
Abstract
Oxidative stress plays a key role in the pathogenesis of aging and many metabolic diseases; therefore, an effective antioxidant therapy would be of great importance in these circumstances. Nutritional, environmental, and chemical factors can induce the overproduction of the superoxide anion radical in both the cytosol and mitochondria. This is the first and key event that leads to the activation of pathways involved in the development of several metabolic diseases that are related to oxidative stress. As oxidation of essential molecules continues, it turns to nitrooxidative stress because of the involvement of nitric oxide in pathogenic processes. Once peroxynitrite forms, it damages via two distinctive mechanisms. First, it has direct toxic effects leading to lipid peroxidation, protein oxidation, and DNA damage. This mechanism involves the induction of several transcription factors leading to cytokine-induced chronic inflammation. Classic antioxidants, including vitamins A, C, and E, have often failed to exhibit beneficial effects in metabolic diseases and aging. Melatonin is a multifunctional indolamine that counteracts virtually all pathophysiologic steps and displays significant beneficial actions against peroxynitrite-induced cellular toxicity. This protection is related to melatonin’s antioxidative and antiinflammatory properties. Melatonin has the capability of scavenging both oxygen- and nitrogen-based reactants, including those formed from peroxynitrite, and blocking transcriptional factors, which induce proinflammatory cytokines. Accumulating evidence suggests that this nontoxic indolamine may be useful either as a sole treatment or in conjunction with other treatments for inhibiting the biohazardous actions of nitrooxidative stress.
Similar content being viewed by others


Introduction
The free-radical theory of aging has gained acceptance and matured in the 50 years since it was proposed by Harman in 1956 (1,2). In addition to free radicals, other cellular reactive byproducts of normal metabolism can lead to biomolecule damage. This had led to the formation of the “oxidative damage theory,” which suggests reactive oxygen species (ROS) are responsible for the accumulation of age-related cellular debris, and that this damage is an important contributor to aging and/or chronic diseases (3). In contrast to a century ago, when infectious diseases were a major cause of mortality, today noninfectious chronic diseases are the most likely causes of death. Disease epidemics (for example, cardiovascular diseases, type 2 diabetes, obesity, and metabolic syndrome) have become major contributors to the burden of disease and are presently emerging, or accelerating, in most developing countries (4). The baby-boomer generation is advancing in age, and the geriatric population in the developed countries will increase tremendously in the next few decades (5). More than 50% of the American public is aware of antiaging therapies, and most people currently use some form of these therapies (6). In excess of 70% of the American population uses dietary supplements daily, most commonly antioxidant vitamin and mineral supplements (7). Likewise, while the use of alternative therapies is on the rise, progress is being made in understanding aging, and the public has more access to this information (through media and the internet) than ever before.
In theory, antiaging interventions should modify the biochemical and molecular events causing aging and chronic diseases, correct physiological changes responsible for symptoms and signs of aging, and decrease the susceptibility to disease associated with aging. Vitamin A and its precursor β-carotene, vitamin C, and vitamin E are the most frequently used antioxidant vitamins. The idea that the pathophysiologic processes caused exclusively by oxygen-derived free radicals could presumably be alleviated by conventional antioxidants, such as vitamins E or C, and/or intracellular enzymatic antioxidants seems worthy of investigation. Several studies, however, have found that antioxidants offered little protection against aging or chronic diseases.
Investigators in the Physicians’ Health Study evaluated data from 83,639 U.S. male physicians, of whom 29% were taking vitamin E, vitamin C, or multivitamin supplements on a self-selected basis (8). The the study report concluded that these supplements were not associated with a significant reduction in total cardiovascular diseases or coronary heart disease mortality. Another prospective study revealed that vitamins E and C and certain carotenoids did not reduce the risk of stroke in 43,738 men 40 to 75 years old who had no cardiovascular disease or diabetes (9). The Heart Outcome Prevention Evaluation trial (10), a randomized controlled trial of patients 55 years old or older who had cardiovascular disease or diabetes, demonstrated that taking 400 IU vitamin E daily for an average of 4.5 years had no influence on cardiovascular outcomes or nephropathy. A randomized, double-blind, placebo-controlled trial of vitamin E revealed, interestingly, that vitamin E supplementation resulted in increased blood pressure in type 2 diabetic patients (11). The authors of this study concluded that the mechanism for this increase remains unknown but appears to be independent of changes in oxidative stress. A metaanalysis of a large body of published data revealed that antioxidant vitamins do not significantly reduce risk of cardiovascular death (12). Moreover, an additional metaanalysis of 11 trials revealed that high doses of vitamin E resulted in slightly but significantly higher rates of all-cause mortality compared with placebo (13). A recent systematic review and metaanalysis that included 68 randomized trials with 232,606 participants (385 publications) also showed that antioxidant vitamins may increase all-cause mortality (14) (Table 1). Oxidative stress is often an early and key event that activates numerous pathways involved in aging and development of chronic diseases. On the basis of published literature, a novel molecular approach to resolve the apparent controversy and to protect cells from oxidative stress is warranted (15).
Nitrooxidative Stress: A Possible Explanation of the Controversy
A theoretical explanation for why antioxidant vitamins do not protect as expected against disease relates to the fact that oxidative stress is more complex than previously realized. This complexity was revealed by the discovery of nitric oxide (NO) and the nitric oxide synthase (NOS) family. A vital, ubiquitous molecule, NO has numerous roles in regulating physiological processes. Because of affinity of NO for the superoxide anion radical  , neither enzymatic nor pharmacologic levels of conventional antioxidants successively compete with NO for
, neither enzymatic nor pharmacologic levels of conventional antioxidants successively compete with NO for  and thus antioxidants do not prevent the occurrence of high peroxynitrite (ONOO−) levels (16). These findings, particularly the discovery of the involvement of ONOO− as a causative reactant, have expanded the oxidative stress theory into new areas (17).
and thus antioxidants do not prevent the occurrence of high peroxynitrite (ONOO−) levels (16). These findings, particularly the discovery of the involvement of ONOO− as a causative reactant, have expanded the oxidative stress theory into new areas (17).
