
Abstract
The purpose of this study was to evaluate the usefulness of the in vitro biorelevant gastrointestinal transfer (BioGIT) system in assessing the impact of dose and formulation on early exposure by comparing in vitro data with previously collected human plasma data of low solubility active pharmaceutical ingredients. Eight model active pharmaceutical ingredients were tested; Lu 35-138C (salt of weak base in a HP-beta-CD solution, three doses), fenofibrate (solid dispersion, tablet, two doses), AZD2207 EQ (salt of weak base, capsule, three doses), posaconazole (Noxafil® suspension, two doses), SB705498 (weak base, tablets vs. capsules), cyclosporine A (Sandimmun® vs. Sandimmun® Neoral), nifedipine (Adalat® capsule vs. Macorel® tablet), and itraconazole (Sporanox® capsule vs. Sporanox® solution). AUC0–0.75h values were calculated from the apparent concentration versus time data in the duodenal compartment of the BioGIT system. Differences in AUC0–0.75h values were evaluated versus differences in AUC0–1h and in AUC0–2h values calculated from previously collected plasma data in healthy adults. Ratios of mean AUC0–0.75h, mean AUC0–1h, and mean AUC0–2h values were estimated using the lowest dose or the formulation with the lower AUC0–0.75h value as denominator. The BioGIT system qualitatively identified the impact of dose and of formulation on early exposure in all cases. Log-transformed mean BioGIT AUC0–0.75h ratios correlated significantly with log-transformed mean plasma AUC0–1h ratios. Based on this correlation, BioGIT AUC0–0.75h ratios between 0.3 and 10 directly reflect corresponding plasma AUC0–1h ratios. BioGIT system is a valuable tool for the assessment of the impact of dose and formulation on early exposure to low solubility drugs.
Similar content being viewed by others


INTRODUCTION
Early exposure, after oral administration, is clinically relevant in situations where rapid onset of drug action is desired. Relevant examples include therapies for breakthrough pain, analgesia, and insomnia (e.g., 1). When a lipophilic active pharmaceutical ingredient (API) is administered orally, early exposure is controlled by the performance of the drug product in the stomach and the upper small intestine, i.e., the dissolution process. In the case of a weak base or an enabling formulation, there is a potential for the API to maintain supersaturated concentrations in the upper small intestine. In cases where luminal supersaturation is aimed and/or luminal precipitation is likely, in vitro drug product performance is usually evaluated on a sequential basis by simulating the transfer from the stomach into the small intestine (2,3,4,5).
The biorelevant gastrointestinal transfer (BioGIT) system is a recently proposed in vitro setup for evaluating the impact of gastrointestinal transfer on drug product performance in the fasted state (4). The design of BioGIT has been based on luminal data, after administration of highly permeable APIs to healthy adults, i.e., the conditions in the duodenal compartment take the transport of a highly permeable API via the epithelium of upper small intestine and the transit through the upper small intestine into account (4). BioGIT comprises commercially available equipment and has been shown to be useful in reproducing concentrations measured in the upper small intestine after administration of lipophilic weak bases formulated as solutions, immediate release products, and/or disintegrating enabling formulations (6,7). With BioGIT, evaluation of concentrations in the upper small intestine during the first hour after drug administration is possible without the need of in silico modeling.
In this study, we evaluated for the first time the usefulness of BioGIT data in assessing the impact of dose and formulation on early exposure of eight model lipophilic APIs by comparing data with previously collected plasma data in healthy adults. A diverse range of formulations, including enabling formulations, were employed. Physicochemical and biopharmaceutical characteristics of the model APIs along with the doses and formulations tested in the present investigation are presented in Table I.
