
Abstract
It is well known that hydrogen plays a key role in p-type doping of GaN. It is believed that H passivates substitutional Mg during growth by forming a Mgs-N-Hi complex; in subsequent annealing, H is removed, resulting in p-type doping. Several open questions have remained, however, such as experimental evidence for other complexes involving Mg and H and difficulties in accounting for the relatively high-temperature anneal needed to remove H. We present first principles calculations in terms of which we show that the doping process is in fact significantly more complex. In particular, interstitial Mg plays a major role in limiting p-type doping. Overall, several substitutional/interstitial complexes form and can bind H, with vibrational frequencies that account for hitherto unidentified observed lines. We predict that these defects, which limit doping efficiency, can be eliminated by annealing in an atmosphere of H and N prior to the final anneal that removes H.
Similar content being viewed by others


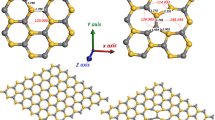
Introduction
GaN is the most promising wide-gap material for blue-green optoelectronics, but further improvements are needed to enhance performance and reliability for commercial applications. A major issue is to increase p-type conductivity. It has been established that H enhances the incorporation of dopants such as Mg, but it must be removed by post-growth annealing to activate the dopants.[1,2] In general, only a relatively small fraction of the total Mg is activated.[3,4]
Theory has offered a simple account of the process[5,6]: In the absence of H, Mg shallow acceptors are compensated by N vacancies (VN +) and Ga interstitials (Gai +++), both of which are donors and have low formation energies in p-type material.[7] When H is present, it passivates substitutional Mg (MgGa −) by forming a MgGa-N-H complex.[5] The Fermi level rises to the midgap region and the formation of VN + and Gai +++ is suppressed. Subsequent annealing removes H and activates MgGa −. Theory predicted[5] the vibrational frequency of the MgGa-N-H complex at 3360 cm−1 and, experiments[4] have since found a line at 3125 cm−1 whose intensity decreases during annealing. Nevertheless, there are unambiguous indications that the doping process is more complex. The temperature needed to remove H (∼700°C) is much higher than expected from the calculated energy to break the MgGa-N-H bond (∼1.5 eV).[5] Photoluminescence, infra-red, and Raman data show clearly that other Mg-related defects are present.[1,3,4] In particular, in material grown by molecular beam epitaxy (MBE), where typically only 10% of Mg is electrically active, infrared and Raman lines in the 2000-cm−1 range have been attributed to direct Mg-H bonds.[3] Theory so far has not offered any potential candidates for these complexes.
Here, we report first-principles calculations of key defect reactions and show that p-type doping is in fact a significantly more complex process. In particular, we show that, in addition to MgGa-N-H, interstitial Mg (Mgi) and its complexes play a major role in controlling the process. In Ga-rich growth conditions, compensation of MgGa − occurs mainly though the formation of Mg substitutional-interstitial pairs with a configuration (MgGa-N-Mg)+ analogous to MgGa-N-H (Fig. 1a). Other defects that play a role are VN +, Mgi ++, Gai +++, (MgGa-N-Ga+)++, and MgGa-VN. This plethora of substitutional/interstitial (SI) Mg-related complexes accounts for the high degree of Mg incorporation in electrically inactive forms. When H is present, all these complexes are passivated, forming direct Ga-H and Mg-H bonds. The calculated vibrational frequencies of these bonds are in the 2000-cm−1 range and provide an identification for infrared and Raman lines that have remained unexplained.[3] Finally, our analysis yields a testable prediction: For samples that show the vibrational signatures of hydrogenated SI complexes, a pre-anneal in an atmosphere of N and H followed by annealing in a N atmosphere would significantly enhance Mg activation.

Schematic representation of some complexes studied in this work: a) MgGa-N-H complex; b) MgiH2; c) MgGa-N-Gai-H2; and d) MgGa-N-Mgi-H.