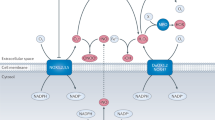
It is presumed that initially ROS production reduced the endothelial NOS (eNOS)-derived NO within endothelial cells while activating inducible NOS (iNOS), which causes almost 1000-fold higher NO production than does eNOS under physiologic circumstances. iNOS is predominantly expressed in inflammatory cells such as macrophages, although epithelial cells from affected tissues also express the enzyme. Intensified expression of iNOS has been detected in virtually all cell types tested, including macrophages, fibroblasts, chondrocytes, osteoclasts, and epithelial cells, and results in the production of much NO in animals and patients with inflammatory diseases (18–21). The level of iNOS expression is well correlated with the degree of inflammation. A controversy arises from observations reporting both cytotoxic and cytoprotective effects of NO. In cases in which NO was found to be cytotoxic, it was questioned whether NO exerted these effects directly or indirectly, or through the formation of more reactive species such as the ONOO−. Thus, it is believed that oxygen-based free radicals are only an initial step in the pathophysiologic mechanisms of aging and chronic diseases (22). The combination of elevated NO plus excess  with the formation of high levels of ONOO− is the proverbial intracellular “devil’s triangle” (Figure 1).
with the formation of high levels of ONOO− is the proverbial intracellular “devil’s triangle” (Figure 1).

Organization of the “devil’s triangle” within the targeted cell. UV, ionizing irradiation, smoking, hyperglycemia, dyslipidemia and chronic inflammation cause excess  and NO production via several means. Normally,
and NO production via several means. Normally,  is readily degraded to H2O by intracellular enzymatic antioxidants. In the presence of abundant
is readily degraded to H2O by intracellular enzymatic antioxidants. In the presence of abundant  and iNOS-derived NO, the biochemically interaction of these substances inevitably produces vast amounts of ONOO− thereby reducing the degradation of
and iNOS-derived NO, the biochemically interaction of these substances inevitably produces vast amounts of ONOO− thereby reducing the degradation of  by SOD. In early stages of oxidative stress, if iNOS is not fully activated, several conventional antioxidants including vitamin E and C diminish the damage via scavenging
by SOD. In early stages of oxidative stress, if iNOS is not fully activated, several conventional antioxidants including vitamin E and C diminish the damage via scavenging  and thereby prevent the activation of the “devil’s triangle.” Unlike the classic antioxidant vitamins, melatonin is the only currently available molecule which is known to block all aspects of the “devil’s triangle” as well as to activate antioxidative enzymes at RNA expression level, thereby preventing the loss of essential cellular antioxidative ezymes.
and thereby prevent the activation of the “devil’s triangle.” Unlike the classic antioxidant vitamins, melatonin is the only currently available molecule which is known to block all aspects of the “devil’s triangle” as well as to activate antioxidative enzymes at RNA expression level, thereby preventing the loss of essential cellular antioxidative ezymes.
Changing the Nature of Oxidative Stress: The “Devil’s Triangle”
In the last two decades, NO produced in excess was found to be a toxic molecule that damages and even kills cells. In the presence of excess  and abundant NO, the latter vital molecule shows its dark side. Neither
and abundant NO, the latter vital molecule shows its dark side. Neither  nor NO is particularly toxic by itself because efficient processes exist that minimize their accumulation (16).
nor NO is particularly toxic by itself because efficient processes exist that minimize their accumulation (16).  is rapidly removed by the superoxide dismutases (SOD), with the isoenzymes of this molecule being located in the mitochondria, cytosol, and extracellular compartments. NO normally is rapidly removed by its fast diffusion through tissues into red blood cells (23), where it is quickly converted to nitrate by a reaction with oxyhemoglobin. This process limits the biological half-life of NO in vivo to less than a second. However, when both
is rapidly removed by the superoxide dismutases (SOD), with the isoenzymes of this molecule being located in the mitochondria, cytosol, and extracellular compartments. NO normally is rapidly removed by its fast diffusion through tissues into red blood cells (23), where it is quickly converted to nitrate by a reaction with oxyhemoglobin. This process limits the biological half-life of NO in vivo to less than a second. However, when both  and NO are generated within a few molecular diameters of each other, they combine spontaneously to form ONOO− in a reaction that occurs at a diffusion-limited rate. Basically, every time NO and
and NO are generated within a few molecular diameters of each other, they combine spontaneously to form ONOO− in a reaction that occurs at a diffusion-limited rate. Basically, every time NO and  collide, they form ONOO−. NO is the only known biological molecule that reacts faster with
collide, they form ONOO−. NO is the only known biological molecule that reacts faster with  and is produced in such high concentrations that it outcompetes endogenous SOD, hence creating the “devil’s triangle” (see Figure 1). Consequently, from a biological viewpoint, the reaction of
and is produced in such high concentrations that it outcompetes endogenous SOD, hence creating the “devil’s triangle” (see Figure 1). Consequently, from a biological viewpoint, the reaction of  with NO to form ONOO− is inevitable. Any of several antioxidants may have beneficial effects in acute conditions that lead to this situation (for example, acute hyperglycemia, ischemia-reperfusion, myocardial infarction). In case of chronic oxidative stress, however, total iNOS activation leads to massive ONOO− generation, against which ordinary antioxidants provide little protection. Thus, in conditions leading to chronic oxidative stress, such as chronic hyperglycemia, dyslipidemia, tobacco smoking, and prolonged drug use, which are known to produce oxidative damage, conventional antioxidants become ineffective.
with NO to form ONOO− is inevitable. Any of several antioxidants may have beneficial effects in acute conditions that lead to this situation (for example, acute hyperglycemia, ischemia-reperfusion, myocardial infarction). In case of chronic oxidative stress, however, total iNOS activation leads to massive ONOO− generation, against which ordinary antioxidants provide little protection. Thus, in conditions leading to chronic oxidative stress, such as chronic hyperglycemia, dyslipidemia, tobacco smoking, and prolonged drug use, which are known to produce oxidative damage, conventional antioxidants become ineffective.
