
Abstract
Mosapride citrate (MC) is a poorly soluble short half-life drug with more pronounced absorption in the stomach. The present study aimed to incorporate MC co-crystals with enhanced solubility into 3D-printed floating tablets. MC co-crystals were prepared via the green method using Saccharin sod. as a co-former at a (1:1) molar ratio. The prepared co-crystals were assessed for solubility, FTIR, thermal behavior, and SEM. Then, it was incorporated into zero % infill 3D-printed tablets of different configurations at two thickness levels by the FDM printing technique. Printed tablets were evaluated for dimensions, weight deviation, friability, and in vitro floating behavior. Drug release and kinetic of the MC release were also assessed. Solubility study of the co-crystals showed a significant (p value < 0.05) increased solubility over pure MC. FTIR and thermal behavior confirmed hydrogen bonding formation during co-crystallization. The obstructed particles had an erratic protrusion form, similar to a nodule, as illustrated by SEM. The printed tablets showed acceptable physicochemical properties. Tablets floated for about ≥ 12 h without floating lag time. In vitro drug release exhibited variable extended release profiles with different lag times depending on the configuration indicating that the tablet’s wall thickness and surface area were the factors manipulated to control drug release. Kinetic analysis of the release data displayed intermediate kinetics between zero-order and diffusional kinetics. The intragastric extended release profile for MC co-crystals of improved solubility could be successfully, economically, and quickly developed utilizing the 3D printing technique.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drugs may have variable solid forms, which may exhibit variable physicochemical as well as biological properties, including thermal stability, solubility, and bioavailability [1]. Drug efficacy depends primarily on three factors which are potency (dose), solubility, and permeability. Potency is an intrinsic drug property that cannot be easily manipulated. On the other hand, solubility and permeability could be modulated. The formation of other solid forms, such as solid eutectic compositions, salts, co-crystals, and co-amorphous, is an important strategy to modify the drug solubility [2, 3]. Crystallization is defined as a process of redundant disciplined arrangement of molecules to form a unit crystal [4]. Co-crystals are formed when correlative atoms of different structures are solidified into one crystal such that when these co-crystals dissolve, they will give back their original starting materials [5]. In addition to improving the active pharmaceutical ingredient (API) solubility, co-crystals can be used to ameliorate taste, stability, flowability, and bioavailability [6]. Co-crystals could be prepared by various methods, of which the solvent-free method has gained great interest as an attractive green tool. This is because these methods can produce co-crystals with limited amounts (liquid-assisted grinding) or even no need for solvent (neat grinding). Co-crystal formation by liquid-assisted grinding is not fully understood, but it could be assumed that the added liquid acts as a lubricant that facilitates components’ molecular diffusion [3].
Mosapride citrate (MC) is a third-generation prokinetic agent that is used for the treatment of a variety of gastrointestinal diseases such as acid reflux, irritable bowel syndrome, and vomiting accompanying chronic gastritis [7]. It acts as a selective serotonin (5-HT4) receptor agonist, where it facilitates the release of acetylcholine from cholinergic neurons [7]. Difficulties in the pharmaceutical formulation of MC could be attributed to its poor solubility and wettability which in turn result in variation in its bioavailability [8]. The solubility of MC has been improved by several techniques in previous studies [9, 10]. Crystallization was recognized by its ability to improve important drug properties (physicochemical as well as biological) due to the inclusion of another molecular agent (co-former), without affecting intrinsic drug activity [3]. The co-crystal formation could be attributed to hydrogen bond formation. In the present study, MC contains both hydrogen donor and acceptor groups which would form co-crystals via interaction with co-formers through hydrogen bonding [9]. Saccharin sod. contains both hydrogen donor and acceptor groups, so it has been abundantly used as a co-former for pharmaceutical co-crystal fabrication [11].
Marketed MC tablets are absorbed orally with T1/2 of about 1.5 h and Tmax in the range of 0.5–1 h. Hence, multiple daily administration is required, which could impair patient compliance [12]. Therefore, a sustained-release MC formulation would be recommended to reduce dosing frequency and enhance patients’ compliance [13]. Several techniques were used to prolong the MC duration of action [14]. However, MC exhibits variable absorption in various GIT segments with more pronounced absorption in the stomach, making conventional extended-release formulations unsuitable for MC [10, 15]. Hence, the development of a floating system for MC co-crystals with enhanced solubility would combine the benefits of enhanced bioavailability together with the decrease in dosing frequency.
Traditional gastric floating drug delivery systems (GFDDS) have the advantage of prolonging gastric retention time together with extending the duration of the drug [16]. Traditional GFDDS is generally achieved by the use of swelling polymers, microporous components, or gas-generating components. So, formulating GFDDS generally requires a considerable number of excipients in a complicated process. In addition, less dense materials used to assist floating behavior may not be compatible with alkaline materials [17]. Hence, new techniques are being studied for designing GFDDS. One of the recent trials was to take the advantage of fabricating complex configurations by three-dimensional (3D) printing to develop low-density tablets for gastric floating. 3D printing is reported as a quick technology of prototyping that is able to fabricate particular shapes of complex interior structures quickly and accurately.