Method
The calculations were performed in the framework of density functional theory and the local density approximation for exchange and correlation, using ultrasoft pseudopotentials and a plane-wave basis set[8] The Ga 3d electrons and the Mg 2p electrons were treated as valence electrons. Additional details for the pseudopotentials can be found in Ref. 9. The energy cutoff for the basis set was chosen at 25 Ry after studies showed convergent results. Supercells of 32 atoms where used in general but some key results were tested in a 64 cubic cell to ensure convergence with supercell size. Such supercells were also found to be well converged in similar cases by earlier investigators.[9,10]
We used three special k points corresponding to the hexagonal symmetry[11] in the wurtzite phase and the {.5,.5,.5} special k point in the cubic structure. Results for cubic and wurtzite GaN are generally the same (within 0.1 eV) except in some cases where topology matters. Those cases will be discussed explicitly. All atoms in the supercell were relaxed until the force on each atom was less than 0.15 eV/A. The calculation of the H-related normal modes involved a partial dynamical matrix that included the H atoms and their neighbors. The normal modes were calculated in the harmonic approximation. In a second step, the anharmonic corrections for the transition from the ground to the first excited state of the stretch modes were calculated in a standard way[12] using the coefficients of the polynomial that fits the energy as a function of the amplitude along the normal mode trajectory.
Results
Much of the theoretical literature on defects and impurities in GaN has so far focused on formation energies. However, in a compound semiconductor the formation energies of point defects are functions of the chemical potential of one of the host atoms and the Fermi energy. In the presence of impurities, formation energies of defect complexes are also functions of the impurity chemical potentials. To avoid much of this complexity, we carried out our analysis by examining key sequences of defect reactions that can be studied by using the same supercell containing identical atomic species and excess charges. The resulting reaction energies are more accurate than absolute formation energies of individual defects, especially charged ones. Values for the latter are available in the literature.[5-7,9,10] We will use these values in our analysis, especially the more accurate calculations that treat the Ga d electrons as valence electrons.
We start with the doping process in the absence of H. For p-type material and Ga-rich conditions, VN + and Gai +++ have low formation energies and comparable concentrations.[7] These native defects are deep donors and their electrons recombine with holes so that p-type doping is thwarted. We have taken the next step and investigated the possible reactions of VN + and Gai +++ with MgGa −. We found the following:
a) MgGa-VN pairs form with a small gain in energy (0.4 eV) with respect to the isolated point defects.
b) (MgGa-N-Gai)++ complexes form with a large gain in energy (1.6 eV). These complexes have a structure very similar to that of the MgGa-N-H (Fig.1a) complexes that have been studied extensively by Van de Walle and Neugebauer. In particular, the Gai occupies the antibonding site on the N side of a MgGa-N bond. Once the complex (MgGa-N-Gai)++ forms, additional energy is gained by an exchange between the MgGa − and Gai +++ to yield interstitial Mg (Mgi ++). The additional energy gain is 1.7 eV, for a net gain of 3.3 eV.
Once Mgi ++ are available, they pair up with isolated MgGa − to form (MgGa-N-Mgi)+ complexes. This pairing yields an extra energy gain of 0.7 eV, for a large net energy gain of 4.0 eV. (Table I).

Relative formation energies of different defects in p-type GaN in Ga-rich conditions. A doped crystal with a concentration of sustitutional MgGa − and a Gai +++ or VN+ are the zero energy references as indicated.
We emphasize that the results of Table 1 are obtained with identical supercells (same atoms, same charge states) in each case so that the relative energies listed are very accurate. For an absolute comparison of the energies of interstitial- and vacancy-related complexes one needs the absolute formation energies of Gai +++ and VN +, which represent a major challenge. Boguslawski et al. have reported the most accurate calculations that current capabilities allow and found the two energies to be comparable.[7] Note, however, that, even if the formation energy of Gai +++ were to be larger than that of VN + by as much as 2.5 eV,[5] the SI complexes just described would have to be taken into account to properly describe the physics of p-type doping.