How Is Peroxynitrite Harmful?
Once ONOO− forms, it can act via two distinct mechanisms. First, ONOO− has direct toxic effects leading to lipid peroxidation, protein oxidation, and DNA damage. The second mechanism involves the induction of several transcription factors, including nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1), leading to cytokine-induced chronic inflammation (Figure 2). This cascade, once activated, causes the release of proinflammatory cytokines, including tumor necrosis factor α (TNF-α) and interleukin 1β, which induce widespread inflammation. During this process, several adhesion molecules and monocyte chemoattractant proteins also become involved, widening the inflammatory response and vascular events (17,24).

Consequences of the “devil’s triangle” (92). ONOO− exerts its harmful effects directly and indirectly. It causes activation of transcriptional factors leading to proinflammatory gene expression. As a result, nitrooxidative stress becomes an inflammatory process. Interactions between transcriptional factors and proinflammatory products induce a vicious circle. These cytokines spread the inflammatory message through the circulation. Unless excess  and iNOS-derived NO production are terminated (for example, through normalizing of blood glucose level, cessation of smoking), this mechanism continues to create damage within cells (for example, endothelial dysfunction). Moreover, ONOO− directly damages all macromolecules including lipids, proteins, and DNA. It is well known that both oxygen and nitrogen-based reactants directly damage mitochondria. The majority of NAD+ consumed by PARP also slows the rate of glycolysis and mitochondrial respiration, and eventually leads to cellular dysfunction. Antioxidant vitamins have no effect on ONOO−-induced subsequent cellular damage. Melatonin, however, is a unique molecule which possesses beneficial effects in virtually all steps of the pathophysiologic process. Melatonin blocks proinflammatory cytokine production by reducing NF-κB and AP-1 translocation into the nucleus and directly inhibits iNOS and inflammatory enzyme COX-2. These features break the vicious circle and alleviate the inflammation. Melatonin preserves cellular energy by several means and has been shown to prevent all types of nitrooxidative damage at the mitochondrial level.
and iNOS-derived NO production are terminated (for example, through normalizing of blood glucose level, cessation of smoking), this mechanism continues to create damage within cells (for example, endothelial dysfunction). Moreover, ONOO− directly damages all macromolecules including lipids, proteins, and DNA. It is well known that both oxygen and nitrogen-based reactants directly damage mitochondria. The majority of NAD+ consumed by PARP also slows the rate of glycolysis and mitochondrial respiration, and eventually leads to cellular dysfunction. Antioxidant vitamins have no effect on ONOO−-induced subsequent cellular damage. Melatonin, however, is a unique molecule which possesses beneficial effects in virtually all steps of the pathophysiologic process. Melatonin blocks proinflammatory cytokine production by reducing NF-κB and AP-1 translocation into the nucleus and directly inhibits iNOS and inflammatory enzyme COX-2. These features break the vicious circle and alleviate the inflammation. Melatonin preserves cellular energy by several means and has been shown to prevent all types of nitrooxidative damage at the mitochondrial level.
A direct toxic effect of ONOO− at the site of its production involves an intriguing process that decides the fate of cells. ONOO− is per se not a radical but is a powerful nitrosating agent. ONOO− interacts with and covalently modifies all major types of biomolecules including membrane lipids, thiols, proteins, and DNA (17,25). In addition, ONOO− can yield the hydroxyl (OH•) and nitrogen dioxide  radicals (less than 30% yield). Although this is a minor process in biology, OH• is a potent reactant and oxidizes relevant targets, including amino acids (tyrosine, phenylalanine, histidine), sugars, and lipids (26,27). The generation of ONOO− also decreases the availability of NO for G-protein stimulation and vasodilatation, thus further contributing to endothelial dysfunction and leading to elevated blood pressure. In addition, ONOO− can inhibit SOD as well as other antioxidant molecules and systems, a process that leads to positive feedback cycles of intracellular oxidant generation and increased radical damage (28). ONOO− activates matrix metalloproteinases (MMPs) (29,30) and triggers the expression of selectins and cellular adhesion molecules via enhancement of NF-κB activation, thereby promoting proinflammatory responses.
radicals (less than 30% yield). Although this is a minor process in biology, OH• is a potent reactant and oxidizes relevant targets, including amino acids (tyrosine, phenylalanine, histidine), sugars, and lipids (26,27). The generation of ONOO− also decreases the availability of NO for G-protein stimulation and vasodilatation, thus further contributing to endothelial dysfunction and leading to elevated blood pressure. In addition, ONOO− can inhibit SOD as well as other antioxidant molecules and systems, a process that leads to positive feedback cycles of intracellular oxidant generation and increased radical damage (28). ONOO− activates matrix metalloproteinases (MMPs) (29,30) and triggers the expression of selectins and cellular adhesion molecules via enhancement of NF-κB activation, thereby promoting proinflammatory responses.
The mutagenic properties of ONOO− induced modified products have also been determined (31,32). Several studies have shown that NO itself does not induce DNA single-strand breaks in vitro in plasmid DNA (33,34), whereas exposure of plasmid DNA to preformed ONOO− (35) or NO plus  generated concurrently induces such strand breaks (36). Single-strand breakage can be induced by treatment with very low concentrations of ONOO−, indicating that this agent is a potent inducer of this type of damage to DNA (37). DNA cleavage caused by ONOO− was observed at almost every nucleotide, with a small preference for guanine residues. Furthermore, it has been reported that ONOO− inactivates several enzymes that are critically involved in the repair of DNA damage. These observations suggest additional pathways by which ONOO− may be associated with not only elevated DNA damage but also impairment of DNA repair capacity (38).
generated concurrently induces such strand breaks (36). Single-strand breakage can be induced by treatment with very low concentrations of ONOO−, indicating that this agent is a potent inducer of this type of damage to DNA (37). DNA cleavage caused by ONOO− was observed at almost every nucleotide, with a small preference for guanine residues. Furthermore, it has been reported that ONOO− inactivates several enzymes that are critically involved in the repair of DNA damage. These observations suggest additional pathways by which ONOO− may be associated with not only elevated DNA damage but also impairment of DNA repair capacity (38).