The ability to design a novel low-density internal structure of different configurations was achieved using 3D-printing technology [18]. The 3D printing utilizes many techniques, of which fused deposition modeling (FDM) attracts the pharmaceutical formulators’ interest. This could be due to its low cost, easy operation, and the ability to produce a hollow entity. FDM typically works by forcing out polymeric filaments that have consistent, well-defined diameters through robotically actuated nozzles where the extruded filaments then solidify on a plate [19]. Drug release control from tablets printed by FDM technology can be done by modifying several factors, including configuration, drug load, or polymer selection [20]. However, FDM suffers from the limitation of being unsuitable for APIs sensitive to temperature changes and the need for incorporation of API into the filament as a preceding step of manufacture. Recently, a syringe was utilized to incorporate pastes or liquids containing drugs that are sensitive to the temperature inside the systems [21]. Therefore, this printer can save time, energy, and space in comparison with the traditional process.
The primary motivation for this study was to prepare MC co-crystals of improved solubility via the green method. The second motivation was to incorporate HPMC K4M gel containing the MC co-crystals into floating 3D-printed tablets of different configurations at two thickness levels using the FDM technique. The effect of configuration and thickness on the floating behavior and drug release was further studied to ensure drug release in the upper part of the stomach.
Materials and Methods
Materials
Mosapride citrate was obtained from Sigma-Aldrich (Germany). The available marketed tablets (mosapride 5 mg, Western Pharmaceutical Industries) were purchased. FDM printer filament made of biodegradable thermoplastic polylactic acid (PLA) (melting point 220 °C, density 1.25 g/cm3) was supplied by Leon3D (Spain). El-GOMHOURIA Company, Cairo (Egypt), was the source of ethyl alcohol and hydrochloric acid. Saccharin sodium and hydroxypropyl methylcellulose METHOCEL™ K4M were obtained from EIPICO Company, Cairo (Egypt). Analytical-grade reagents and chemicals were employed for all other processes.
Preparation of Mosapride Citrate Co-crystals
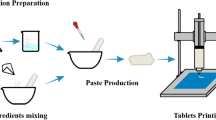
The liquid-assisted grinding technique was used to prepare MC co-crystals since it is a simple and costly effective method that gives co-crystals of high yields. In addition, this method saves time being independent of the solubility of the primary components. Based on dissolution data (see supplementary file), drug and co-former (Saccharin sod.) were blended at (1:1) molar ratio in mortar and pestle for 15 min. Enhanced molecular diffusion was attained by the inclusion of a small amount of liquid, which acts as a catalyst for encouraging co-crystal preparation. A few drops of ethanol (the maximum amount of ethanol was 0.2 mL) were added dropwise to moisten the mixture of the drug and co-former during grinding [22]. The resultant co-crystal powder was stored in air-tight containers until use.
Characterization of the Prepared Mosapride Citrate Co-crystal Powder
Solubility Study
The method of shake flask was used for studying the solubility of pure MC powder, physical mixture (MC and Saccharin sod.), and the MC co-crystals in 0.1 N HCl and phosphate buffer of pH 6.8 [23]. Excess amounts of MC, the physical mixture, and the MC co-crystals were dissolved individually in 10 mL of 0.1 N HCl and phosphate buffer of pH 6.8. The samples were shaken on a water bath shaker (Gallen Kamp (England)) at 37 ± 0.5 °C for 24 h. Solutions were then centrifuged for 30 min at 3000 rpm (Remi Laboratory Centrifuge R32A, Remi Equipment, Bombay, India), and 2 mL of supernatant was diluted to 50 mL with the same medium and examined spectrophotometrically using UV-Visible spectrophotometer (6405 UV/Vis, Jenway, Staffordshire, UK) at 264 nm [24]. One-way analysis of variance (ANOVA) was used for statistical analysis using the GraphPad Prism 6 software (Otago information technology system, New Zealand), and a significance level of p < 0.05 was set.
Fourier Transform Infrared Spectroscopy
Predicting the formation of co-crystals was done with the aid of Fourier transform infrared (FTIR), a useful analytical tool that reveals the presence of distinct functional groups in the co-crystals. Pure MC, Saccharin sod., a mixture of the two substances, and the co-crystals were all scanned by FTIR spectrophotometer (VERTEX 70 (Perkin-Elmer, FTS-1710, Beaconsfield, UK). Hydraulic pressure was used to compress the KBR-dispersed samples into discs. The spectrum was then recorded once the pellet was positioned in the light path. The wavenumber range of (4000 to 400 cm−1) was used to acquire the spectra of each sample [25].
Thermal Analysis Utilizing Differential Scanning Calorimetry
To verify the development of co-crystals, the molecular state of MC in the co-crystals was investigated using differential scanning calorimetry (DSC). A differential scanning calorimeter (Shimadzdu, model DSC-50) (Japan) was used to analyze the samples (pure MC, Saccharin sod., a mixture of the two substances, and the co-crystals). For analysis, about 5 mg of each sample was heated in a sealed aluminum pan over a temperature range of 0 to 300 °C at a scanning rate of 5 °C/min while under a nitrogen purge at a rate of 30 mL/min. The standard was based on pure indium [26].
