by grants from the National Institutes of Health (GM38032 and AA11290) to CH Lang and Pennsylvania Department of Health using Tobacco Settlement Funds (AA Kazi). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kazi, A.A., Lang, C.H. PRAS40 Regulates Protein Synthesis and Cell Cycle in C2C12 Myoblasts. Mol Med 16, 359–371 (2010). https://doi.org/10.2119/molmed.2009.00168
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2009.00168