
Abstract
We present density–functional theory studies for a variety of surfaces and extended defects in GaN. According to previous theoretical studies1{10\(\bar 1\)0} type surfaces are electrically inactive. They play an important role in GaN since similar configurations occur at open–core screw dislocations and nanopipes as well as at the core of threading edge dislocations. Domain boundaries are found to consist of four–fold coordinated atoms and are also found to be electrically inactive. Thus, except for full–core screw dislocations which possess heavily strained bonds all investigated extended defects do not induce deep states into the band–gap. However, electrically active impurities in particular gallium vacancies and oxygen related defect complexes are found to be trapped at the stress field of the extended defects.
Similar content being viewed by others

1. Introduction
GaN has recently been the subject of considerable interest due to its optoelectronic properties. In particular the wide band gap (3.4 eV for wurtzite GaN) makes blue light applications feasible. Defect–induced electronic states in the band gap can significantly alter the optical performance. This fact becomes extremely important in laser devices, where parasitic components in the emission spectrum are highly undesirable. Moreover, point defects could be trapped in the stress field of extended defects giving rise to charge accumulated in the vicinity. The resulting electrostatic field leads to electron scattering which will severely affect the electron mobility (see Look and Sizelove2 for a recent model). Therefore, there is considerable interest in understanding the microstructure of extended defects in GaN and their interaction with point defects.
In this paper we present the geometries, energetics and electrical properties of extended defects in GaN using an ab initio local density–functional (LDF) cluster method, AIMPRO, and a self–consistent charge density–functional tight–binding method SCC–DFTB. The latter can be used in large supercells and enables the formation energy of the defects to be found. Details of the methods and their application to GaN and oxygen related defect complexes in GaN have been given previously3–5 and will not be repeated here.
2. Threading Dislocations in GaN
A. Threading Screw Dislocations
We consider first a screw dislocation with a full core3. Full core screw dislocations have recently been observed by Xin et al. using the high resolution Z–contrast imaging technique6. The presence of atoms so close to the dislocation axis leads to severely strained bond lengths distorted by as much as 0.4 Å. Consequently it is not surprising that such dislocations possess deep gap states ranging from E v +0:9 to Ev+1:6 eV and shallow states around Ec − 0:2 eV. An analysis of these gap states revealed that the states above Ev are localised on N core atoms, whereas those below E c are localised on both Ga and N core atoms. The strong distortion of the bonds of the core atoms leads to a high line energy within our calculations.
A similar calculation was then carried out with the hexagonal core of the screw dislocation removed giving a core with a narrow opening of ≈7.2 Å. The atoms on the walls adopt three fold coordinations similar to those found on the (10\(\bar 1\)0) surface. Thus Ga (N) atoms develop sp 2 (p 3) hybridisations which lower the surface energy and clean the gap from deep states1. Removing further material from the core resulted in a significantly higher line energy. Thus our calculations allow only diameters very much smaller than the diameter of nanopipes and we suppose that their formation is not due to Frank’s mechanism.
B. Threading Edge Dislocations
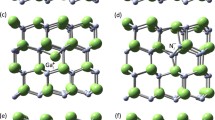
The relaxed core of a threading edge dislocation with Burgers vector a[1\(\bar 1\)10]/3 is shown in Fig. 1. It consists of a line of Ga and N atoms which have 3-fold coordination. With respect to the perfect lattice the distance between columns (1/2) and (3/4) [and the equivalent on the right] are 9 % contracted while the distance between columns (9/10) and (7/8) [and the equivalent on the right] are 13 % stretched. This atomic geometry for the threading edge dislocation has recently been confirmed by Xin et al. 6 using atomic resolution Z–contrast imaging. Consistent with our calculation they determined a contraction (stretching) of 15±10 % of the distances between the columns at the dislocation core. See table 1 for the details of the geometry. The three-fold coordinated Ga (N) atoms (labelled 1 and 2 in Fig. 1 move in such a way to enhance sp 2 and p 3 hybridisation, respectively. This leads to empty Ga lone pairs pushed towards E c, and filled lone pairs on N atoms lying near Ev, in a manner identical to the (10\(\bar 1\)0) surface. Thus threading edge dislocations are then also electrically inactive except possibly for shallow levels. It seems that the small bond distortions within the core makes it energetically uneconomic for the core of the threading edge dislocation to be open. We obtain a line energy for the edge dislocation which is by more than a factor of two lower than that found for the screw dislocation. This is mainly due to the lower elastic energy arising from the shorter Burgers vector.

