
Abstract
Patients with chronic hepatitis C frequently have serum and hepatic iron overload, but the mechanism is unknown. Recently identified hepcidin, exclusively synthesized in the liver, is thought to be a key regulator for iron homeostasis and is induced by infection and inflammation. This study was conducted to determine the hepatic hepcidin expression levels in patients with various liver diseases. We investigated hepcidin mRNA levels of liver samples by real-time detection-polymerase chain reaction; 56 were hepatitis C virus (HCV) positive, 34 were hepatitis B virus (HBV) positive, and 42 were negative for HCV and HBV (3 cases of autoimmune hepatitis, 7 alcoholic liver disease, 13 primary biliary cirrhosis, 9 nonalcoholic fatty liver disease, and 10 normal liver). We analyzed the relation of hepcidin to clinical, hematological, histological, and etiological findings. Hepcidin expression levels were strongly correlated with serum ferritin (P < 0.0001) and the degree of iron deposit in liver tissues (P < 0.0001). Hepcidin was also correlated with hematological parameters (vs. hemoglobin, P = 0.0073; vs. serum iron, P = 0.0012; vs. transferrin saturation, P < 0.0001) and transaminase levels (P = 0.0013). The hepcidin-to-ferritin ratio was significantly lower in HCV+ patients than in HBV+ patients (P = 0.0129) or control subjects (P = 0.0080). In conclusion, hepcidin expression levels in chronic liver diseases were strongly correlated with either the serum ferritin concentration or degree of iron deposits in the liver. When adjusted by either serum ferritin values or hepatic iron scores, hepcidin indices were significantly lower in HCV+ patients than in HBV+ patients, suggesting that hepcidin may play a pivotal role in the pathogenesis of iron overload in patients with chronic hepatitis C.
Similar content being viewed by others


Introduction
Iron is an essential element for all living organisms, being a requirement in a wide range of metabolic processes including DNA synthesis, oxygen transport, and energy production, but excess iron can be harmful to the organism, in part through the generation of oxygen radicals, and is potentially lethal (1). Therefore, iron homeostasis must be tightly regulated in all organisms. Recent work has established the importance of the peptide hormone hepcidin in iron homeostasis as a negative regulator of iron release into the system by duodenal enterocytes and reticuloendothelial macrophages (2,3). Hepcidin binds to the iron exporter ferroportin, which results in ferroportin internalization and degradation (4). In addition to its response to iron homeostasis, hepcidin is induced by inflammation (5), an effect believed to be dependent on cytokine production (6); how hepcidin levels are kept in balance through upstream signaling pathways is still under investigation.
Iron accumulation in the liver, where hepcidin is exclusively synthesized, is common in patients with chronic liver diseases (7), especially in patients with chronic hepatitis C virus (HCV) infection (8,9). Increased hepatic iron concentration is present in 10% to 36% of patients with chronic hepatitis C (8,9), and hepatic iron overload is even more common among patients with end-stage liver disease due to hepatitis C (10,11). Excess iron deposition in the liver is known to be hepatotoxic and may exacerbate liver injury (12) and be resistant to interferon-based therapy in patients with chronic hepatitis C (13,14); however, little is known about the mechanism of iron accumulation in the liver. We previously reported that transferrin receptor 2 (TfR2), which was recently identified as the second receptor for transferrin (15), was higher in the liver of patients with chronic hepatitis C compared than those with chronic hepatitis B (16). TfR2 function was thought at first to be consistent with its homology to classical transferrin receptor-TfR1 (15,17); iron uptake by the hepatocytes [TfR2 has limited tissue distribution, with prominent expression in the liver, especially in hepatocytes (15,18)]. But a disabling mutation in the TfR2 gene in humans (19,20) and mice (18) leads to significant hepatic iron accumulation despite an absence of expression of TfR2, suggesting that iron uptake by the hepatocytes is not a principal role of TfR2, and that TfR2 has a important but unknown role for maintenance of iron homeostasis. Recently, Kawabata et al. (21) demonstrated that hepcidin is downregulated in TfR2 mutant mice, suggesting that TfR2 may be the upstream sensor for hepcidin production in the pathway of iron homeostasis.
In view of these considerations, we decided to examine the gene expression of hepcidin in liver samples from patients with various liver conditions. In addition, we assessed the relationship of hepcidin gene expression with clinical, hematological, histological, and etiological findings.