MATERIALS AND METHODS
Materials
Lu 35-138C crystalline salt (hydrochloric salt ≈ 100% purity) and aqueous solution (1 mg Lu 35-138 base equivalent per mL) were kindly provided by H. Lundbeck A/S (Valby, Denmark). Excipients in the solution were 10% (w/v) hydroxypropyl-beta-cyclodextrin (HP-β-CD) and 10 ppm neohespiridine dihydrochalcone. Fenofibrate crystalline powder (purity > 99%, Midas Pharma Ingelheim, Germany) and fenofibrate tablets (54 and 160 mg/tablet) containing drug in solid dispersion were kindly provided by Abbvie (AbbVie Deutschland GmbH & Co, Knollstrasse, Germany). AZD2207 EQ crystalline salt (hemi-1,5-naphthalenedisulfonate salt, 98% purity) and AZD2207 EQ capsules (0.5, 5, and 45 mg AZD2207 base equivalent/cap) were kindly provided by AstraZeneca R&D Gothenburg, (Mölndal, Sweden). Excipients in capsules were lactose monohydrate and sodium stearyl fumarate. SB705498 crystalline free base (100% purity), immediate release tablets (25 mg/tablet), and capsules (200 mg/cap) were manufactured and donated by GSK (Ware, UK). Each capsule contained two 100 mg immediate release tablets and anhydrous lactose as a filler. Excipients in the SB705498 tablets were lactose monohydrate, cellulose microcrystalline, sodium starch glycolate, magnesium stearate, and hydroxypropyllmethylcellulose. Cyclosporine A (cyclosporine) crystalline powder and commercially available self-emulsifying drug delivery systems, Sandimmun® capsules (100 mg/cap) and Sandimmun Neoral® capsules (25 and 100 mg/cap), were kindly provided by Novartis (Basel, Switzerland). Nifedipine crystalline powder was obtained from Sigma Aldrich (St. Louis, USA). Soft gelatin capsules (Adalat®, 10 mg/cap) were obtained from Bayer S.p.A. (Milan, Italy) and immediate release tablets (Macorel®, 10 mg/tab) were obtained from Elpen S.A. (Athens, Greece). Posaconazole crystalline powder (purity 100%) was kindly provided by the Chemical Research Division of MSD (Merck & Co., Inc., Kenilworth, NJ, USA). Itraconazole crystalline powder (purity 100%) was kindly provided by Janssen Research and Development (Beerse, Belgium). Sodium phosphate monobasic, sodium hydroxide, sodium chloride, ammonium formate, and pepsin from porcine gastric mucosa (15.8% protein) were purchased from Sigma Aldrich (St. Louis, USA). Hydrochloric acid was from Panreac Co. (Barcelona, Spain). Acetonitrile, 2-propanol, trifluoroacetic acid, formic acid, and water (HPLC grade) were also from Sigma Aldrich (St. Louis, USA). SIF® Powder Original was kindly donated by biorelevant.com (Surrey, UK).
Methods
Equilibrium Solubility Measurements in Biorelevant Media
Equilibrium solubility measurements were performed in Level III FaSSGF and Level II FaSSIF based on the recently proposed four levels of simulation of luminal composition, Level 0, Level I, Level II, and Level III (17). Level III FaSSGF simulates the pH, buffer capacity, total bile salt concentration, total (lyso)phosphatidylcholine concentration and pepsin concentration in the bulk gastric contents after administration of a glass of water in the fasted state. Level II FaSSIF simulates similar characteristics in the fasted upper small intestine without taking into account the presence of proteins or enzymes (17).
Excess amounts of Lu 35-138C, fenofibrate, AZD2207 EQ, posaconazole, SB705498, cyclosporine, or itraconazole were placed into 5-mL vials. Three milliliters of Level III FaSSGF (Level I FaSSGF for posaconazole) or Level II FaSSIF (17) were added to each vial. Vials were placed in a shaking incubator (75 oscillations/min, 37°C) until equilibrium was reached (16–24 h, data not shown). At equilibrium, samples were filtered through a regenerated cellulose 0.2-μm filter and concentrations were measured. Adsorption of APIs to the filter had been evaluated and found to be negligible in all cases. Measurements were performed in triplicate.
BioGIT Experiments
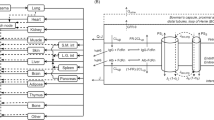
BioGIT is an open in vitro setup simulating drug dissolution and transfer from the fasted stomach into the upper small intestine, after drug administration with a glass of water (4, Fig. 1). The initial volume of the gastric compartment is 250 mL [a mini vessel with 500 mL capacity from Erweka (Heusenstamm, Germany) is used]. The duodenal volume is maintained at 40 mL during the entire experiment [a mini vessel with 100 mL capacity from Distek (New Brunswick, NJ, USA) is used]. The mini-paddles in gastric and duodenal compartments rotate at 75 rpm. The emptying of contents of the gastric compartment (on a volume basis) follows first-order kinetics with a half-life of 15 min (4). Experiments are performed at 37°C for 45 min using a three-channel peristaltic pump (Reglo ICC pump, part ISM 4308, Ismatec®). Incoming flow rates are changed every 10 min and sampling is performed at midpoint (4).