Top view (in [0001]) of the relaxed core of the threading edge dislocation (\({\rm{b}} = {1 \over 3}\left[ {1\bar 210} \right]\)). This geometry has recently been con rmed using the atomic resolution Z–contrast imaging technique6. The three fold coordinated atoms 1 (Ga) and 2 (N) adopt a hybridasation similar to the (10\(\bar 1\)0) surface atoms.

Bond lengths, min-max (average) in Å and bond angles, min-max (average) in for the most distorted atoms at the core of the threading edge dislocation (\(\left( {{\text{b}} = \frac{1}{3}\left[ {1\bar 210} \right]} \right)\)). Atom numbers refer to Fig. 1.
In AlN, Wright et al. 7 have found, using a supercell geometry, that edge dislocations have an empty band of levels ∼ E c – 2 eV, while in n-type material, the core contains VA1 defects. The very much larger band gap of this material might be responsible for the differences of the properties of the edge dislocations from those in GaN.
To summarise, the density functional calculations reveal that the threading screw dislocations in their pure, i.e. impurity free form can exist with full cores and narrow open cores with diameters up to ≈7.2 Å. While full core screws are electrically active, open core screws induce no deep gap states. Threading edge dislocations in wurtzite GaN are stable with full cores and electrically inactive. However, the strained and ‘dangling’ bonds present in their cores could permit impurities and intrinsic defects to be trapped there.
3. Interaction of Oxygen with dislocations
A. Oxygen and Open Core Screw Dislocations
There is experimental evidence that oxygen acts as a donor in bulk GaN8 and total energy calculations show that O sits on a N site9. Since the internal surfaces of screw dislocations are very similar to those of the low energy (10\(\bar 1\)0) surface, we investigated10 the likely surface sites for oxygen replacing N atoms. We found that the energy of a neutral ON defect is 0.8 eV lower at the relaxed (10\(\bar 1\)0) surface. This shows that there is a tendency for O to segregate to the surface. The added oxygen has an additional electron occupying a state near Ec. The defect has therefore a high energy and would attract acceptors resulting in a neutral complex. One possible acceptor, other than added dopants, would be a gallium vacancy (VGa) which acts as a triple acceptor and has been calculated to have a low formation energy in n–type GaN11 , 12. Consequently, we suppose that the surface oxygen concentration could be sufficiently large, and the oxygen atoms sufficiently mobile, that the three N neighbours of VGa at the (10\(\bar 1\)0) surface are replaced by O forming the VGa–(ON)3 defect.
Our calculations10 showed that VGa–(ON )3 is more stable at the surface than in the bulk by 2.15 eV. Two O neighbours of the surface vacancy lie below the surface and each is bonded to three Ga neighbours, but the surface O is bonded to only two subsurface Ga atoms in a normal oxygen bridge site. The defect is electrically inactive with the O atoms passivating the vacancy in the same way as VH4 in Si.
The question then arises as to the in uence of the defect on the growth of the material. Growth over the defect must proceed by adding a Ga atom to the vacant site but this leaves three electrons in shallow levels near the conduction band resulting in a very high energy. This suggests that the defect can stabilise the surface and thus inhibit growth. From this we can conclude that such defects lead to the formation of nanopipes if we assume that during growth of the epilayers, either nanopipes with very large radii are formed which gradually shrink when their surfaces grow out, or there is a rapid drift of oxygen to a preexisting nanopipe. In either case the concentration of oxygen and VGa–(ON )3 defects increases at the walls of the nanopipe. The maximum concentration of this defect would be reached if 50% (100%) of the first (second) layer N atoms were replaced by O and further growth then would be prevented. It is, however, likely that far less than the maximum concentration is necessary to stabilise the surface and make further shrinkage of the nanopipe impossible. Provided oxygen could di use to the surface fast enough, the diameter and density of the holes would be related to the initial density of oxygen atoms in the bulk. This model requires that the walls of the nanopipe are coated with oxygen although the initial stages of formation of the pipe are obscure.
In conclusion, we have shown that oxygen has a tendency to segregate to the (10\(\bar 1\)0) surface and forms stable and chemically inert VGa–(ON)3 defects. These defects increase in concentration when the internal surfaces grow out. When a critical concentration of the order of a monolayer is reached, further growth is prevented. This model leads to nanopipes with (10\(\bar 1\)0) walls coated with GaO and supports the suggestions of Liliental-Weber et al. that nanopipes are linked to the presence of impurities13.