Materials and Methods
Liver Samples
We used 132 liver samples in this study. Fifty-six samples were obtained from patients with chronic HCV infection (HCV+ group; positive serum HCV RNA and negative serum hepatitis B surface antigen, or HBsAg), and 34 were from patients with chronic hepatitis B virus (HBV) infection [HBV+ group; positive serum HBsAg and negative serum HCV antibody]. Forty-two samples were obtained from patients without HCV or HBV infections (HCV−HBV− group; 3 cases of autoimmune hepatitis, 7 alcoholic liver disease with ethanol intake > 80 g/day [range, 95–155 g/day] in the 10 years that preceded the hospital admission, 13 primary biliary cirrhosis, 9 nonalcoholic fatty liver disease, and 10 normal liver). In cases of normal liver, surgically resected liver specimens were obtained during operation for metastatic liver cancer, while the other liver specimens were obtained by needle biopsy for diagnosis of chronic liver diseases. Liver biopsy was performed under stable conditions, without fever of unknown etiology, and before any interferon therapy. Social drinkers were included in HCV+ and HBV+ groups. Serum iron parameters were determined by routine automated laboratory methods on the day of liver tissue sampling. History of blood transfusions, use of iron-containing medications, and daily consumption of alcohol were investigated in all patients. Informed consent was obtained from each patient, and the study was approved by the Mie University Ethics Committee and carried out according to the guidelines of the 1975 Declaration of Helsinki.
Histological Evaluation
Liver biopsy specimens were divided in two parts. One portion was fixed in buffered formalin and embedded in paraffin for histological examination, and the other was immediately frozen and stored at −80°C for RNA extraction. Hematoxylin & eosin, Masson’s trichrome, and Perls’ Prussian blue staining for iron were performed. Liver histology was evaluated by two pathologists who were blinded to clinical conditions or hepcidin expression levels. Liver specimens were scored for stage of liver fibrosis and grade of inflammatory activity according to the classification of Desmet et al. (22). The histological quantification of iron was done according to Deugnier et al. (23) by scoring iron separately within hepatocytes (hepatic iron score, or HIS, 0 to 36), sinusoidal cells (sinusoidal iron score, or SIS, 0 to 12), and portal tracts or fibrotic tissue (portal iron score, or PIS, 0 to 12). The total iron score (TIS), 0 to 60, was the sum of these scores. This score is highly correlated with the biochemical hepatic iron index and hepatic iron concentration measured by atomic absorption spectrophotometry in patients with chronic liver diseases (24,25).
mRNA Preparation and Reverse Transcription
mRNA was extracted from liver tissue using the SV RNA Isolation System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, liver tissue samples were homogenized in a Dounce glass homogenizer with lysis buffer (4 M guanidine thiocyanate, 10 mM Tris-HCl, pH 7.5, 0.97% β-mercaptoethanol). Dilution buffer (350 µL) was added to the homogenized solution and incubated at 70°C for 3 min. After separating by centrifugation, the supernatant was precipitated using ethanol. RNA was treated with DNase (Boehringer Mannheim, Mannheim, Germany) to remove any contaminating genomic DNA. Precipitated RNA was purified by spin column assembly and dissolved in 100 µL DEPC-treated water containing 10 mM DTT and 200 U/mL RNase inhibitor using a siliconized tube. The amount of mRNA was determined by spectrophotometry. The cDNA was generated by reverse-transcription of 2 µg adjusted RNA, with random hexamers and Moloney murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA, USA) for 30 min at 48°C.