Poly(ADP-ribose) Polymerase Activation
ONOO− induces both apoptosis and necrosis of cells. More highly elevated exposure of this agent is associated with necrosis rather than apoptosis (28,39). In this mechanism, activation of the DNA repair enzyme poly(ADP ribose)polymerase-1 (PARP-1), a member of PARP enzyme family, mediates ONOO−-induced necrosis. PARP-1 detects and signals DNA strand breaks induced by a variety of genotoxic insults, including ionizing radiation, alkylating agents, oxidants (essentially OH* and ONOO−), and free radicals (40–44). When strand breaks occur at the time of binding to DNA, PARP transfers ADP-ribose units from the respiratory coenzyme nicotinamide adenine dinucleotide (NAD+) to various nuclear proteins. From a physiological viewpoint, PARP-1 activity and poly(ADP-ribosyl)ation reactions are implicated in DNA repair, genomic stability maintenance, gene-transcription regulation, and DNA replication. An important function of PARP-1 is to allow DNA repair and cell recovery under conditions associated with a low level of DNA damage. In case of severe DNA injury, over-activation of PARP-1 depletes the cellular stores of NAD+, an essential cofactor in principal energy production mechanisms including the glycolytic pathway, the tricarboxylic acid cycle, and the mitochondrial electron transport chain (ETC). As a result, the loss of NAD+ leads to a marked reduction in the cellular pools of ATP, resulting in cellular dysfunction and death via the necrotic pathway (28,39). This scenario is known as the “suicide hypothesis” of PARP activation and seems to be a regulatory mechanism to eliminate cells after irreversible DNA injury. A vast amount of experimental evidence has established that the PARP-1 pathway of cell death plays a pivotal role in tissue injury and organ dysfunction in numerous disease processes (41,45,46).
Based on the proposed mechanisms, several steps may be selected as potential pharmacological targets to block the hazards of free radicals: reduce excess ROS production; prevent intracellular enzymatic antioxidant depletion; stimulate intracellular enzymatic antioxidants; inhibit iNOS overactivation; increase eNOS-produced NO bioavailability; detoxify nitrogen-based species; inhibit (normalize) NF-κB and AP-1 activation and/or lower NF-κB binding to DNA; reduce proinflammatory cytokines such as TNF-α; limit adhesion molecule production; prevent lipid peroxidation and/or repair damaged lipids; limit protein oxidation and nitration; block PARP-1 activation, thereby preserving NAD+ and cellular energy; and reduce DNA damage and/or promote its repair. Identification of these pharmacological targets suggests the need for a multifunctional agent with antioxidant, iNOS-inhibitory, and ONOO−-scavenging properties. An endogenously produced indolamine, melatonin, is the one molecule that has these characteristics. This indolamine has a variety of means by which it influences the physiology of organisms; some of these actions are receptor-mediated whereas others are receptor-independent (47,48).
Melatonin: A Versatile Antioxidant
A very large body of evidence indicates that melatonin is a major scavenger of both oxygen- and nitrogen-based reactive molecules (49–53), including ONOO− (54–56). Melatonin has scavenging actions at both physiologic and pharmacologic concentrations. Not only melatonin but also several of its metabolites can detoxify free radicals and their derivatives (57–59). Studies also reveal that melatonin eliminates the decomposition products of ONOO−, including OH•,  , and the carbonate radical
, and the carbonate radical  in the presence of physiological carbon dioxide concentrations (60–62). Melatonin also supports several intracellular enzymatic antioxidant enzymes, including SOD and glutathione peroxidase (GSH-Px) (63,64). Moreover, melatonin induces the activity of γ-glutamylcysteine synthetase, thereby stimulating the production of another intracellular antioxidant, glutathione (GSH) (65). A number of studies have shown that melatonin is significantly better than the classic antioxidants in resisting free-radical–based molecular destruction. In these in vivo studies, melatonin was more effective than vitamin E (66–68), β-carotene (69), and vitamin C (69–71), and superior to garlic oil (72). Beneficial antioxidant effects of melatonin have been recently shown in clinical settings for several chronic diseases, including patients with rheumatoid arthritis (73), elderly patients with primary essential hypertension (74), and females with infertility (75).
in the presence of physiological carbon dioxide concentrations (60–62). Melatonin also supports several intracellular enzymatic antioxidant enzymes, including SOD and glutathione peroxidase (GSH-Px) (63,64). Moreover, melatonin induces the activity of γ-glutamylcysteine synthetase, thereby stimulating the production of another intracellular antioxidant, glutathione (GSH) (65). A number of studies have shown that melatonin is significantly better than the classic antioxidants in resisting free-radical–based molecular destruction. In these in vivo studies, melatonin was more effective than vitamin E (66–68), β-carotene (69), and vitamin C (69–71), and superior to garlic oil (72). Beneficial antioxidant effects of melatonin have been recently shown in clinical settings for several chronic diseases, including patients with rheumatoid arthritis (73), elderly patients with primary essential hypertension (74), and females with infertility (75).
Several antioxidants reportedly preserve the activities of SOD and/or GSH-Px. These effects are indirect, however, owing to their ability to scavenge free radicals and protect the protein from damage. Melatonin, on the other hand, possesses genomic actions and regulates the expression of several genes, including those for SOD and GSH-Px. Melatonin influences both antioxidant enzyme activity and cellular mRNA levels for these enzymes under physiological conditions and during elevated oxidative stress (63), possibly through epigenetic mechanisms (76–78). The occurrence of these two features in a single molecule is unique for an antioxidant, and both actions protect against pathologically-generated free radicals.