Schematic representation of the BioGIT system. F1 and F2 are the incoming flow rates and F is the outgoing flow rate; F=F1 + F2
Due to limited availability of tablets, experiments with SB705498 tablets were performed in duplicate. All other experiments were performed in triplicate.
Contents of Gastric Compartment
For Lu 35-138C, three dose levels were investigated (Table I): 15, 40, or 75 mL of the HP-β-CD solution were brought to the gastric compartment containing 235, 210, and 175 mL Level III FaSSGF, respectively. For fenofibrate, two dose levels were investigated (Table I); at each dose level, one solid dispersion tablet was brought to the gastric compartment containing 250 mL Level III FaSSGF. For AZD2207 EQ, three dose levels were investigated (Table I); capsules were brought to the gastric compartment containing 250 mL Level III FaSSGF. For SB705498, one dose in the form of tablets or capsules (Table I) was brought to the gastric compartment containing 250 mL Level III FaSSGF. One dose of cyclosporine in the form of Sandimmun® capsules or Sandimmun Neoral® capsules as indicated in Table I was brought to the gastric compartment containing 250 mL Level III FaSSGF. For nifedipine, one dose in the form of Adalat® capsule or Macorel® tablet (Table I) was brought to the gastric compartment containing 250 mL Level III FaSSGF.
Contents of Duodenal Compartment at Time Zero and Contents of Reservoir Compartment
Level II FaSSIF was employed in the duodenal compartment in order to simulate fasted state conditions in the upper small intestine. A series of phosphate buffer solutions containing sodium chloride, sodium taurocholate, and phosphatidylcholine were employed in the reservoir compartment so that the composition of contents in the duodenal compartment (pH, buffer capacity, osmolality, sodium taurocholate concentration, and phosphatidylcholine concentration) remained unaltered during an experiment (4).
Sample Treatment
Upon collection, each sample from the duodenal compartment was immediately filtered through regenerated cellulose filters (Titan 3, 17 mm, 0.2 μm, SUN SRi, Rockwood, USA). Adsorption on to the filter had been evaluated and found to be negligible in all cases. The filtrate was divided in two portions. The first was immediately diluted with mobile phase (so that precipitation during subsequent handling was avoided) for measuring apparent concentration. The second portion was used for measuring apparent equilibrium solubility by incubating it (37°C, 75 oscillations/min) in the presence of excess of solid material of compounds until equilibrium was reached. The duration of incubation for achieving equilibration was evaluated with preliminary experiments (16–24 h, data not shown). In experiments with Lu 35-138C, specifically, upon collection, each sample was divided in two parts. In the first part, total Lu 35-138 amount per volume (solid and in solution, expressed in μg/mL) was measured. The second part was filtered immediately and treated as described above for all other APIs.
Analytical Methods
Quantification of the model compounds was performed with HPLC and the chromatographic conditions are presented in Table II.
Data Treatment
AUC0–0.75h values were calculated from the apparent concentration versus time data in the duodenal compartment of the BioGIT, after each experiment, using data at 5, 15, 25, 35, and 45 min. AUC0–0.75h was used to estimate early exposure. The same procedures were applied also to BioGIT data collected previously with Noxafil® suspension at two posaconazole dose levels, 40 mg (6) and 400 mg (4), and with itraconazole (200 mg) using two different products, Sporanox® capsules (100 mg/cap; spray-dried solid dispersion in HPMC) and Sporanox® HP-β-CD solution (10 mg/mL) (6).