Quantification of Hepatic Hepcidin mRNA Expression Levels by RTD-PCR
The hepatic mRNA levels of hepcidin were determined by TaqMan real-time detection-polymerase chain reaction (RTD-PCR). RTD-PCR primers and probes were designed using Primer Express software (Applied Biosystems, Tokyo, Japan), which spanned introns to avoid coamplification of genomic DNA. The sequences were forward primer 5′-TTCCCCATCTGCATTTTCTG-3′, reverse primer 5′-TCTACGTCTTGCAGC ACATCC-3′, and FAM/TAMURA probe 5′-TGCGGCTGCTGTCATCGATCAA-3′. cDNA of liver tissue (5 µL) was incubated with 20.75 µL TaqMan Master Mix (Perkin Elmer, Yokohama. Japan) (8% glycerol; 10× TaqMan buffer; 10 mM each dCTP, dATP, and dGTP; 20 mM dUTP; 0.01 U/µL AmpErase uracil N-glycosylase; 25 mM MgCl2; and 0.025 U/µL AmpliTaq Gold DNA polymerase), 10 µM forward primer, 10 µM reverse primer, and 5 µM probe. The reaction mixture was brought up to a final volume of 50 µL with RNase-free distilled water. The amplification was performed using the ABIPRISM 7700 sequence detection system (Applied Biosystems). Amplification conditions were 2 min at 50°C, 10 min at 95°C, 53 cycles of 15 s at 95°C, and then 1 min at 60°C. Data were analyzed using Sequence Detector 1.6 software (Applied Biosystems). The results for hepcidin mRNA are expressed as the amount relative to that of GAPDH mRNA quantified simultaneously in each liver sample. Experiments were performed in triplicate, and the amount of RNA was calculated from a standard curve drawn using serial dilutions of total RNA extracted from a wedge-resected liver specimen. Standard samples were run in parallel during each analysis. This method is able to measure hepcidin mRNA linearly from 102 to more than 105/GAPDH mRNA.
Statistical Analysis
Data are expressed as the median and range or mean ± standard deviation. Categorical variables were compared using the chi-squared test or Fisher exact test. Continuous variables were compared using the Student t test, one-way factorial ANOVA test, Wilcoxon rank-sum test, and Kruskal-Wallis test. Correlation was assessed by Spearman rank correlation. Two-sided P values < 0.05 were considered statistically significant. Calculations were performed using StatView of JMP software (SAS Institute).
Results
Clinical Characteristics of the Patients
Clinical characteristics of patients in this study according to viral infection (HCV+, HBV+, and HCV−HBV−) are shown in Table 1. There was no statistical difference in age and sex distribution in the three groups. Serum hemoglobin, iron levels, transferrin saturation, and ferritin levels were significantly higher in HCV+ group than in the HCV−HBV− non-iron overload subgroup, but there was no statistical difference between the HCV+ group and the HCV−HBV− iron overload subgroup (elevated serum ferritin levels, > 220 ng/mL for men and > 100 ng/mL for women). Regarding liver histological findings, patients with chronic viral infection (HCV+ and HBV+) had more progressive grading and staging scores than patients without viral infection (HCV−HBV−). Iron deposition in the liver was prominent in chronic HCV-infected patients, and TIS scores were significantly higher in HCV+ than in HBV+ or HCV−HBV− non-iron overload patients.
Correlation between Hepatic Hepcidin mRNA Expression Levels and Clinical Findings
Hepatic mRNA expression levels of hepcidin were measurable in all 132 patients. Correlation of clinical findings with hepatic hepcidin expression levels, normalized by GAPDH mRNA, was evaluated, and the results summarized in Table 2. Patients’ age was not related to hepcidin expression levels. Hepcidin levels were significantly higher in men than in women (10,900 ± 13,100, median 6590, vs. 5820 ± 10,700, median 2600, P = 0.0012, Wilcoxon rank-sum test). Hepcidin levels were not significantly different between the HCV+, HBV+, and HCV−HBV− groups (HCV+, 7790 ± 7850, median 5730; HBV+, 8950 ± 13,900, median 3290; HCV−HBV−, 9560 ± 15,700, median 3150) (Figure 1). Serum alanine aminotransferase (r = 0.281, P = 0.0013) (Figure 2a) and aspartate aminotransferase (r = 0.249, P = 0.0043) levels were marginally correlated with hepcidin mRNA levels when all patients were included in the statistical evaluation.

Hepatic mRNA expression levels of hepcidin in various liver diseases. Hepatic hepcidin mRNA expression levels were compared between HCV+, HBV+, and HCV−HBV− groups. Graphs depict the median (line within the box), 25th to 75th percentiles (upper and lower border of the box), and 10th and 90th percentiles (whiskers).

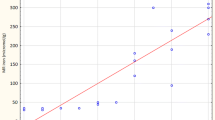
Correlations between hepatic hepcidin mRNA expression levels and clinical laboratory and histological data in patients with various liver diseases. Relationship between hepcidin mRNA expression levels and serum alanine aminotransferase levels (a), serum iron levels (b), transferrin saturation (c), serum ferritin levels (d), and total iron score of liver tissues (e) in 132 patients. ○, HCV+; □, HBV+; △, HCV−HBV−.