Of particular interest is the possible role of melatonin as a bioenergetic agent that can improve and maintain mitochondrial function. The majority of molecular oxygen (O2) inhaled and eventually taken up by cells is processed in the mitochondrial ETC, where it is converted to water after its four-electron reduction. During this reductive process, however, partially reduced species of O2 produce ROS and RNS. A specific isoform of NOS, mitochondrial NOS, is a constitutively expressed enzyme proposed to exist in mitochondria, where it produces NO within mitochondria. NO is a physiological regulator of respiration and also the rate of ATP synthesis. Elevation of NO levels higher than the physiological concentrations may inhibit complexes I, III, and IV. In this eventuality, mitochondrial electron transfer reactions are compromised and electron leakage is exaggerated, leading to increased formation of  and all downstream oxidants (79). We recently reviewed the mechanisms by which melatonin improves mitochondrial respiration and increases ATP synthesis under physiological and poisonous conditions (80). Consequently, the antioxidant and free-radical-scavenging capacities of melatonin protect proteins of the ETC and mtDNA from ROS/RNS-induced oxidative damage. This protective effect limits the loss of intramitochondrial GSH, improves ETC activity, and reduces mtDNA damage. Melatonin’s actions at the mtDNA level also increase the expression of complex IV and the activity of complex I and complex IV of the ETC (81).
and all downstream oxidants (79). We recently reviewed the mechanisms by which melatonin improves mitochondrial respiration and increases ATP synthesis under physiological and poisonous conditions (80). Consequently, the antioxidant and free-radical-scavenging capacities of melatonin protect proteins of the ETC and mtDNA from ROS/RNS-induced oxidative damage. This protective effect limits the loss of intramitochondrial GSH, improves ETC activity, and reduces mtDNA damage. Melatonin’s actions at the mtDNA level also increase the expression of complex IV and the activity of complex I and complex IV of the ETC (81).
Melatonin against iNOS and ONOO−
In many inflammatory processes, ONOO− rather than other reactive molecules is the predominant molecule that determines the fate of cells. Once formed by the coupling of NO and  , ONOO− cannot be removed or scavenged by vitamins E or C, or by other conventional antioxidants. As a multifunctional antioxidant, however, melatonin and its metabolites have unique features not shared by the usual antioxidants, including iNOS-inhibitory (82–86) and ONOO−-scavenging (49,54,62,87,88) properties. These features of melatonin, apart from direct antioxidative effects, have been documented in chemical-induced hyperglycemia (89–91) and other circumstances such as colitis (84), liver and lung damage (83), and alkylating-agent toxicity (55,56,92,93). Thus, melatonin is the only currently available molecule known to block all aspects of the “devil’s triangle.”
, ONOO− cannot be removed or scavenged by vitamins E or C, or by other conventional antioxidants. As a multifunctional antioxidant, however, melatonin and its metabolites have unique features not shared by the usual antioxidants, including iNOS-inhibitory (82–86) and ONOO−-scavenging (49,54,62,87,88) properties. These features of melatonin, apart from direct antioxidative effects, have been documented in chemical-induced hyperglycemia (89–91) and other circumstances such as colitis (84), liver and lung damage (83), and alkylating-agent toxicity (55,56,92,93). Thus, melatonin is the only currently available molecule known to block all aspects of the “devil’s triangle.”
Melatonin has been shown to ameliorate inflammation by blocking transcriptional factors and TNF-α (94,95). A large body of evidence confirms that these cytokines induce formation of free radicals and promote iNOS activity and transcriptional factor activation within cells. These events inevitably induce a vicious cycle of cellular damage. In the case of ONOO−-induced DNA damage, PARP overactivates in a genome-repair process and consumes NAD+ as a substrate, causing an energy crisis within cells, leading to their eventual necrosis. Preservation of NAD+ and cellular energy production may facilitate PARP to repair the DNA damage rather than blocking PARP; melatonin preserves cellular energy production (96–98) and protects against DNA damage (99).
Under physiologic conditions in resting cells, eNOS-derived NO suppresses both iNOS and cyclooxygenase-2 (COX-2) expression by reducing NF-κB translocation into the nucleus. However, it is well documented that NO derived from iNOS during inflammatory processes further potentiates COX-2 activity through the NF-κB pathway (100), thereby exaggerating the inflammatory process. This effect is not unexpected given that ONOO− directly activates COX-2 as well (101). In the case of chronic inflammation, inhibition of COX-2 and iNOS (rather than COX-2 only) would be beneficial in reducing the severity of inflammation. A recent, intriguing report (102) suggests that neither tryptophan nor serotonin, but only melatonin, inhibits COX-2 and iNOS transcriptional activation.
Another advantage of melatonin over classical antioxidants is its lack of prooxidative actions. All classical antioxidants are potential electron donors and they exhibit both reduced and oxidized forms. Once they donate an electron to neutralize a free radical, they are transformed from a reduced to an oxidized state. Usually, the oxidized form will be regenerated to the reduced state through the mechanism known as redox reaction or recycling. In this pathway, the recycling of vitamin C or vitamin E occurs at the expense of GSH. In many cases, however, GSH is a better antioxidant than either vitamin C or vitamin E (103). Because these antioxidants are electron donors and exhibit redox reactions, their oxidized forms also can oxidize other molecules. Therefore, classical antioxidants are prooxidants. Melatonin sacrifices itself and does not participate in redox cycling after scavenging free radicals. As previously mentioned, melatonin not only does not consume cellular GSH, it also preserves or even increases the content of GSH in tissues. Thus, melatonin is classified as a suicidal or terminal antioxidant (104).
Concluding Remarks
Melatonin has been administered in both physiological and pharmacological amounts to humans and animals, and there is widespread agreement that it is a nontoxic molecule (105). In pregnant rats, the maternal lowest-no-observed-effect level was found to be 200 mg/kg/day, and the developmental no-observed-adverse-effect level was ≥200 mg/kg/day (106). Melatonin is easily synthesized in a pharmacologically pure form and is inexpensive and affordable; thus, because of its versatility in protecting against nitrooxidative stress and reducing inflammation, melatonin could have significant potential to improve public health (107).
Disclosure
We declare that the authors have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Harman D. (2006) Free radical theory of aging, an update: increasing the functional life span. Ann. N. Y. Acad. Sci. 1067:10–21.