Surface Morphology Analysis
Analytical scanning electron microscopy (SEM) (JSM-6360A, JEOL, Tokyo, Japan) was employed to examine the topography of the MC co-crystals. A double-sided adhesive tape was affixed to a stub of aluminum, and a small amount of the sample was sprinkled on top. Under an argon atmosphere, the stubs were coated with platinum resulting in a thickness of about 10 A° and scanned using SEM.
Development of 3D-Printed Floated Tablets
Preparation of Printable Gel
Hydroxypropyl methylcellulose K4M powder was added to 100 mL hot water (70 °C) and mixed thoroughly until a uniform dispersion was attained, yielding a 1% w/v 3D printable gel. After the mixture was mixed, it was chilled with ice and placed in the refrigerator for 24 h where a homogeneous gel solution free of air bubbles and aggregates was formed [27]. Finally, the MC co-crystals were added until complete distribution.
3D Printing Process
Zero % infill tablets of different configurations (cube, cylinder, sphere) were printed utilizing FDM and syringe-based techniques using REGEMAT 3D V1 printer (Regemat 3D S.L., Spain) previously loaded with PLA filaments. PLA filaments were selected for the printing process since it shows optimum mechanical properties, sustained release behavior, and variable density that can be adjusted for the floating purpose [28,29,30]. Both ULTIMAKER CURA, a slicing program, and SOLIDWORKS, a design program, were employed in the production of the 3D-printed systems. Syringes with a 0.3 mm internal diameter and a 0.7 mm external diameter were used to house the printable gel. Three-dimensional printed floating tablets were created using FDM with the following printing parameters: layer thickness (0.4 or 0.8 mm), perimeter speed 25 mm/s, infill speed 8 mm/s, feeding rate 1.2 mm/s, and bed’s temperature 60 °C. Tablets were digitally developed and extruded layer by layer on a glass slide. Twenty-one layers, each with four sealing perimeters, were extruded to make the structures. After extruding the initial two layers, the third layer features parallel lines spaced 1.2 mm apart. In the fourth layer, the extruder follows the same procedure, except this time, the lines run perpendicular to those in the first three levels. A 1.2 mm by 1.2 mm square mesh was generated in this manner. Up until the 18th layer, the same procedure was used. As the extruder withdrew, the printable gel (0.5 mL) was introduced into the printed structure using a syringe. Three additional layers were further built by an FDM extruder to complete the printed tablets [27]. To build the structure, every step, including the use of the FDM extruder and the syringes, was done by hand. After the third layer, the structure was dried.
Drug Content, Features, and Physical Parameters of Printed Tablets
The MC content of the printed tablets was carried out after crushing utilizing UV-spectrophotometer (6405 UV/Vis, Jenway, Staffordshire, UK) [24, 31]. The content of the crushed printed tablets was individually dissolved in 0.1 N HCl (pH 1.2) in a 100-mL volumetric flask and stirred for 15 min. After sufficient dilution with 0.1 N HCl, spectrophotometric absorbance was examined at 264 nm [24]. Measurements were carried out in triplicate.
A digital caliper (made by VWR in Stockholm, Sweden) was used to measure the tablets’ thickness and diameter. Also, the test for brittleness was done in a digital tablet brittleness machine (VEEGO model: FT-2D, India). Measurements were carried out in triplicate. An electronic balance (Mettler, J 100, (Switzerland)) was used to measure the weight of twenty tablets after drying.
Floating Study of 3D-Printed Tablets
3D tablets were tested for floating in vitro under both static and dynamic settings. Static investigations used a beaker containing 900 mL of 0.1 N HCl to mimic stomach juice. In the dynamic investigations, tablets were tested in USP dissolution equipment II (paddle type) (DA6D, Bombay-400–069, India) at a speed of 75 rpm and 900 mL of 0.1 N HCl. Each tablet’s floating lag time was monitored, where the total floating time was the period of time the tablet was on the surface of the medium [32].
In Vitro Mosapride Citrate Release from 3D-Printed Tablets
The drug release from both 3D-printed floating tablets and the marketed available tablets was studied using USP dissolution equipment II (paddle type) (DA6D, Bombay-400-069, India) at a speed of 75 rpm. The dissolution medium was simulated gastric fluid (0.1 N HCl) in a volume of 900 mL kept at 37 °C ± 0.5 °C. At 1, 2, 3, 4, 5, 7, and 8 h; 5 mL aliquots of the media were removed and promptly exchanged with an equivalent volume of fresh dissolving medium. The filtered samples were examined spectrophotometrically at 264 nm utilizing a UV-spectrophotometer (6405 UV/Vis, Jenway, Staffordshire, UK) [24]. Measurements were carried out in triplicate.