B. Oxygen and Edge Dislocations
The VGa–(ON)3 defect considered above is electrically inactive at a (10\(\bar 1\)0) surface site but defects like VGa–(ON)2 and VGa−ON act as single and double acceptors, respectively. If these were trapped in the strain field of a dislocation, then we would expect the dislocation to appear electrically active5. Indeed we nd the energy of VGa–(ON)n to be much lower at the dislocation core than in the bulk. This is because a neighbouring pair of three-fold coordinated Ga and N atoms are removed from the core and an O atom is inserted into the N site. The oxygen atom then lies in bridge site between two Ga atoms in a normal bonding configuration. Assuming Ga–rich growth conditions, O in equilibrium with Ga2O3 and n–type material the resulting formation energies of these defects lie between -2 and -3 eV. Thus the core of the dislocation will spontaneously oxidise if oxygen is mobile. In any event, we anticipate that the core will contain electrically active donor and acceptor pairs possibly giving rise to a negatively charged dislocation line. This is in agreement with recent temperature–dependent Hall–effect measurements2.
In conclusion, the density functional calculations show that in wurtzite GaN the stress field of threading edge dislocations is likely to trap gallium vacancies and oxygen as well as their complexes resulting in a negatively charged dislocation line in n–type material.
4. Domain boundaries on {11\(\bar 2\)0} planes
In addition to dislocations, two kinds of domain boundaries have also been observed14–19. They lie on {11\(\bar 2\)0} and {10\(\bar 1\)0} planes and following Xin et al. 19 are denoted by DB–I and DB–II respectively. Domain boundaries are either described in terms of a double position boundary (DPB) [otherwise termed a stacking mismatch boundary (SMB)] consisting of a different stacking sequence across the boundary, or an inversion domain boundary (IDB) which is characterised by a polarity inversion across the boundary. Domain boundaries of type DB–II have been explored extensively using transmission electron microscopy (TEM)14 , 15 , 17 , 16 and theoretically by Northrup et al. 20 using the scf–LDA method with a plane wave basis.
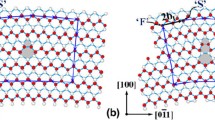
In contrast to DB–II type boundaries which originate at the epilayer substrate interface the DB–I type boundaries found in a GaN sample grown by molecular beam epitaxy (MBE) on GaP extend only a short distance along the c–axis19. A high resolution Z–contrast image down [0001] reported by Xin et al. 21 shows clearly that DB–I has a horizontal displacement of \(R_h = 1/2\left\langle {10\bar 10} \right\rangle \). This con guration which is also called prismatic stacking fault is composed of four– and eight– fold rings along the fault. For an illustration see Fig. 2.
We investigated several models22 corresponding to this horizontal displacement and found a double position boundary to possess the lowest domain wall energy. This double position boundary has an additional vertical displacement of 1/2〈0001〉 giving a total displacement of 1/2〈10\(\bar 1\)1〉 as derived from TEM by Xin et al. 19 and is therefore denoted by DPB* –I. All atoms along the boundary are four–fold coordinated and form Ga–N bonds across the boundary (see Fig. 2). Since Ga–N bonds are very strong DPB* –I has a clearly defined spacing of ∼ 1.90 Å between the {11\(\bar 2\)0} planes at the boundary. Some of the bonds are quite distorted which makes that DPB* –I induces shallow electronic states ∼ 0.35 eV above VBM in the band gap. However, these states are not deep enough to be responsible for the yellow luminescence which is centred at ≈2.2 eV and observed in n–type GaN. On the other hand point defects may segregate to the DPB* –I boundary and change the electrical properties. We evaluated the formation energy of VGa 3− at the domain boundary and found it to be lower by 1.1 eV at pos. 3 with respect to a position in a bulk like environment. This suggests that if Ga vacancies di use easily in GaN a lot of them will be trapped at DPB* –I where they would introduce deep acceptor states and can act as electron traps, in agreement with recent electron energy loss spectroscopy (EELS) measurements by Natusch et al. 23.

Top view along [0001] (left) and side view along [10\(\bar 1\)0] (right) of the DPB* –I structure which has a total displacement of 1/2〈10\(\bar 1\)1〉. DPB* –I is found to possess the lowest wall energy of domain boundaries on {11\(\bar 2\)0} planes. Atom numbers 1 (2) refer to Ga (N) atoms in eight–fold rings close to the boundary, whereas atom numbers 3 (4) refer to Ga (N) atoms in four–fold rings with bonds across the boundary.
5. Summary
We have presented density-funcitonal calculations for a variety of extended defects observed in wurtzite GaN. All stable structures consist of four–fold coordinated atoms or possess pairs of three–fold coordinated Ga and N atoms which adopt energetically favorable sp 2 and p 3 positions as at the {10\(\bar 1\)0} surface. Apart from full–core screw dislocations which have very strained bonds and therefore induce deep states into the band gap, all other extended defects seem to possess only shallow gap states. However, point defects and impurities are likely to be trapped in the stress fields and could alter the electrical properties.