Serum bilirubin concentrations were marginally correlated with hepcidin levels (r = 0.239, P = 0.0065), but serum albumin and hyaluronic acid were not correlated (Table 2). Hepcidin expression levels were correlated with hemoglobin concentrations (r = 0.235, P = 0.0073), serum iron levels (r = 0.283, P = 0.0012) (Figure 2b), and serum transferrin saturation (r = 0.360, P < 0.0001) (Figure 2c). Serum ferritin levels were strongly and positively correlated with hepatic hepcidin mRNA expression levels in various patients with liver disease (r = 0.832, P < 0.0001) (Figure 2d). In this study, 4 patients (3 HCV−HBV− and 1 HCV+;enclosed by open square in Figure 2d), had relatively low hepcidin expression levels with severe hyperferritinemia, suggesting the possibility of hepcidin dysregulation in these patients. Clinical features of these cases are summarized in Table 3. When the exceptional cases were excluded from analysis because of possible hemochromatosis traits, the correlation statistic was improved to r = 0.916 and P < 0.0001 (n = 128). There were no significant correlations between hepatic hepcidin mRNA levels and inflammatory activity score or fibrosis staging in liver biopsy specimens. A significant positive correlation was found between hepatic hepcidin mRNA expression levels and the degree of iron deposition in the liver as evaluated by TIS score (r = 0.457, P < 0.0001) (Figure 2e).
Comparison of mRNA Expression Levels of Hepcidin in Patients with Various Liver Diseases
Hepcidin expression has been reported to increase in response to iron overload in an experimental setting (2,26). Our findings of strong positive correlations between hepatic hepcidin mRNA expression levels and serum ferritin and hepatic iron levels also suggest feedback to hepcidin expression against iron overload in humans, and the ratio of hepcidin levels per iron overload may be constant in various liver conditions. To evaluate the relative amounts of hepcidin in relation to iron overload, we calculated the ratio of liver hepcidin mRNA/serum ferritin levels in each patient. The ratio of hepcidin/serum ferritin was significantly lower in the HCV+ group (26.8 ± 15.4, median 23.3) than in the HBV+ group (46.5 ± 41.0, median 31.6; P = 0.0129) or the HCV−HBV− group (44.7 ± 33.7, median 34.0; P = 0.0080) (Figure 3a). The relative amount of hepatic hepcidin per hepatic iron deposition (the ratio of hepcidin mRNA/TIS) was also significantly lower in the HCV+ group (1280 ± 2160, median 773) than in the HBV+ group (2920 ± 4600, median 1130; P = 0.0392) or the HCV−HBV− group (3320 ± 4210, median 1870; P = 0.0098) (Figure 3b). The ratios of hepcidin/serum ferritin and hepcidin/TIS were not statistically different between the HBV+ and HCV−HBV− groups. These results indicate that hepatic hepcidin expression levels in relation to body iron store are lower in HCV-infected patients compared with HBV-infected or uninfected patients.

Relative hepatic mRNA expression levels of hepcidin in various liver diseases. (a) The ratio of hepatic hepcidin mRNA/serum ferritin was calculated in each patient and compared between HCV+, HBV+, and HCV−HBV− groups. (b) The ratio of hepatic hepcidin mRNA/hepatic total iron score was calculated in each patient and compared between HCV+, HBV+, and HCV−HBV− groups. Graphs depict the median (line within the box), 25th to 75th percentiles (upper and lower border of the box), and 10th and 90th percentiles (whiskers).
Discussion
Hepcidin, exclusively synthesized in the liver, was originally isolated from human serum and urine as having an antimicrobial activity (27,28). The lack of hepcidin expression in knockout mice leads to iron overload (29), and conversely, overexpression of hepcidin in transgenic mice causes severe iron deficiency (30). Moreover, hepcidin mutations are associated with a new type of severe juvenile hemochromatosis not related to HFE mutations (31). In normal mice, iron overload increases and iron deficiency decreases hepatic mRNA expression of hepcidin; change in hepcidin expression is associated with inverse changes in intestinal iron absorption (26,32). Recently, it was demonstrated that hepcidin can bind ferroportin, the major cellular iron exporter protein, inducing ferroportin internalization and degradation and resulting in reduced iron efflux from enterocytes (4). Thus, hepcidin is thought to be a major negative regulatory hormone for iron homeostasis.