Kinetic Modelling of Mosapride Citrate Release
Several kinetic models of drug release were enforced to in vitro release data to better understand the pattern of MC release from 3D-printed floating tablets (zero-order, first-order, Higuchi, and Korsmeyer–Peppas models). The kinetic parameters and correlation coefficient were also calculated.
Results and Discussion
Characterization of the Prepared Mosapride Citrate Co-crystal Powder
Solubility Study
Co-crystallization is a simple technique that is known to improve drug solubility. This technique was already employed to improve the solubility of different drugs in several previous studies [33]. Pure MC had a solubility of 0.98 mg/mL and 0.71 mg/mL at 37 °C in 0.1 N HCl and phosphate buffer of pH 6.8 respectively. The solubility of the physical with Saccharin sod. mixture was found to be 0.97 mg/mL and 0.702 mg/mL at 37 °C in 0.1 N HCl and phosphate buffer of pH 6.8 respectively. The solubility of the co-crystals was 14.5 mg/mL and 11.6 mg/mL at 37 °C in 0.1 N HCl and pH 6.8 phosphate buffer respectively. Statistical analysis revealed that co-crystallization of MC with Saccharin sod. had significantly (p value < 0.05) increased the MC solubility in both media over the pure drug. Results confirm co-crystal formation between MC and the used co-former. The enhanced co-crystal solubility can be explained by the release of MC particles from the crystal lattice and the solvation of such released particles. The altered crystal structure by co-crystal formation resulted in decreased crystal lattice energy which in turn enhanced drug solubility. In addition, improved solubility could be related to the increasing solvent affinity towards MC in the presence of Saccharin sod. (reduction in solvation energy) [34].
Fourier Transform Infrared Spectroscopy
FTIR is an important analytical tool for confirmation of co-crystal formation by observing variations in the frequencies of certain functional groups in the resulting co-crystals compared to the individual ingredients. The intermolecular hydrogen bonding between the drug and the co-former can be determined by FTIR. The formation of co-crystals involves non-covalent interactions (hydrogen bonding) between the drug and the used co-formers. The formation of a hydrogen bond (HB) depends on the presence of a hydrogen donor that can give hydrogen (like phenolic O−H, carboxylic O–H, and amine N−H) to hydrogen receptor (like the amidic N−C=O and a carboxylic C=O oxygen atom) [35].
Saccharin sod. molecule has both carbonyl bonds which can act as a strong hydrogen acceptor as well as (N–H) bond which can act as strong donors. The occurrence of these two bonds allows the molecule to form an imide homodimer with the subsequent formation of co-crystals with other molecules [34]. MC molecule contains a carbonyl bond which can act as a strong hydrogen acceptor. This carbonyl group has the ability to form a bond with the (N–H) hydrogen donor group of Saccharin sod. Also, the MC molecule contains a (N–H) bond which acts as a strong hydrogen donor and may have the ability to interact with the strong hydrogen acceptor of Saccharin sod. The authors predict the formation of co-crystals as presented in Fig. 1.

Theoretical probability of hydrogen bond formation between Mosapride citrate and Saccharin sod. molecules
FTIR spectra of pure MC, Saccharin sod., physical mixture, and the MC co-crystals are presented in Fig. 2. FTIR spectra of pure MC exhibited a characteristic forked absorption band at about 3443.18 and 3379.54 cm−1 corresponding to (−NH2) stretching vibration. There was also another forked absorption band at about 3334.22 and 3226.23 cm−1 corresponding to the stretching vibration of tertiary amine (–CONH). A characteristic absorption band at 1723.08 cm−1 could be due to the carbonyl amido (–CONH) stretching vibration. Other several sharp bands were observed at 1628.49 cm−1 (C–N stretching), 1546.53 cm−1 (C–H stretching), 1249.64 cm−1 (C–Cl stretching), and 1214.21 cm−1(C–F stretching). The observed spectra confirmed the purity of MC [9].

FTIR spectra of pure Mosapride citrate, Saccharin sod., physical mixture, and the MC co-crystals powder
FTIR spectrum of Saccharin sod. showed several characteristic bands. The bands observed were at 3250–3550 cm−1 and 1720 cm−1 corresponding to the cyclic imide (N−H) bond and the (C=O) bond of the secondary amine respectively. Bands corresponding to asymmetric and symmetric stretches of the (SO2) group were observed at 1333 and 1173 cm−1. This observation agrees with findings observed by previously published data [34]. However, when MC and Saccharin sod. were mixed physically, their spectra were found to be merely additions of the spectra of the two distinct compounds, with essentially no modifications in the characteristic peaks. FTIR spectrum of co-crystals differs markedly from that of the physical mixture and reveals the disappearance of the characteristic bands for MC at 3334.32 and 3226.23 cm−1 corresponding to tertiary amine (–CONH) stretching vibration. Also, there was a decrease in the intensity of the MC carbonyl amido (–CONH) characteristic stretching vibration. The characteristic bands of Saccharin sod. at 3550 cm−1 and at 1720 cm−1 disappeared. These changes in the FTIR peaks of the drug along with Saccharin sod. confirmed hydrogen bond formation during co-crystallization [36].