Harman D. (2003) The free radical theory of aging. Antioxid. Redox. Signal. 5:557–61.
de Grey AD. (2006) Free radicals in aging: causal complexity and its biomedical implications. Free Radic. Res. 40:1244–9.
O’Keefe JH Jr, Cordain L. (2004) Cardiovascular disease resulting from a diet and lifestyle at odds with our Paleolithic genome: how to become a 21st-century hunter-gatherer. Mayo Clin. Proc. 79:101–8.
Diczfalusy E. (2006) Our common future: a rapidly growing and rapidly aging humankind. Aging Male 9:125–34.
Eisenberg DM, et al. (1998) Trends in alternative medicine use in the United States, 1990–1997: results of a follow-up national survey. JAMA 280:1569–75.
Woo JJ. (2007) Adverse event monitoring and multivitamin-multimineral dietary supplements. Am. J. Clin. Nutr. 85:323S–4S.
Muntwyler J, Hennekens CH, Manson JE, Buring JE, Gaziano JM. (2002) Vitamin supplement use in a low-risk population of US male physicians and subsequent cardiovascular mortality. Arch. Intern. Med. 162:1472–6.
Ascherio A, et al. (1999) Relation of consumption of vitamin E, vitamin C, and carotenoids to risk for stroke among men in the United States. Ann. Intern. Med. 130:963–70.
Lonn E, et al. (2002) Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 25:1919–27.
Ward NC, et al. (2007) The effect of vitamin E on blood pressure in individuals with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. J. Hypertens. 25:227–34.
Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. (2003) Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 361:2017–23.
Miller ER 3rd, et al. (2005) Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 142:37–46.
Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. (2007) Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA 297:842–57.
Ceriello A. (2006) Controlling oxidative stress as a novel molecular approach to protecting the vascular wall in diabetes. Curr. Opin. Lipidol. 17:510–8.
Beckman JS, Koppenol WH. (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 271:C1424–37.
Pacher P, Beckman JS, Liaudet L. (2007) Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87:315–424.
Weidig P, McMaster D, Bayraktutan U. (2004) High glucose mediates pro-oxidant and antioxidant enzyme activities in coronary endothelial cells. Diabetes Obes. Metab. 6:432–41.
Cooke CL, Davidge ST. (2002) Peroxynitrite increases iNOS through NF-kappaB and decreases prostacyclin synthase in endothelial cells. Am. J. Physiol. Cell Physiol. 282:C395–402.
Stockklauser-Farber K, Ballhausen T, Laufer A, Rosen P. (2000) Influence of diabetes on cardiac nitric oxide synthase expression and activity. Biochim. Biophys. Acta 1535:10–20.
Moncada S, Palmer RM, Higgs EA. (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43:109–42.
Dedon PC, Tannenbaum SR. (2004) Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 423:12–22.
Joshi MS, et al. (2002) Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc. Natl. Acad. Sci. U. S. A. 99:10341–6.
Forstermann U, Munzel T. (2006) Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113:1708–14.
Demicheli V, Quijano C, Alvarez B, Radi R. (2007) Inactivation and nitration of human superoxide dismutase (SOD) by fluxes of nitric oxide and superoxide. Free Radic. Biol. Med. 42:1359–68.
Alvarez B, Radi R. (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25:295–311.
Alvarez B, Radi R. (2001) Peroxynitrite decay in the presence of hydrogen peroxide, mannitol and ethanol: a reappraisal. Free Radic. Res. 34:467–75.
Szabo C. (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol. Lett. 140–141:105–12.
Okamoto T, et al. (2001) Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J. Biol. Chem. 276:29596–602.
Wu J, et al. (2001) Enhanced vascular permeability in solid tumor involving peroxynitrite and matrix metalloproteinases. Jpn. J. Cancer Res.. 92:439–51.
Whang YE, et al. (1998) Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. U. S. A. 95:5246–50.
Juedes MJ, Wogan GN. (1996) Peroxynitrite-induced mutation spectra of pSP189 following replication in bacteria and in human cells. Mutat. Res. 349:51–61.
Masuda M, Nishino H, Ohshima H. (2002) Formation of 8-nitroguanosine in cellular RNA as a biomarker of exposure to reactive nitrogen species. Chem. Biol. Interact. 139:187–97.
Tamir S, deRojas-Walker T, Wishnok JS, Tannenbaum SR. (1996) DNA damage and genotoxicity by nitric oxide. Methods Enzymol. 269:230–43.
Yoshie Y, Ohshima H. (1997) Nitric oxide syner-gistically enhances DNA strand breakage induced by polyhydroxyaromatic compounds, but inhibits that induced by the Fenton reaction. Arch. Biochem. Biophys. 342:13–21.
Chaturvedi N, et al. (1998) Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus. Lancet 351:28–31.
Yermilov V, Yoshie Y, Rubio J, Ohshima H. (1996) Effects of carbon dioxide/bicarbonate on induction of DNA single-strand breaks and formation of 8-nitroguanine, 8-oxoguanine and base-propenal mediated by peroxynitrite. FEBS Lett. 399:67–70.
Chien YH, Bau DT, Jan KY. (2004) Nitric oxide inhibits DNA-adduct excision in nucleotide excision repair. Free Radic. Biol. Med. 36:1011–7.
Virag L, Szabo E, Gergely P, Szabo C. (2003) Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol. Lett. 140–141:113–24.
Szabo C. (2006) Poly(ADP-ribose) polymerase activation by reactive nitrogen species—relevance for the pathogenesis of inflammation. Nitric Oxide 14:169–79.
Korkmaz A, Yaren H, Topal T, Oter S. (2006) Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch. Toxicol. 80:662–70.
Korkmaz A, Topal T, Oter S. (2007) Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis; implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol. Toxicol. 23:303–12.
Yaren H, et al. (2007) Lung toxicity of nitrogen mustard may be mediated by nitric oxide and peroxynitrite in rats. Res. Vet. Sci. 83:116–22.