References
J. E. Northrup and J. Neugebauer, Phys. Rev. B 53, 10477 (1996).
D.C. Look and J.R. Sizelove, submitted to PRL 1998.
J. Elsner, R. Jones, P.K. Sitch, V.D. Porezag, M. Elstner, Th. Frauenheim, M.I. Heggie, S. Öberg and P.R. Briddon, Phys. Rev. Lett. 79, 3672 (1997).
M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, Th. Frauenheim, S. Shuhai and G. Seifert, Phys. Rev. B 58, 7260 (1998).
J. Elsner, R. Jones, M. Haugk, Th. Frauenheim, M.I. Heggie, S. Öberg and P. R. Briddon, Phys. Rev. B 58, 12571 (1998).
Y. Xin, S. J. Pennycook, N. D. Browning, P. D. Nelist, S. Sivananthan, F. Omnès, B. Beaumont, J.–P. Faurie and P. Gibart, Appl. Phys. Lett. 72, 2680 (1998).
A. F. Wright and J. Furthmüller, Appl. Phys. Lett. 72, 3467 (1998).
C. Wetzel, T. Suski, J. W. Ager, E. R. Weber, E. E. Haller, S. Fischer, B. K. Meyer, R. J. Molnar and P. Perlin, Phys. Rev. Lett. 78, 3923 (1997).
J. Neugebauer and C.G. Van de Walle, Festkörperprobleme 35, 25 (1996).
J. Elsner, R. Jones, M. Haugk, R. Gutierrez, Th. Frauenheim, M. I. Heggie, S. Öberg and P. R. Briddon, Appl. Phys. Lett. in press (1998).
J. Neugebauer and C. Van de Neugebauer, Appl. Phys. Lett. 69, 503 (1996).
P. Bougulawski, E.L. Briggs and J. Bernholc, Phys. Rev. B 51 R17255 (1995).
Z. Liliental–Weber, Y. Chen, S. Ruvimov and J. Washburn, Phys. Rev. Lett. 79, 2835 (1997)
Z. Sitar, M. J. Paisley, B. Yan, and R. F. Davis, Mater. Res. Soc. Symp. Proc. 162, 537 (1990).
S. Tanaka, R. Scott Kern, and R. F. Davis, Appl. Phys. Lett. 66, 37 (1995).
D. J. Smith, D. Chandrasekhar, B. Sverdlov, A. Botchkarev, A. Salvador, and H. Morkoc, Appl. Phys. Lett. 67, 1830 (1995).
B. N. Sverdlov, G. A. Martin, H. Morkoc, and D. J. Smith, Appl. Phys. Lett. 67, 2063 (1995).
J.–L. Rouvière, M. Arlery, A. Bourret, R. Niebuhr, and K. Bachem, Inst. Phys. Conf. Series 146, 285 (1995).
Y. Xin, P. D. Brown and C.J. Humphreys, Appl. Phys. Lett. 70, 1308 (1997).
J. E. Northrup, J. Neugebauer and L. T. Romano, Phys. Rev. Lett. 77, 103 (1996).
Y. Xin, S. J. Pennycook, N. D. Browning, P. D. Nellist, S. Sivananthan, J.-P. Faurie and P. Gibart, Nitride Semicond. 482 781, edited by F.A. Ponce, S.P. Den Baars, B.K. Meyer, S. Nakamura, S. Strite, Mat. Res. Soc., Pennsylvania (1998).
J. Elsner, M. Kaukonen, M. I. Heggie, M. Haugk, Th. Frauenheim and R. Jones, Phys. Rev. B in press (1998).
M.K.H. Natusch, G.A. Botton, R.F. Broom, P.D. Brown, D.M. Tricker and C.J. Humphreys, Nitride Semiconductors 482 763, edited by F.A. Ponce, S.P. Den Baars, B.K. Meyer, S. Nakamura, S. Strite, Mat. Res. Soc., Pennsylvania (1998).
Author information
Authors and Affiliations
Rights and permissions
About this article

Cite this article
Elsner, J., Frauenheim, T., Haugk, M. et al. Extended Defects in GaN: a Theoretical Study. MRS Internet Journal of Nitride Semiconductor Research 4 (Suppl 1), 250–256 (1999). https://doi.org/10.1557/S1092578300002544
Published:
Issue Date:
DOI: https://doi.org/10.1557/S1092578300002544