Thermal Analysis Using Differential Scanning Calorimetry (DSC)
DSC was performed to study the thermal behavior of the MC co-crystals relative to the individual components and the physical mixture. DSC thermogram provides a useful tool that confirms co-crystallization via alteration in the characteristic peaks of the co-crystals as compared to the drug and co-former since, in more than 50% of cases, co-crystals exhibited a melting point lower than that of both drug and co-former [1]. DSC thermograms of Pure MC, Saccharin sod., physical mixture, and the MC co-crystals are presented in Fig. 3. DSC thermogram of MC displayed two sharp endothermic peaks at 110 °C and 188 °C, corresponding to its melting behavior. Saccharin sod. also displayed an endothermic peak at 130 °C. Similar thermal events for MC and Saccharin sod. were obtained in the physical mixture. For the co-crystals, there was a drastic reduction the in the melting point of MC. In addition, the peak corresponding to Saccharin sod. was not detected which confirms the formation of co-crystals [37]. Differences observed in the melting behavior of prepared co-crystals in comparison with the pure MC could be explained by altered molecular arrangement in the prepared co-crystals. This alteration results in a new crystalline arrangement as well as altered thermodynamic properties. This could be due to non-covalent physical interaction through hydrogen bonding between polar functional groups of the drug and Saccharin sod.

DSC thermograms of pure Mosapride citrate, Saccharin sod., physical mixture, and the MC co-crystals powder
Surface Morphology Using Scanning Electron Microscopy (SEM)
Scanning electron microscopy was performed to examine the morphology of the co-crystal particles. Several SEM images of the co-crystals were taken, and the clearest one is presented in Fig. 4. In this figure, MC co-crystals were seen as obstructed particles with an erratic protrusion form, similar to a nodule. This shape of MC co-crystals may be caused by the grinding action. The surface of the MC co-crystals was found to be rough in nature.

Scanning electron microscopy for the MC co-crystals powder
Evaluation of the 3D-Printed Floating Tablets
Drug Content and Physical Properties of 3D-Printed Tablets
Tablets of different configurations (cube, cylinder, sphere) and thickness levels (0.4 & 0.8 mm) were obtained by 3D printing technique. The 3D-printed tablets were evaluated for MC content, dimensions, weight variation, and friability (Table I).
The average percent drug content was 95.90% ± 2.95–97.16% ± 2.26. All the investigated 3D tablets complied with the pharmacopeial requirements for their content uniformity, which was found to be within ± 10%. The diameter of the 3D tablets was 13.94 mm ± 0.21–10.10 mm ± 0.20. The volume was 520 mm3 ± 0.23–625 mm3 ± 0.19. The weight of 3D tablets was 225.7 mg ± 0.32–258.6 mg ± 0.13. Also, the friability of tablets was measured to indicate the physical strength of the printed tablet. The presence of cracked, cleaved, or broken tablets after tumbling the tablets fails the test. The friability of the 3D tablets was less than 1% indicating good mechanical strength. Results indicated that FDM printing represents a precise, accurate 3D printing system that can produce structures with the ability to hold the content from the syringe.
Floating Behavior of 3D-Printed Tablets
In the design of a floating drug delivery system, two main criteria have to be considered. First, the floating duration which has to be extended for more than 4–8 h to ensure retention of the floating system in the stomach without entering the small intestine. The second main concern is the time taken by the delivery system to float (floating lag time). Minimum floating lag time reduces the risk of entry of the floating system into the small intestine. Floating systems with a floating lag time of more than 1 h after immersion might pass to the small intestine since human gastric residence time is about 1–3 h. Hence, the in vitro floating studies of 3D floating tablets were performed under both static and dynamic conditions [38]. In the floating study, the printed 3D tablets with different configurations showed immediate floating (floating lag time = 0 min) after being immersed in media under either static or dynamic conditions. In addition, all tablets were found to float for more than 12 h (surface floating duration ≥ 12 h) as presented in Fig. 5. On the other hand, the marketed tablet did not float after immersion in 0.1 N HCl. Nevertheless, the disturbance effect produced by the dissolution apparatus is not exactly the same for stomach movement. So, further study of the floating behavior in humans would be recommended.

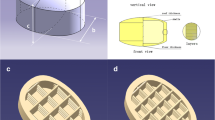
Photographs of a 3D-printed tablet with different configurations and their floating behavior
In Vitro Mosapride Citrate Release from 3D-Printed Tablets
The release behavior of mosapride citrate from zero % infill 3D-printed tablets of various configurations at two thickness levels and the available marketed tablets (mosapride 5 mg, Western Pharmaceutical Industries) were conducted in 0.1 N HCl (pH 1.2) for 8 h. As presented in Figs. 6 and 7, 3D-printed tablets with higher wall thickness displayed integrity for a longer duration before MC release in comparison with 3D-printed tablets of a lower wall thickness. The 3D-printed tablets with a wall thickness of 0.8 mm started MC release at the end of the 3rd h, whereas those with a wall thickness of 0.4 mm started MC release at the end of the 1st h. Similar results were obtained by Maroni et al., where two compartment capsular devices, each having a different thickness, showed increased lag phase proportionally with an increased compartment thickness [39]. In another research directed by Reddy et al., the authors printed tablet shells with different thickness levels and reported that as the shell thickness increased, the lag time before drug release was increased. This phenomenon can be explained by the fact that the higher wall thickness would prevent penetration of dissolution media inside the 3D-printed tablets for a longer time [40].