To get an estimate of the absorption process during a period similar to the period in which the in vitro data were collected, AUC0–1h was used as metric of early exposure in vivo. However, the number of plasma samples during the first hour after administration was limited, and hence to reduce variability of the estimated early exposure in vivo (22), AUC0–2h values were also calculated. The impact of dose and/or formulation on AUC0–0.75h was evaluated versus the impact of dose and/or formulation on AUC0–1h and AUC0–2h values calculated from previously measured individual plasma concentration versus time data, after single dose administrations of identical doses, formulations, and strength of dose units (unless otherwise specified) to fasted healthy adults with a glass of water. AUC values until the last quantifiable concentration in plasma (AUC0–tlast) were also calculated. In all cases, AUCs were estimated with the linear trapezoidal rule.
For Lu 35-138, individual plasma data, after single dose administrations of 15, 40, and 75 mg Lu 35-138 equivalent of Lu 35-138C as HP-β-CD solution to 6, 5, and 7 fasted volunteers, respectively, were available from H. Lundbeck A/S (data on file). AUC0–1h and AUC0–2h estimations were based on plasma concentrations at 0.5 and 1 h post-dosing and at 0.5, 1, 1.5 and 2 h post-dosing, respectively. Median Tmax values were 5, 4, and 3.5 h, after the 15, 40, and 75 mg dose, respectively.
For fenofibrate, individual plasma data, after single dose administrations under fasting conditions of one 54 mg and one 160 mg solid dispersion tablet to 20 and 19 volunteers, respectively, were available from Abbvie (data on file). AUC0–1h and AUC0–2h estimations were based on plasma concentrations at 1 h post-dosing and at 1 and 2 h post-dosing, respectively. Median values for Tmax, after the 54 mg dose administration and after the 160 mg dose administration, were identical, 2.0 h.
For AZD2207, individual plasma data, after single dose administrations of 8, 30, and 100 mg AZD2207 equivalent of AZD2207 EQ capsules to 6 fasted volunteers were available from AstraZeneca (data on file). AUC0–1h and AUC0–2h estimations were based on plasma concentrations at 0.33, 0.65, and 1 h post-dosing and at 0.33, 0.65, 1, and 2 h post-dosing, respectively. Median Tmax values were 2, 4.5, and 2.75 h, after the 8 mg, the 30 mg, and the 100 mg dose, respectively.
For posaconazole, plasma data, after single dose administrations of 40 mg (1 mL, Noxafil® suspension, 40 mg/mL) to 5 fasted volunteers and 400 mg (10 mL, Noxafil® suspension, 40 mg/mL) to 5 fasted volunteers, were extracted from published mean plasma profiles (23,24) using WebPlotDigitizer 4.0 (TX, USA). The formulation and doses that had been administered were identical to those previously used for collecting the BioGIT data (6). AUC0–1h and AUC0–2h estimations were based on mean plasma concentrations at 0.5 and 1 h post-dosing and at 0.5, 1, 1.5, and 2 h post-dosing, respectively. Tmax values were 3.5 h (median value, KULeuven data on file) and 6 h (from the mean plasma profile) after the 40 and 400 mg dose, respectively.
For SB705498, individual plasma data, after single dose administrations of tablets and capsules to 19 and 12 fasted volunteers, respectively, were available (GSK data on file). AUC0–1h estimations were based on plasma concentrations at 0.25, 0.5, 0.75, and 1 h post-dosing for the tablets and at 0.5 and 1 h post-dosing for the capsules. AUC0–2h estimations were based on plasma concentrations at 0.25, 0.5, 0.75, 1, 1.5, and 2 h post-dosing for the tablets and at 0.5, 1, 1.5, and 2 h post-dosing for the capsules. Median Tmax values were 2 and 1.5 h, after the administration of the tablet and capsule formulation, respectively.
For cyclosporine, individual plasma data, after single dose administrations of 300 mg Sandimmun® capsules (3 × 100 mg/cap) and of 180 mg Sandimmun® Neoral capsules (3 × 60 mg/cap) to 24 healthy fasted adults on a crossover basis have previously been published (25). It is acknowledged, therefore, that, in the present investigation, Sandimmun® Neoral capsules were tested at a slightly lower dose level (175 mg, Table I), compared with the dose level previously used for collecting the plasma data (180 mg), due to unavailability of the 60 mg capsule strength. AUC0–1h and AUC0–2h estimations were based on plasma concentrations at 0.25, 0.5, 0.75, and 1 h post-dosing and at 0.25, 0.5, 0.75, 1, 1.5. and 2 h post-dosing, respectively. Median Tmax values were 2 and 1.5 h, after the Sandimmun® capsules and the Sandimmun® Neoral capsules, respectively.