The clear conclusion then is that the entire family of SI defects play a role in controlling doping. Under equilibrium Ga-rich conditions in the absence of other impurities, (MgGa-N-Mgi)+ is the dominant defect responsible for thwarting p-type doping, followed by Mgi ++, and (MgGa-N-Ga)++. In p-type material under equilibrium N-rich conditions, Gai are suppressed in favor of other native defects[5,7] that do not exchange with MgGa. On the other hand, if thermodynamic equilibration does not occur during low-temperature growth, the formation of defects is governed by kinetics so that any or all stable defects may form irrespective of their formation energies.
We turn now to the case when H is present. First, we confirmed the known result that MgGa −binds an H atom in a MgGa-N-H configuration. In addition, the SI complexes bind one or two H atoms, depending on their formal charge, and Gai +++ binds up to three H atoms. In particular, we carried out detailed calculations on the following complexes (MgGa-N-Gai-H)+, MgGa-N-Gai-H2, (MgiH)+, MgiH2, and MgGa-N-Mgi-H. In this context we note that the distinction between cubic and hexagonal GaN is important: the geometries of some of the hydrogenated SI complexes are quite different in the two isomorphs (we will discuss the geometries later in the paper). From here on we will confine our discussion to hexagonal GaN, which is the material that is most commonly used for experiments and device fabrication.
There is a substantive difference between MgGa-N-H and the hydrogenated SI (SI-H) com-plexes: In MgGa-N-H, H acts as a donor constituent, whereas in the SI-H complexes H acts as an acceptor constituent. The latter have occupied energy levels in the gap which is consistent with higher formation energies (Table II). Nevertheless, as we shall see below, the SI-H complexes play a key role in the non-equilibrium processing that is needed to achieve p-type doping.

Relative formation energies of H-compensated defects (in eV). The H chemical potential is chosen to be the formation energy of H2 in an interstitial site.
Infrared and Raman spectra[3] have found several lines in the vicinity of 2150 cm−1 that were initially attributed to MgGa-H stretch modes because they where absent in control samples without Mg.[3] Subsequent theory, however, found that H does not bind directly to MgGa , but rather forms the MgGa-N-H complex whose calculated vibrational frequency is 3360 cm−1.[5] In recent experiments, Gotz et al.[4] detected the 3125-cm−1 infrared line that corresponds to MgGaN-H and observed it decrease as a function of annealing that activates Mg acceptors. They did not report measurements in the 2000-cm−1 range. Earlier work had proposed that VN + may be the dominant compensating defect. Upon hydrogenation, VN + traps an H atom but their calculated frequency is less than 600 cm−1.[6] Accordingly, the identification of the vibrational lines in the vicinity of 2150-cm−1 has remained an important open problem.
We calculated the vibrational frequencies of several SI-H complexes and indeed found Mg-H and Ga-H in the vicinity of 2000-cm−1. The uncertainty in the calculations is ∼200 cm−1 so that identification of the observed defects cannot be made by simply comparing theoretical and experimental vibrational frequencies. Symmetry and other considerations, however, help narrow the choices. We discuss here the strongest candidates to account for the experimental data.
MgiH2 is a stable complex with the Mgi near the hexagonal (O) interstitial site[7] (see Fig. 1b) and the two H’s are along the c direction. The point symmetry is C3v. There is no reflection symmetry along the c direction and the two Mg-H bond lengths are different, namely 1.55 A and 1.78 A . The two” spring constants” are also different and, as a result, the two H’s oscillate almost independently of each other. The short Mg-H bond has a stretch mode at 2001 cm−1. The long Mg-H bond has a stretch mode at 1570 cm−1. The wag-mode frequencies are 1370 cm−1 and 1200 cm−1 respectively. The entire ``molecule” oscillates with frequencies lower than 500 cm−1.