MC release from 3D-printed floating tablets (n = 3) with different configurations and thicknesses of 0.4 mm and marketed tablet in 0.1 N HCl

MC release from 3D printed floating tablets (n = 3) with different configurations and thicknesses of 0.8 mm and marketed tablet in 0.1 N HCl
Moreover, the tablet configuration has been shown to control the drug release, where % MC released was 84.76 ± 0.43, 89.82 ± 0.61, and 71.89 ± 0.63% at 8 h for the cube, cylinder, and sphere tablets of 0.4 mm wall thickness respectively. While the % MC released at 8 h for the cube, cylinder, and sphere tablets of 0.8 mm wall thickness was 75.7 ± 0.43, 77.78 ± 0.61, and 63.83 ± 0.63%. The order of MC released from 3D-printed floating tablets was cylinder > cube > sphere. This could be explained by variation in the surface area of the 3D-printed tablets where a higher surface area value gives higher drug release, as shown in (Table I). The bigger surface area admits effortless entry of the dissolution media inside the 3D-printed tablets, resulting in a faster release of MC. Reddy et al., who used FDM 3D printing for the development of core-shell gastro retentive floating pulsatile drug delivery systems, reported that a diminish in the surface area caused a slower drug release from the printed systems [40]. Xiaowen et al., who fabricated three types of tablets enclosing drug-containing gels, reported variation in the rate of drug release along with variation in the surface area of tablets [41]. Marketed tablets, on the other hand, showed fast release behavior with > 70% of MC was released in the 1st hour. It was observed that the 3D-printed floating tablets exhibited more controlled drug release in comparison with the available marketed tablet which displayed complete drug release within 2 h. This confirms quite control for the drug release by the 3D-printed floating tablets where the drug is released in the upper gastric region after the polymer is dissolved.
Kinetic Modelling of Mosapride Citrate Release
Linear regression was used to study dissolution data to predict the order for drug release. The model of the highest correlation coefficient (r2) (Table II) was determined as the best-fit kinetic model. The zero-order model gave the highest R2 values for all of the 3D-printed tablets. This observation comes along with findings observed by Huanbutta and Sangnim who developed zero-order drug-release gastro-retentive floating tablets produced by 3D printing technology [32].
Also, Korsmeyer–Peppas model was used to study the release mechanism. The obtained diffusional exponent (n) value can define the mechanism of drug release. The 3D-printed tablet showed the Fickian diffusion release model as the best fit with the Korsmeyer–Peppas model; (r2 = 0.0646–0.5015) and exponent values (n) ≤ 0.5. The obtained results indicated that the drug is released with diffusional kinetics, as indicated by the Korsmeyer time exponent. In general, this kinetic can be considered suitable for sustained release. This drug release model might have the ability to stabilize drug plasma levels which would maximize therapeutic efficacy and minimize drug overdose side effects and toxicity.
Conclusion
In this study, co-crystals of MC were propitiously developed via the green method. The resultant co-crystals had significantly (p value < 0.05) enhanced MC solubility (in 0.1 N HCl and pH 6.8 phosphate buffer) in comparison with the pure drug. The formation of co-crystals was endorsed by thermal analysis and FTIR which furnished proof for hydrogen bonding during co-crystallization. SEM clarified nodular-like irregular protrusion-shaped particles.
The 3D-printed floating tablets of different configurations with zero % infill were propitiously printed via FDM technique using PLA filaments. The printed tablets displayed acceptable physicochemical properties with an adequate floating duration of more than 12 h in the absence of floating lag time. Quite control for drug release was also propitiously accomplished by manipulating the tablet’s wall thickness and surface area. In this concern, the duration of the lag phase before drug release can be increased by increasing the tablets’ wall thickness levels. Also, different configurations had different surface areas which resulted in diversity in the release profiles. MC was released in the following order: cylinder > cube > sphere for both wall thicknesses. The drug was released with release kinetics between zero order as well as diffusional drug release, as indicated by the Korsmeyer time exponent. The obtained drug release pattern could stabilize drug concentrations in plasma. The gained results proved the ability of the FDM printing technique to fabricate 3D-printed systems that can hold the content from the syringe and can regulate the drug release in the upper part of the stomach. The authors believe this would be a significant step forward to the broader application of 3D printing drug design and manufacture, particularly in areas of clinical development.
Data Availability
The authors confirmed that the data supporting the findings of this study are available within the article and its supplementary material. Raw data that support findings of this study are available from corresponding author upon reasonable request.
References
Schultheiss N, Newman A. Pharmaceutical cocrystals and their physicochemical properties. Cryst Growth Des. 2009;9(6):2950–67.