In the present study, we investigated hepcidin expression in the liver of patients with chronic liver diseases and normal subjects and analyzed correlations between clinical parameters and hepatic hepcidin expression levels. Hepcidin mRNA levels were strongly and positively correlated with serum ferritin levels and the degree of hepatic iron accumulation as assessed by TIS. These results are consistent with presently elucidated hepcidin functions; when iron storage increases, serum ferritin elevates, and hepatic hepcidin is upregulated, leading to decreased intestinal iron absorption for maintenance of iron homeostasis. Our study demonstrated a relationship between serum hemoglobin, iron, transferrin saturation levels, and hepcidin mRNA levels, supporting the hypothesis of an impact of anemia and/or hypoxia on hepcidin expression, as reported in mice (5,33). However, we did not find a significant correlation between red blood cell count or hematocrit levels and hepcidin levels in our study, suggesting additional regulatory mechanisms of hepcidin may exist. Serum alanine aminotransferase and aspartate aminotransferase levels were positively correlated with hepatic hepcidin expression levels, indicating that hepatic inflammatory status also may influence the expression levels of hepcidin in patients with chronic liver disease, although the histological inflammatory activity score was not related to hepcidin levels. We examined the relationship between parameters reflecting hepatic function and hepcidin expression. Although serum albumin levels and hepatic fibrosis status (histological staging and hyaluronic acid) were not correlated with hepcidin levels, serum bilirubin was positively correlated with hepcidin. This correlation may be caused by the positive relationship of serum bilirubin levels and accumulation of hepatic iron in our patients (bilirubin vs. TIS, r = 0.305, P = 0.0005; Spearman). Further study is necessary to determine the participation of hepcidin in liver function.
From these results, that several clinical factors were associated with hepatic hepcidin expression, it is suggested that multiregulatory mechanisms act to alter hepatic expression of hepcidin. First, our results of a strong positive relationship between hepatic hepcidin expression and serum ferritin and liver TIS leads to the idea that hepcidin is regulated by body-stored iron, most strongly. Second, hepcidin expression is regulated by sensing the iron amount in circulation, because hepcidin levels were also correlated with serum iron and transferrin saturation levels. It is reported that hepcidin expression is greatly diminished in TfR2-mutated hemochromatosis patients (34) and mice (21) despite elevated iron stores. Thus, TfR2 appears to be an upstream regulator of hepcidin and is required for hepcidin to respond appropriately to changes in serum transferrin saturation. Therefore, TfR2 may act as a communicator between iron status in serum and hepcidin production.
Third, inflammatory status in the liver may influence hepatic hepcidin expression. Hepcidin is the key mediator of anemia due to chronic inflammation (3,5), and its mRNA expression is increased in response to inflammatory stimuli such as lipopolysaccharide or interleukin-6 (IL-6) (6,33). Recently, it was demonstrated that Kuppfer cells, which release IL-6, are required for the activation of hepcidin synthesis during inflammation in the liver, but not for regulatory activity by iron conditions (35). The positive correlation between hepatic hepcidin levels and serum aminotransferase levels in our study also supports the existence of this regulatory pathway, although serum IL-6 levels were not evaluated in our study. Thus, there are at least three major, distinct mechanisms for regulation of hepcidin: by body-stored iron condition, serum iron status, and inflammatory condition.
Previous studies demonstrated that chronic HCV infection is frequently associated with elevated serum and liver iron storage markers (8–11). In our study, serum iron levels and transferrin saturation in HCV+ patients were significantly higher than those without viral infection, and TIS was significantly higher in the HCV+ group than in the HBV+ or HCV− HBV− groups. Hepatocellular iron uptake may be upregulated during chronic inflammation. It has been previously demonstrated that inflammatory cytokines enhance TfR1-mediated iron uptake by hepatocytes (36). However, considering that the serum aminotransferase levels and hepatic inflammatory score are not statistically different between HCV+ and HBV+ groups and that TIS is more prominent in HCV+ than in HBV+ patients, chronic hepatic inflammation alone does not seem to be responsible for hepatic iron accumulation in patients with chronic hepatitis C. HCV infection itself seems to have a direct influence on hepatic iron accumulation. In this study, hepcidin expression in relation to serum ferritin and the hepatic TIS were significantly lower in the HCV+ group than in the HBV+ or HCV−HBV− groups, suggesting that upregulation of hepatic hepcidin expression by increased body-stored iron may be relatively diminished in the HCV-infected liver. We previously reported that TfR2 expression in the liver was significantly higher in the HCV+ patients than in the HBV+ patients (16), although its clinical implication for iron accumulation in the liver of patients with chronic hepatitis C was unknown. It was reported that TfR2 disabling mutations in humans (34) and mice (21) decrease hepcidin expression. Therefore, at first we expected that hepcidin expression would be higher in HCV patients (higher TfR2 expression group) than the other patients; the result was opposite. Because hepcidin and TfR2 expression in the liver were inversely correlated in our study (data not shown), upregulation of TfR2 may involve the downregulation of hepcidin in the liver of patients with chronic hepatitis C. Further in vitro study is required to clarify the distinct interaction between TfR2, hepcidin, and the degree of intracellular iron content in hepatocytes during chronic HCV infection.