Turning back to the short-bond stretch mode, we note that the movement of the atoms is in the c direction, making it difficult to detect by infrared absorption if the wave vector of the incident light is oriented in the c direction. These considerations and the calculated frequencies make the short-bond stretch mode of the MgiH2 complex an excellent candidate for the Raman lines[3] at 2151 cm−1 and 2185 cm−1 that are not seen by infrared experiments. The long-bond stretch mode frequency is too small to explain the second peak of the pair in the experimental data. The second peak in the pair might be due to an anharmonic coupling between the stretch mode and the vibration of the whole molecule. The frequencies of these” molecular” modes are low enough for their first excited states to be populated at room temperature. If this is the case, the relative amplitude of these peaks should be a function of the temperature.
We now consider the complex MgGa-N-Gai-H2. In this defect, the H’s (see Fig. 1c) are again located on either side of Gai (see Fig. 1b). Again we found a short Ga-H bond (1.50 A) and a long Ga-H bond (1.82 A). The symmetry of the defect is no longer C3v because of the presence of MgGa. Consequently the Gai-H bonds are not oriented exactly in the c direction so that the stretch modes are accessible to infrared spectroscopy when the wave vector of the light is in the c direction. We obtained a frequency of 2270 cm−1 for the short-bond stretch mode and 1500 cm−1 for the long-bond stretch mode. The high-frequency mode is a strong candidate to explain the second pair of peaks observed experimentally at 2168 cm−1 and 2219 cm−1. Here again the pair of excitations might be due to the influence of anharmonic couplings.
Finally, in Fig. 1d we show schematically the structure of MgGa-N-Mgi-H. The stretch mode frequency of this complex is calculated to be 1320 cm−1. We are not aware of experimental data in this range of frequencies. Measurements in the range 1000-2000 cm−1 would be highly desirable to test the theoretical predictions.
We now discuss the implications of the above results on understanding the doping process during growth by MBE or chemical vapor deposition (CVD). During growth by MBE, the temperatures are relatively low (∼800°C) and H is present unintentionally.[3] Experiments have shown that p-type doping is achieved with about 10% of the Mg in the sample being active. In material with very high Mg content (1020 cm−3), moderate H content and low doping efficiency, distinct infrared and Raman lines have been observed in the vicinity of 2200 cm−1,[3] which we have now identified as due to SI-H complexes. We note that our analysis, having identified these SI complexes as limiting doping efficiency, also suggests a process to eliminate the undesirable SI-H complexes and increase doping efficiency: Annealing in a H-rich atmosphere or, better yet, in an atmosphere rich in both H and N, will lead to the conversion of the SI-H complexes to MgGa-N-H complexes. This result follows naturally from the fact the SI-H complexes have larger formation energies (Table 2). The N atmosphere helps by suppressing formation of Gai and VN, both of which are detrimental to p-type doping,[5,7] and favors formation of VGa whose presence helps convert Mgi to MgGa. A subsequent anneal in a N-rich atmosphere would activate Mg as in the case of the CVD-grown films[1] (see also below).
During CVD growth of GaN, H is introduced in ample quantities at relatively high temperatures (∼1000 °C). If the material were fully equilibrated, the dominant hydrogenated defect would indeed be MgGa-N-H as suggested by earlier theory[5] and confirmed by Table 2. There are strong indications, however, that the material is generally not fully equilibrated.
In their pioneering and extensive studies of CVD GaN, Nakamura et al.[1] found photoluminescence peaks which they attributed to Mg-H complexes. These experiments determine the energy levels of the underlying defects but provide no structural information. Theoretical results on energy levels are not reliable for identification purposes. It would be highly desirable to correlate the luminescence data with infrared and/or Raman data to facilitate identification.