Miroshnyk I, Mirza S, Sandler N. Pharmaceutical co-crystals-an opportunity for drug product enhancement. Expert Opin Drug Deliv. 2009;6(4):333–41.
Solares-Briones M, Coyote-Dotor G, Páez-Franco JC, Zermeño-Ortega MR, de la O Contreras CM, Canseco-González D, et al. Mechanochemistry: a green approach in the preparation of pharmaceutical cocrystals. Pharmaceutics. 2021;13(6):790.
Aulton ME, Taylor MEG. Aulton’s pharmaceutics: the design and manufacture of medicines. Elsevier Health Sciences; 2013;933.
Almarsson Ö, Peterson ML, Zaworotko M. The A to Z of pharmaceutical cocrystals: a decade of fast-moving new science and patents. Pharm Pat Anal. 2012;1(3):313–27.
Jones W, Motherwell WDS, Trask AV. Pharmaceutical cocrystals: an emerging approach to physical property enhancement. MRS Bull. 2006;31(11):875–9.
Liu Z, Sakakibara R, Odaka T, Uchiyama T, Uchiyama T, Yamamoto T, et al. Mosapride citrate, a novel 5‐HT4 agonist and partial 5‐HT3 antagonist, ameliorates constipation in parkinsonian patients. Movement Disorders: Official Journal of the Movement Disorder Society. 2005;20(6):680–6.
Sakashita M, Mizuki Y, Yamaguchi T, Miyazaki H, Sekine Y. Pharmacokinetics of the gastrokinetic agent mosapride citrate after intravenous and oral administrations in dogs and monkeys. Arzneim-ForschungDrug Res. 1993;43(8):864–6.
Ali AA, Sayed OM. Preparation and characterization of mosapride citrate inclusion complexes with natural and synthetic cyclodextrins. Pharm Dev Technol. 2011;18(5):1042.
Liu K, Meng Z, Li Y, Liu J, Xu Y, Wang Y, et al. Preparation and evaluation of mosapride citrate dual-release dry suspension. AAPS PharmSciTech. 2019;20(4):155.
Banerjee R, Bhatt PM, Ravindra NV, Desiraju GR. Saccharin salts of active pharmaceutical ingredients, their crystal structures, and increased water solubilities. Cryst Growth Des. 2005;5(6):2299–309.
Chae JW, Song BJ, Baek IH, Yun HY, Ma JY, Kwon KI. Effects of food intake on pharmacokinetics of mosapride in beagle dogs. J Vet Pharmacol Ther. 2015;38(5):497–9.
Goyal S, Agarwal G, Agarwal S, Karar PK. Oral sustained release tablets: an overview with a special emphasis on matrix tablet. Am J Adv Drug Deliv. 2017.
Kim HJ, Lee SH, Lim EA, Kim JS. Formulation optimization of solid dispersion of mosapride hydrochloride. Arch Pharm Res. 2011;34(9):1467.
Li Y. Intestinal absorption characters of mosapride citrate using caco-2 cells and single-pass intestinal perfusion methods. Chin Pharm J. 2017:1519–24. https://doi.org/10.11669/cpj.2017.17.009
Awasthi R, Kulkarni GT. Decades of research in drug targeting to the upper gastrointestinal tract using gastroretention technologies: where do we stand? Drug Deliv. 2016;23(2):378–94.
Sathish D, Himabindu S, Kumar YS, Shayeda null, Rao YM. Floating drug delivery systems for prolonging gastric residence time: a review. Curr Drug Deliv. 2011;8(5):494–510.
Li P, Zhang S, Sun W, Cui M, Wen H, Li Q, et al. Flexibility of 3D extruded printing for a novel controlled-release puerarin gastric floating tablet: design of internal structure. AAPS PharmSciTech. 2019;20(6):236.
Verstraete G, Samaro A, Grymonpré W, Vanhoorne V, Van Snick B, Boone MN, et al. 3D printing of high drug loaded dosage forms using thermoplastic polyurethanes. Int J Pharm. 2018;536(1):318–25.
Martinez PR, Goyanes A, Basit AW, Gaisford S. Fabrication of drug-loaded hydrogels with stereolithographic 3D printing. Int J Pharm. 2017;532(1):313–7.
Khaled SA, Burley JC, Alexander MR, Roberts CJ. Desktop 3D printing of controlled release pharmaceutical bilayer tablets. Int J Pharm. 2014;461(1):105–11.
Garbacz P, Wesolowski M. Benzodiazepines co-crystals screening using FTIR and Raman spectroscopy supported by differential scanning calorimetry. Spectrochim Acta A Mol Biomol Spectrosc. 2020;234: 118242.
Jouyban A. Handbook of solubility data for pharmaceuticals. CRC Press; 2009. https://doi.org/10.1201/9781439804889
Sankar PDS, Sujana K, Preetham DN. Development and validation of spectrophotometric method for the estimation of mosapride in bulk and pharmacutical formulation. Asian J Pharm Clin Res. 2013;189–91.