Korkmaz A, et al. (2005) Peroxynitrite may be involved in bladder damage caused by cyclophosphamide in rats. J. Urol. 173:1793–6.
de la Lastra CA, Villegas I, Sanchez-Fidalgo S. (2007) Poly(ADP-ribose) polymerase inhibitors: new pharmacological functions and potential clinical implications. Curr. Pharm. Des. 13:933–62.
Korkmaz A, et al. (2008) Effects of poly(ADP-ribose) polymerase inhibition in bladder damage caused by cyclophosphamide in rats. Exp. Biol. Med. (Maywood) 233:338–43.
Tan DX, et al. (2003) Melatonin: a hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J. Pineal Res. 34:75–8.
Reiter RJ, et al. (2007) Medical implications of melatonin: receptor-mediated and receptor-independent actions. Adv. Med. Sci. 52:11–28.
Reiter RJ, Tan DX, Burkhardt S. (2002) Reactive oxygen and nitrogen species and cellular and organismal decline: amelioration with melatonin. Mech. Ageing Dev. 123:1007–19.
Reiter RJ, Tan DX, Allegra M. (2002) Melatonin: reducing molecular pathology and dysfunction due to free radicals and associated reactants. Neuro. Endocrinol. Lett. 23(Suppl 1):3–8.
Reiter RJ, et al. (2003) Melatonin: detoxification of oxygen and nitrogen-based toxic reactants. Adv. Exp. Med. Biol. 527:539–48.
Lopez-Burillo S, Tan DX, Mayo JC, Sainz RM, Manchester LC, Reiter RJ. (2003) Melatonin, xanthurenic acid, resveratrol, EGCG, vitamin C and alpha-lipoic acid differentially reduce oxidative DNA damage induced by Fenton reagents: a study of their individual and synergistic actions. J. Pineal Res. 34:269–77.
Sudnikovich EJ, et al. (2007) Melatonin attenuates metabolic disorders due to streptozotocin-induced diabetes in rats. Eur. J. Pharmacol. 569:180–7.
Gilad E, Cuzzocrea S, Zingarelli B, Salzman AL, Szabo C. (1997) Melatonin is a scavenger of peroxynitrite. Life Sci. 60:PL169–74.
Ucar M, et al. (2007) Melatonin alleviates lung damage induced by the chemical warfare agent nitrogen mustard. Toxicol. Lett. 173:124–31.
Topal T, et al. (2005) Melatonin ameliorates bladder damage induced by cyclophosphamide in rats. J. Pineal Res. 38:272–7.
Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. (2007) One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 42:28–42.
Reiter RJ, Tan DX, Terron MP, Flores LJ, Czarnocki Z. (2007) Melatonin and its metabolites: new findings regarding their production and their radical scavenging actions. Acta. Biochim. Pol. 54:1–9.
Reiter RJ, et al. (2008) Biogenic amines in the reduction of oxidative stress: melatonin and its metabolites. Neuro Endocrinol. Lett. 29:391–8.
Peyrot F, Houee-Levin C, Ducrocq C. (2006) Melatonin nitrosation promoted by NO*2; comparison with the peroxynitrite reaction. Free Radic. Res. 40:910–20.
Zhang H, Squadrito GL, Uppu R, Pryor WA. (1999) Reaction of peroxynitrite with melatonin: a mechanistic study. Chem. Res. Toxicol. 12:526–34.
Soung DY, et al. (2004) Peroxynitrite scavenging activity of indole derivatives: interaction of indoles with peroxynitrite. J. Med. Food 7:84–9.
Rodriguez C, et al. (2004) Regulation of antioxidant enzymes: a significant role for melatonin. J. Pineal Res. 36:1–9.
Reiter RJ, Tan DX, Maldonado MD. (2005) Melatonin as an antioxidant: physiology versus pharmacology. J. Pineal Res. 39:215–6.
Winiarska K, Fraczyk T, Malinska D, Drozak J, Bryla J. (2006) Melatonin attenuates diabetes-induced oxidative stress in rabbits. J. Pineal Res. 40:168–76.
Baydas G, Canatan H, Turkoglu A. (2002) Comparative analysis of the protective effects of melatonin and vitamin E on streptozocin-induced diabetes mellitus. J. Pineal Res. 32:225–30.
Wahab MH, Akoul ES, Abdel-Aziz AA. (2000) Modulatory effects of melatonin and vitamin E on doxorubicin-induced cardiotoxicity in Ehrlich ascites carcinoma-bearing mice. Tumori 86:157–62.
Montilla P, et al. (2001) Melatonin versus vitamin E as protective treatment against oxidative stress after extra-hepatic bile duct ligation in rats. J. Pineal Res. 31:138–44.
Hsu C, Han B, Liu M, Yeh C, Casida JE. (2000) Phosphine-induced oxidative damage in rats: attenuation by melatonin. Free Radic. Biol. Med. 28:636–42.
Gultekin F, Delibas N, Yasar S, Kilinc I. (2001) In vivo changes in antioxidant systems and protective role of melatonin and a combination of vitamin C and vitamin E on oxidative damage in erythrocytes induced by chlorpyrifos-ethyl in rats. Arch. Toxicol. 75:88–96.
Rosales-Corral S, et al. (2003) Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta pep-tide in rat brain: a comparative, in vivo study versus vitamin C and E. J. Pineal Res. 35:80–4.
Anwar MM, Meki AR. (2003) Oxidative stress in streptozotocin-induced diabetic rats: effects of garlic oil and melatonin. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 135:539–47.
Forrest CM, Mackay GM, Stoy N, Stone TW, Darlington LG. (2007) Inflammatory status and kynure-nine metabolism in rheumatoid arthritis treated with melatonin. Br. J. Clin. Pharmacol. 64:517–26.
Kedziora-Kornatowska K, et al. (2008) Antioxidative effects of melatonin administration in elderly primary essential hypertension patients. J. Pineal Res. 45:312–7.
Tamura H, et al. (2008) Oxidative stress impairs oocyte quality and melatonin protects oocytes from free radical damage and improves fertilization rate. J. Pineal Res. 44:280–7.
Tomas-Zapico C, Coto-Montes A. (2005) A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J. Pineal Res. 39:99–104.