It is possible that, by empirical means, suppression of SI-H defects is currently achieved in the growth of high-quality GaN. Nevertheless, systematic infrared and Raman data in both the 2000-cm−1 and 3000-cm−1 range would help unravel further the complexity of p-type doping. For example, SI-H defects are a natural candidate to account for the fact that annealing CVD material to remove H requires higher temperatures than theory can account for in terms of breaking the H bonds in the MgGa-N-H complexes.[5] The rate-limiting step for Mg activation may very well be determined by the conversion of SI-H complexes to MgGa-N-H. We, therefore, propose the following: CVD samples should be checked for the presence of SI-H complexes via their infrared signatures in the 2000-cm−1 range. If present, a preanneal in a H and N atmosphere followed by annealing in an N atmosphere would again produce optimum activation.
Conclusions
We have shown that complexes involving self-interstitials and interstitial impurities play a major role in the p-type doping process. The theoretical prediction of these defects provides an identification of the infrared and Raman lines observed in MBE-grown material. The combined experimental and theoretical information suggests that the control of these defects is the ultimate key in achieving higher p-type conductivity. Systematic experimental studies, especially using infrared and Raman spectroscopies, guided by the present theoretical results, would offer the promise of achieving higher p-type conductivity. The most important conclusion of the present work is that an annealing stage in H-rich atmosphere followed by annealing in a N-rich atmosphere would generally improve p-type doping efficiency. We anticipate that similar defect complexes may also form in the case of other dopants such as Be and possibly in the case of n-type dopants as well as in other wide-gap semiconductors.
References
S. Nakamura, T. Mukai, and M. Senoh, Appl. Phys Lett. 64, 1687 (1994); S. Nakamura {etet al.}, Jpn. J. Appl. Phys. 35, L74 (1996).
J. A. Van Vechten, Zook, J. D., R. D. Horning and B. Goldenberg, Jpn. J. Appl. Phys. 31, 3662 (1992); M. Johnson and D. P. Bour, Appl. Phys. Lett. 68, 3470 (1996).
M.S. Brandt {etet al.}, Appl. Phys Lett. 64, 2264 (1994); M. S. Brandt {etet al.}, Phys. Rev. B 49, 14758 (1994) and references therein.
W. Goetz, N. M. Johnson, J. Walker, D. P. Bour, and R. A. Street, Appl. Phys. Lett. 68, 667 (1996); W. Goetz, N. M. Johnson, D. P. Bour, M. D. McCluskey, and E. E. Haller, Appl. Phys. Lett. 69, 3725 (1996); W. Goetz, N
J. Neugebauer and C. G. Van de Walle, Phys. Rev. B 50, 8067 (1994); Phys. Rev. Lett. 75, 4452 (1995); ibid., Appl. Phys. Lett. 68, 1829 (1996).
C. G. Van de Walle, Phys Rev. B 56, R10020 (1997).
P. Boguslawski, E. L. Briggs, and J. Bernholc, Phys. Rev. B 51, 17255 (1995).
M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys. 64, 1045 (1992).
F. Bernardini, V. Fiorentini, and A. Bosin, Appl. Phys. Lett. 70, 2990 (1997).
C. H. Park and D. J. Chadi, Phys. Rev. B 55, 12995 (1997).
D. J. Chadi, and M. L. Cohen, Phys. Rev. B 8, 5747 (1973).
L. D. Landau and E. M. Lifshitz, Quantum Mechanics, (Pergamon, Oxford, 1997).
Acknowledgments
The authors would like to thank V. Fiorentini and A. Filippetti for providing us the ultrasoft pseudopotentials and M. Ferconi for related discussions. This work was supported in part by the National Science Foundation Grant DMR9803768 and by the William A. and Nancy F. McMinn Endowment at Vanderbilt University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Reboredo, F.A., Pantelides, S.T. Defect Complexes and Non-Equilibrium Processes Underlying the P-Type Doping of GaN. MRS Internet Journal of Nitride Semiconductor Research 4 (Suppl 1), 508–513 (1999). https://doi.org/10.1557/S1092578300002969
Published:
Issue Date:
DOI: https://doi.org/10.1557/S1092578300002969