Patients with known HFE genetic hemochromatosis expected to exhibit abnormal hepcidin regulation (37,38) were excluded in this study, because no patient had HFE mutations C282Y or H63D. We could not role out the other types of hereditary hemochromatosis completely (caused by mutations of hepcidin, hemojuvelin, TfR2, and ferroportin) (39). In this study, four patients had relatively low hepatic hepcidin expression levels with severe hyperferritinemia, suggesting hemochromatosis traits (Table 3). In Japan, one patient with HFE-hemochromatosis and a few patients with non-HFE-hemochromatosis and ferroportin disease have been reported during the last 10 years (40–43). Therefore, further investigation should be necessary in these patients with hepcidin dysregulation.
Aoki et al. (44) also reported that hepatic hepcidin expression is increased in response to iron overload in patients with chronic hepatitis C. We have extended the measurement of hepcidin expression in HBV+ and noninfected patients and compared it to data from HCV+ patients, showing the relatively low levels of hepcidin in patients with chronic hepatitis C.
In conclusion, we evaluated hepcidin mRNA expression in the liver of patients with various liver conditions. Despite the heterogeneity of our patients, hepcidin levels were related to hepatic and body iron stores, hematological parameters, and serum transaminase levels, suggesting that multiregulatory mechanisms act in hepcidin production. Relatively low levels of hepatic hepcidin expression for the degree of iron burden may be involved in the pathophysiologic mechanism of increased iron overload in patients with chronic hepatitis C, and supplementation of hepcidin may be beneficial for these conditions.
References
Hentze MW, Muckenthaler MU, Andrews NC. (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–97.
Nicoras G, Viatte L, Bennoun M, Beaumont C, Kahn A, Vaulont S. (2002) Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 29:327–35.
Ganz T. (2003) Hepcidin: a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 102:783–8.
Nemeth E et al. (2004) Hepcidin regulates iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–3.
Nicolas G et al. (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 110:1037–44.
Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. (2004) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113:1271–6.
Di Bisceglie AM, Axiotis CA, Hoofnagle JH, Bacon BR. (1992) Measurements of iron status in patients with chronic hepatitis. Gastroenterology 102:2108–13.
Boucher E, Bourienne A, Adams P, Turlin B, Brissot B, Deugnier Y. (1997) Liver iron concentration and distribution in chronic hepatitis C before and after interferon treatment. Gut 41:115–20.
Metwally MA, Zein CO, Zein NN. (2004) Clinical significance of hepatic iron deposition and serum iron values in patients with chronic hepatitis C infection. Am. J. Gastroenterol. 99:286–91.
Tharburn D et al. (2002) The role of iron and haemochromatosis gene mutations in the progression of liver disease in chronic hepatitis C. Gut 50:248–52.
Tung BY, Emond MJ, Bromer MP, Raaka SD, Cotler SJ, Kowdley KV. (2003) Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology 124:318–26.
Bassett SE et al. (1999) Effects of iron loading on pathogenicity in hepatitis C virus-infected chimpanzees. Hepatology 29:1884–92.
Olynyk JK et al. (1995) Hepatic iron concentration as a predictor of response to interferon alfa therapy in chronic hepatitis C. Gastroenterology 108:1104–9.
Souza RM, Freitas LAR, Lyra AC, Moraes CF, Braga EL, Lyra LGC. (2006) Effect of iron overload on the severity of liver histologic alterations and on the response to interferon and ribavirin therapy of patients with hepatitis C infection. Braz. J. Med. Biol. Res. 39:79–83.
Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP. (1999) Molecular cloning of transferrin receptor 2: a new member of the transferrin receptor-like family. J. Biol. Chem. 274:20826–32.
Takeo M, Kobayashi Y, Fujita N, et al. (2005) Up-regulation of transferrin receptor 2 and ferroportin 1 mRNA in the liver of patients with chronic hepatitis C. J. Gastroenterol. Hepatol. 20: 562–9.
Kawabata H, Germain RS, Vuong PT, Nakamaki T, Said JW, Koeffler HP. (2000) Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J. Biol. Chem. 275:16618–25.
Fleming RE et al. (2000) Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. U. S. A. 97:2214–9.
Camaschella C et al. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25:14–5.
Roetto A et al. (2001) New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 97:2555–60.
Kawabata H et al. (2005) Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 105:376–81.
Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. (1994) Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology 19:1513–20.
Deugnier YM et al. (1992) Liver pathology in genetic hemochromatosis: a review of 135 homozygous cases and their bioclinical correlations. Gastroenterology 102:2050–9.
Deugnier YM et al. (1993) Differentiation between heterozygotes and homozygotes in genetic hemochromatosis by means of a histological hepatic iron index: a study of 192 cases. Hepatology 17:30–4.
Silvia TSS et al. (2005) Iron overload in patients with chronic hepatitis C virus infection: clinical and histological study. J. Gastroenterol. Hepatol. 20:243–8.
Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loreal O. (2001) A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 276:7811–9.
Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. (2000) LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 480:147–50.
Park CH, Valore EV, Waring AJ, Ganz T. (2001) Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 276:7806–10.
Nicolas G, Beennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. (2001) Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. U. S. A. 98:8780–5.
Nicolas G et al. (2002) Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc. Natl. Acad. Sci. U. S. A. 99:4596–601.
Roetto A et al. (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 33:21–2.
Frazer DM, Wilkins SJ, Becker EM, Vulpe CD, Mckie AT, Trinder D, Anderson GJ. (2002) Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology 123:835–44.
Lee P, Peng H, Gelbart T, Beutler E. (2004) The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta2-microglobulin-deficient hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 101:9263–5.
Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. (2005) Hepcidin is decreased in TfR2-hemochromatosis. Blood 105:1803–6.
Montosi G et al. (2005) Kuppfer cells and macrophages are not required for hepatic hepcidin activation during iron overload. Hepatology 41:545–52.
Kobune M, Kohgo Y, Kato J, Miyazaki E, Niitsu Y. (1994) Interleukin-6 enhances hepatic transferrin uptake, and ferritin expression in rats. Hepatology 19:1468–75.
Ahmad KA et al. (2002) Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cell Mol. Dis. 29:361–6.
Bridle KR et al. (2003) Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homeostasis. Lancet 361:669–73.
Pietrangelo A. (2005) Non-HFE hemochromatosis. Semin. Liver Dis. 25:450–60.
Sohda T, Okubo R, Kamimura S, Ohkawara T. (2001) Hemochromatosis with HFE gene mutation in a Japanese patient. Am. J. Gastroenterol. 96:2487–8.
Koyama C et al. (2005) Two novel mutations, L490R and V561X, of the transferrin receptor 2 gene in Japanese patients with hemochromatosis. Hematologica 90:302–7.
Koyama C et al. (2005) Three patients with middle-age-onset hemochromatosis caused by novel mutations in the hemojuvelin gene. J. Hepatol. 43: 740–2.
Koyama C et al. (2005) A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern. Med. 44:990–3.
Aoki CA, Rossaro L, Ramsamooj R, Brandhagen D, Burritt MF, Bowlus CL. (2005) Liver hepcidin mRNA correlates with iron stores, but not inflammation, in patients with chronic hepatitis C. J. Clin. Gastroenterol. 39:71–4.
Acknowledgments
This study was supported by a grant-in-aid (no. 18590728, 2006-2007) from the Ministry of Education, Science and Culture of Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fujita, N., Sugimoto, R., Takeo, M. et al. Hepcidin Expression in the Liver: Relatively Low Level in Patients with Chronic Hepatitis C. Mol Med 13, 97–104 (2007). https://doi.org/10.2119/2006-00057.Fujita
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2006-00057.Fujita