Korkmaz A, Reiter RJ. (2008) Epigenetic regulation: a new research area for melatonin? J. Pineal Res. 44:41–4.
Korkmaz A, Sanchez-Barcelo EJ, Tan DX, Reiter RJ. (2008) Role of melatonin in the epigenetic regulation of breast cancer. Breast Cancer Res. Treat. 2008, Jul 1 [Epub ahead of print].
Brown GC. (1992) Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J. 284(Pt 1):1–13.
Reiter RJ, Paredes SD, Korkmaz A, Jou MJ, Tan DX. (2008) Melatonin combats molecular terrorism at the mitochondrial level. Interdisc. Toxicol. 1:137–49.
Leon J, Acuna-Castroviejo D, Escames G, Tan DX, Reiter RJ. (2005) Melatonin mitigates mitochondrial malfunction. J. Pineal Res. 38:1–9.
Gilad E, et al. (1998) Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: role of inhibition of NFkappaB activation. FASEB J. 12:685–93.
Crespo E, et al. (1999) Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysac-charide-induced multiple organ dysfunction syndrome in rats. FASEB J. 13:1537–46.
Dong WG, et al. (2003) Effects of melatonin on the expression of iNOS and COX-2 in rat models of colitis. World J. Gastroenterol. 9:1307–11.
Rodriguez MI, et al. (2007) Chronic melatonin treatment prevents age-dependent cardiac mito-chondrial dysfunction in senescence-accelerated mice. Free Radic. Res. 41:15–24.
Lopez LC, et al. (2006) Identification of an inducible nitric oxide synthase in diaphragm mitochondria from septic mice: its relation with mitochondrial dysfunction and prevention by melatonin. Int. J. Biochem. Cell Biol. 38:267–78.
Teixeira A, et al. (2003) Melatonin protects against pro-oxidant enzymes and reduces lipid peroxidation in distinct membranes induced by the hydroxyl and ascorbyl radicals and by peroxynitrite. J. Pineal Res. 35:262–8.
Mayo JC, et al. (2005) Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), in macrophages. J. Neuroimmunol. 165:139–49.
Rao VS, Santos FA, Silva RM, Teixiera MG. (2002) Effects of nitric oxide synthase inhibitors and melatonin on the hyperglycemic response to streptozotocin in rats. Vascul. Pharmacol. 38:127–30.
Montilla PL, et al. (1998) Oxidative stress in diabetic rats induced by streptozotocin: protective effects of melatonin. J. Pineal Res. 25:94–100.
Korkmaz A, Topal T, Oter S, Tan DX, Reiter RJ. (2008) Hyperglycemia-related pathophysiologic mechanisms and potential beneficial actions of melatonin. Mini Rev. Med. Chem. 8:1144–53.
Sadir S, Deveci S, Korkmaz A, Oter S. (2007) Alpha-tocopherol, beta-carotene and melatonin administration protects cyclophosphamide-induced oxidative damage to bladder tissue in rats. Cell Biochem. Funct. 25:521–6.
Yildirim I, Korkmaz A, Oter S, Ozcan A, Oztas E. (2004) Contribution of antioxidants to preventive effect of mesna in cyclophosphamide-induced hemorrhagic cystitis in rats. Cancer Chemother. Pharmacol. 54:469–73.
Li JH, et al. (2005) Melatonin reduces inflammatory injury through inhibiting NF-kappaB activation in rats with colitis. Mediators Inflamm. 2005:185–93.
Reiter RJ. (2003) Melatonin: clinical relevance. Best Pract. Res. Clin. Endocrinol. Metab. 17:273–85.
Dugo L, et al. (2001) Effect of melatonin on cellular energy depletion mediated by peroxynitrite and poly (ADP-ribose) synthetase activation in an acute model of inflammation. J. Pineal Res. 31:76–84.
Tan DX, et al. (2005) Interactions between melatonin and nicotinamide nucleotide: NADH preservation in cells and in cell-free systems by melatonin. J. Pineal Res. 39:185–94.
Lopez LC, Escames G, Ortiz F, Ros E, Acuna-Castroviejo D. (2006) Melatonin restores the mitochondrial production of ATP in septic mice. Neuro. Endocrinol. Lett. 27:23–630.
Tan DX, et al. (1993) The pineal hormone melatonin inhibits DNA-adduct formation induced by the chemical carcinogen safrole in vivo. Cancer Lett. 70:65–71.
Mollace V, Muscoli C, Masini E, Cuzzocrea S, Salvemini D. (2005) Modulation of prostaglandin biosynthesis by nitric oxide and nitric oxide donors. Pharmacol. Rev. 57:217–52.
Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. (1996) Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 93:15069–74.
Deng WG, Tang ST, Tseng HP, Wu KK. (2006) Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108:518–24.
Regoli F, Winston GW. (1999) Quantification of total oxidant scavenging capacity of antioxidants for peroxynitrite, peroxyl radicals, and hydroxyl radicals. Toxicol. Appl. Pharmacol. 156:96–105.
Tan DX, et al. (2002) Chemical and physical properties and potential mechanisms: melatonin as a broad spectrum antioxidant and free radical scavenger. Curr. Top. Med. Chem. 2:181–97.
Seabra ML, Bignotto M, Pinto LR Jr, Tufik S. (2000) Randomized, double-blind clinical trial, controlled with placebo, of the toxicology of chronic melatonin treatment. J. Pineal Res. 29:193–200.
Jahnke G, et al. (1999) Maternal and developmental toxicity evaluation of melatonin administered orally to pregnant Sprague-Dawley rats. Toxicol. Sci. 50:271–9.
Reiter R, Gultekin F, Flores LJ, Terron MP, Tan DX. (2006) Melatonin: potential utility for improving public health. TAF Prev. Med. Bull. 5:131–58.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Korkmaz, A., Reiter, R.J., Topal, T. et al. Melatonin: An Established Antioxidant Worthy of Use in Clinical Trials. Mol Med 15, 43–50 (2009). https://doi.org/10.2119/molmed.2008.00117
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2008.00117