Basavoju S, Boström D, Velaga SP. Indomethacin–saccharin cocrystal: design, synthesis and preliminary pharmaceutical characterization. Pharm Res. 2008;25(3):530–41.
Tomaszewska I, Karki S, Shur J, Price R, Fotaki N. Pharmaceutical characterisation and evaluation of cocrystals: importance of in vitro dissolution conditions and type of coformer. Int J Pharm. 2013;453(2):380–8.
Linares V, Casas M, Caraballo I. Printfills: 3D printed systems combining fused deposition modeling and injection volume filling. Application to colon-specific drug delivery. Eur J Pharm Biopharm Off J Arbeitsgemeinschaft Pharm Verfahrenstechnik EV. 2019 Jan;134:138–43.
Öblom H, Zhang J, Pimparade M, Speer I, Preis M, Repka M, et al. 3D-Printed isoniazid tablets for the treatment and prevention of tuberculosis-personalized dosing and drug release. AAPS PharmSciTech. 2019;20(2):52.
Azad MA, Olawuni D, Kimbell G, Badruddoza AZ, Hossain MdS, Sultana T. Polymers for extrusion-based 3D printing of pharmaceuticals: a holistic materials–process perspective. Pharmaceutics. 2020;12(2):124.
Tümer EH, Erbil HY. Extrusion-based 3D printing applications of PLA composites: a review. Coatings. 2021;11(4):390.
Morsi NM, Abdelbary GA, Ahmed MA. Preparation and physicochemical characterization of mosapride citrate pharmacosomes. 2014;2014(4):7.
Huanbutta K, Sangnim T. Design and development of zero-order drug release gastroretentive floating tablets fabricated by 3D printing technology. J Drug Deliv Sci Technol. 2019;52:831–7.
Madan JR, Waghmare SV, Patil RB, Awasthi R, Dua K. Cocrystals of apixaban with improved solubility and permeability: formulation, physicochemical characterization, pharmacokinetic evaluation, and computational studies. ASSAY Drug Dev Technol. 2021;19(2):124–38.
Soliman II, Kandil SM, Abdou EM. Gabapentin–saccharin co-crystals with enhanced physicochemical properties and in vivo absorption formulated as oro-dispersible tablets. Pharm Dev Technol. 2020;25(2):227–36.
Al-Dulaimi AF, Al-kotaji M, Abachi FT. Co-crystals for improving solubility and bioavailability of pharmaceutical products. Egypt J Chem. 2022;65(1):81–9.
Garbacz P, Wesolowski M. DSC, FTIR and Raman spectroscopy coupled with multivariate analysis in a study of co-crystals of pharmaceutical interest. Molecules. 2018;23(9):2136.
Saganowska P, Wesolowski M. DSC as a screening tool for rapid co-crystal detection in binary mixtures of benzodiazepines with co-formers. J Therm Anal Calorim. 2018;133(1):785–95.
Ibrahim M, Naguib Y, Sarhan H, Abdelkader H. Gastro-retentive oral drug delivery systems: a promising approach for narrow absorption window drugs. Journal of Advanced Biomedical and Pharmaceutical Sciences. 2019;2(3):98–110.
Maroni A, Melocchi A, Parietti F, Foppoli A, Zema L, Gazzaniga A. 3D printed multi-compartment capsular devices for two-pulse oral drug delivery. J Control Release. 2017;268:10–8.
Reddy Dumpa N, Bandari S, A Repka M. Novel gastroretentive floating pulsatile drug delivery system produced via hot-melt extrusion and fused deposition modeling 3D printing. Pharmaceutics. 2020;12(1):52.
Xu X, Zhao J, Wang M, Wang L, Yang J. 3D printed polyvinyl alcohol tablets with multiple release profiles. Sci Rep. 2019;9(1):12487.
Acknowledgements
I would like to thank everyone in the pharmacy department et al.-Azhar University for the university’s outstanding resources and extensive help with this study. I would like to thank Dr. Mona H. Ibrahim for her prediction about co-crystal formation. Also, I would like to thank Dr. Hatem Hussny Tokhy for his help in SEM work. I am grateful to EIPICO (Egypt) for the kind provision of materials, as well as to Cairo University’s Central Laboratory & Microanalytical Unit for their assistance with the DSC study and FTIR work, respectively.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Omnya M. Amin and Marwa A. Abd El-Fattah participated in all stages of the experiment, from conception to execution to analysis. Hesham El Qady designed and manufactured the zero % infill 3D-printed tablets with various thicknesses. Omnya M. Amin and Marwa A. Abd El-Fattah helped figure out what the data meant and worked on the manuscript. All authors discussed the findings and provided feedback on the text.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amin, O.M., EL Qady, H.N. & Abd El-Fattah, M.A. An Intragastric Delivery Device Employing FDM Technology: 3D-Printed Tablet Containing Green Developed Mosapride-Saccharin Co-crystals. AAPS PharmSciTech 24, 127 (2023). https://doi.org/10.1208/s12249-023-02578-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-023-02578-9