
Abstract
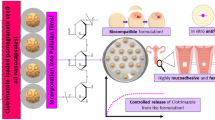
This paper focuses on the development and evaluation of mucoadhesive vaginal gel of fluconazole using nanolipid carriers to enhance tissue deposition in treating vulvovaginal candidiasis. Treatment of vulvovaginal candidiasis includes antimycotic agents prescribed for 1 to 7 days or longer, in relapse either orally or topically. The delivery of fluconazole as nanolipid carriers in vaginal gel can be proposed as suitable alternative to the existing conventional formulations to improve the patient acceptability, compliance and localized drug action. The nanolipid carriers of fluconazole were prepared by phase inversion temperature technique and incorporated into Carbopol 974P as gelling polymer. GRAS excipients selected and optimized were Precirol ATO 5, oleic acid and Kolliphor RH 40 to produce nanolipid dispersions. Stable nanolipid dispersions were developed using sodium dodecyl sulfate as the charge inducer. The optimized nanolipid dispersion of fluconazole had particle size, polydispersity index and zeta potential value of 158.33 ± 2.55 nm, 0.278 ± 0.003 and − 27.33 ± 0.40 mV, respectively and the average entrapment of fluconazole in the lipid carriers was found to be 67.24 ± 0.87%. The optimized vaginal gel had satisfactory mucoadhesive strength and rheological properties to facilitate vaginal application. The fluconazole release from the gel was sustained showing 30.69 ± 1.02% drug deposition in the porcine vaginal mucosa at the end of 8 h with improved antifungal activity against Candida albicans during well diffusion studies. The optimized gel was non-irritant to the vaginal mucosa of female Wistar rats with no signs of erythema or edema.







Similar content being viewed by others


References
Mashburn J. Vaginal infections update. J Midwifery Womens Health. 2012;57(6):629–34. https://doi.org/10.1111/j.1542-2011.2012.00246.x.
Sobel JD, Faro S, Force RW, Foxman B, Ledger WJ, Nyirjesy PR, et al. Vulvovaginal candidiasis: epidemiologic, diagnostic, and therapeutic considerations. Am J Obstet Gynecol. 1998;178(2):203–11. https://doi.org/10.1016/S0002-9378(98)80001-X.
Spampinato C, Leonardi D. Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agents. Biomed Res Int. 2013;2013:1–13. https://doi.org/10.1155/2013/204237.
Valenta C. The use of mucoadhesive polymers in vaginal delivery. Adv Drug Deliv Rev. 2005;57(11):1692–712. https://doi.org/10.1016/j.addr.2005.07.004.
Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today. 2014;9(2):223–43. https://doi.org/10.1016/j.nantod.2014.04.008.
Patravale V, Dandekar P, Jain R. Nanoparticulate drug delivery: perspectives on the transition from laboratory to market; Elsevier 2012, DOI: https://doi.org/10.1533/9781908818195.
Natarajan JV, Nugraha C, Ng XW, Venkatraman S. Sustained-release from nanocarriers: a review. J Control Release. 2014;193:122–38. https://doi.org/10.1016/j.jconrel.2014.05.029.
Ensign LM, Cone R, Hanes J. Nanoparticle-based drug delivery to the vagina: a review. J Control Release. 2014;190:500–14. https://doi.org/10.1016/j.jconrel.2014.04.033.
Shidhaye S, Vaidya R, Sutar S, Patwardhan A, Kadam V. Solid lipid nanoparticles and nanostructured lipid carriers-innovative generations of solid lipid carriers. Curr Drug Deliv. 2008;5(4):324–31. https://doi.org/10.2174/156720108785915087.
de Araújo Pereira RR, Bruschi ML. Vaginal mucoadhesive drug delivery systems. Drug Dev Ind Pharm. 2012;38(6):643–52. https://doi.org/10.3109/03639045.2011.623355.
Das Neves J, Bahia M. Gels as vaginal drug delivery systems. Int J Pharm. 2006;318(1):1–14. https://doi.org/10.1016/j.ijpharm.2006.03.012.
Vanić Ž, Škalko-Basnet N. Nanopharmaceuticals for improved topical vaginal therapy: can they deliver? Eur J Pharm Sci. 2013;50(1):29–41. https://doi.org/10.1016/j.ejps.2013.04.035.
Goodman LS. Goodman and Gilman’s the pharmacological basis of therapeutics. New York: McGraw-Hill; 1996.
Faro S. Systemic vs. topical therapy for the treatment of vulvovaginal candidiasis. Infect Dis Obstet Gynecol. 1994;1(4):202–8. https://doi.org/10.1155/S1064744994000098.
Shen H, Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2006;58(9):1183–91. https://doi.org/10.1211/jpp.58.9.0004.
Joshi M, Patravale V. Nanostructured lipid carrier (NLC) based gel of celecoxib. Int J Pharm. 2008;346(1):124–32. https://doi.org/10.1016/j.ijpharm.2007.05.060.
Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A. Nanostructured lipid carriers (NLC): a potential delivery system for bioactive food molecules. Innovative Food Sci Emerg Technol. 2013;19:29–43. https://doi.org/10.1016/j.ifset.2013.03.002.
Kaur S, Nautyal U, Singh R, Singh S, Devi A. Nanostructure lipid carrier (NLC): the new generation of lipid nanoparticles. Asian Pac J Health Sci. 2015;2:76–93.
Sanad RA, AbdelMalak NS, Badawi AA. Formulation of a novel oxybenzone-loaded nanostructured lipid carriers (NLCs). AAPS PharmSciTech. 2010;11(4):1684–94. https://doi.org/10.1208/s12249-010-9553-2.
Gupta M, Vyas SP. Development, characterization and in vivo assessment of effective lipidic nanoparticles for dermal delivery of fluconazole against cutaneous candidiasis. Chem Phys Lipids. 2012;165(4):454–61. https://doi.org/10.1016/j.chemphyslip.2012.01.006.
Sharma R, Kaur G, Kapoor DN. Fluconazole loaded cubosomal vesicles for topical delivery. Int J Drug Dev Res. 2015;7(1):32–41.
Han F, Yin R, Che X, Yuan J, Cui Y, Yin H, et al. Nanostructured lipid carriers (NLC) based topical gel of flurbiprofen: design, characterization and in vivo evaluation. Int J Pharm. 2012;439(1):349–57. https://doi.org/10.1016/j.ijpharm.2012.08.040.
Aher ND, Nair HA. Bilayered films based on novel polymer derivative for improved ocular therapy of gatifloxacin. Sci World J. 2014;2014:1–9. https://doi.org/10.1155/2014/297603.
Bachhav YG, Patravale VB. Microemulsion based vaginal gel of fluconazole: formulation, in vitro and in vivo evaluation. Int J Pharm. 2009;365(1):175–9. https://doi.org/10.1016/j.ijpharm.2008.08.021.
Khurana S, Jain N, Bedi P. Nanoemulsion based gel for transdermal delivery of meloxicam: physico-chemical, mechanistic investigation. Life Sci. 2013;92(6):383–92. https://doi.org/10.1016/j.lfs.2013.01.005.
Khullar R, Kumar D, Seth N, Saini S. Formulation and evaluation of mefenamic acid emulgel for topical delivery. Saudi Pharm J. 2012;20(1):63–7. https://doi.org/10.1016/j.jsps.2011.08.001.
Fang J-Y, Fang C-L, Liu C-H, Su Y-H. Lipid nanoparticles as vehicles for topical psoralen delivery: solid lipid nanoparticles (SLN) versus nanostructured lipid carriers (NLC). Eur J Pharm Biopharm. 2008;70(2):633–40. https://doi.org/10.1016/j.ejpb.2008.05.008.
Cirri M, Bragagni M, Mennini N, Mura P. Development of a new delivery system consisting in “drug–in cyclodextrin–in nanostructured lipid carriers” for ketoprofen topical delivery. Eur J Pharm Biopharm. 2012;80(1):46–53. https://doi.org/10.1016/j.ejpb.2011.07.015.
Squier CA, Mantz MJ, Schlievert PM, Davis CC. Porcine vagina ex vivo as a model for studying permeability and pathogenesis in mucosa. J Pharm Sci. 2008;97(1):9–21. https://doi.org/10.1002/jps.21077.
Martín-Villena M, Fernández-Campos F, Calpena-Campmany A, Bozal-de Febrer N, Ruiz-Martínez M, Clares-Naveros B. Novel microparticulate systems for the vaginal delivery of nystatin: development and characterization. Carbohydr Polym. 2013;94(1):1–11. https://doi.org/10.1016/j.carbpol.2013.01.005.
Wavikar P, Vavia P. Nanolipidgel for enhanced skin deposition and improved antifungal activity. AAPS PharmSciTech. 2013;14(1):222–33. https://doi.org/10.1208/s12249-012-9908-y.
Mendes A, Silva A, Catita J, Cerqueira F, Gabriel C, Lopes C. Miconazole-loaded nanostructured lipid carriers (NLC) for local delivery to the oral mucosa: improving antifungal activity. Colloids Surf B: Biointerfaces. 2013;111:755–63. https://doi.org/10.1016/j.colsurfb.2013.05.041.
Bassi P, Kaur G. Bioadhesive vaginal drug delivery of nystatin using a derivatized polymer: development and characterization. Eur J Pharm Biopharm. 2015;96:173–84. https://doi.org/10.1016/j.ejpb.2015.07.018.
Kumar L, Reddy M, Shirodkar R, Pai G, Krishna V, Verma R. Preparation and characterisation of fluconazole vaginal films for the treatment of vaginal candidiasis. Indian J Pharm Sci. 2013;75(5):585–90.
Deshkar SS, Patil AT. Development of mucoadhesive gel of fluconazole for vaginal candidiasis. Indo Am J Pharm Res. 2015;5(11):3599–610.
Acknowledgements
The authors wish to acknowledge Glenmark Research Center (Taloja, India), Gattefosse India Pvt. Ltd (Mumbai, India), Signet Chemical Corporation (Mumbai, India), Cremer Oleodivison (Germany), Nikkol (Japan), Abitec Corporation (USA) and Lubrizol (India) for providing the gift samples of drug and excipients.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The experimental protocol for the study was approved by the Animal Ethics Committee of Mumbai Veterinary College (MVC/IAEC/12/2016).
Rights and permissions
About this article

Cite this article
Takalkar, D., Desai, N. Nanolipid Gel of an Antimycotic Drug for Treating Vulvovaginal Candidiasis—Development and Evaluation. AAPS PharmSciTech 19, 1297–1307 (2018). https://doi.org/10.1208/s12249-017-0918-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-017-0918-7