For nifedipine, mean plasma data after single dose administration of one Adalat® capsule to 7 fasted adults and of one Macorel® tablet to 18 fasted adults were extracted from published mean plasma profiles (26,27) using WebPlotDigitizer 4.0 (Texas, USA). Mean AUC0–1h and mean AUC0–2h estimations after Adalat® administration were based on mean plasma concentrations at 0.16, 0.5, 0.75, and 1 h post-dosing and at 0.16, 0.5, 0.75, 1, 1.25, 1.5, 1.75, and 2 h post-dosing, respectively. Mean AUC0–1h and mean AUC0–2h estimations, after Macorel® administration were based on mean plasma concentrations at 0.17, 0.33, 0.5, 0.75 and 1 h post-dosing and at 0.17, 0.33, 0.5, 0.75, 1, 1.33, 1.67, and 2 h post-dosing, respectively. From the mean profiles, Tmax values were observed to be 0.16 and 1.33 h, after the Adalat® capsule and after the Macorel® tablet, respectively.
Individual plasma concentrations of itraconazole and hydro-itraconazole, after single dose administrations of 200 mg itraconazole in the form of Sporanox® capsules and Sporanox® solution to 5 healthy adults on a crossover basis, were provided by KULeuven (28). The dose and formulations that had been administered were identical to those previously used for collecting the BioGIT data (6). AUC0–1h and AUC0–2h estimations were based on individual plasma concentrations at 0.5 and 1 h post-dosing and at 0.5, 1, 1.5, and 2 h post-dosing, respectively, after administration of both formulations. Median Tmax values were 2.5 h and 2 h after Sporanox® capsules and Sporanox® solution, respectively.
The impact of dose on AUC0–0.75h, on AUC0–1h, on AUC0–2h, and on AUC0–tlast was evaluated with a one way ANOVA and the Holm–Sidak post hoc test for Lu 35-138 and AZD2207 and with unpaired t test for fenofibrate. The impact of SB705498 formulation on AUC0–1h, on AUC0–2h, and on AUC0–tlast value was evaluated with unpaired t test. The impact of cyclosporine formulation on AUC0–0.75h was evaluated with unpaired t test whereas the impact of formulation on AUC0–1h and on AUC0–2h was evaluated with paired t test. The impact of posaconazole dose, nifedipine formulation, and itraconazole formulation on AUC0–0.75h was evaluated with unpaired t test. The impact of posaconazole dose and of nifedipine formulation on AUC0–1h, on AUC0–2h, and on AUC0–tlast values could not be evaluated statistically, as complete sets of individual plasma profiles or SD values for each API were not available. Since the normality test failed, the impact of itraconazole formulation on AUC0–1h and on AUC0–2h was evaluated with the Wilcoxon test. All statistical comparisons were performed at the 0.05 level using SigmaPlot for Windows version 11.0 (Systat Software, Inc.).
Statistical Evaluation of the Usefulness of BioGIT Data for the Prediction of Differences in Early Exposure
To allow for a statistical analysis of all available data together, the following normalization was applied: mean AUC0–0.75h values estimated from BioGIT data, and mean AUC0–1h and AUC0–2h values estimated from plasma data were transformed into ratios of mean BioGIT AUC values and ratios of mean plasma AUC values by using the lowest dose (of Lu 35-138C, fenofibrate, AZD2207 EQ, and posaconazole) as the denominator or the formulation (of SB705498, cyclosporine, nifedipine, and itraconazole) with the lower AUC0–0.75h in BioGIT as the denominator. Initially, two Spearman rank order correlations were performed; one between mean BioGIT AUC0–0.75h ratios and mean plasma AUC0–1h ratios and one between mean BioGIT AUC0–0.75h ratios and mean plasma AUC0–2h ratios. The relative standard deviation (RSD) values and standard errors (SE) of the ratios of mean BioGIT AUC0–0.75h values and, depending on the availability of individual plasma levels, of mean plasma AUC0–1h and AUC0–2h values were estimated using standard error propagation methods. RSD values of the ratios of plasma AUC0–1h values or of plasma AUC0–2h values were 4–75 times greater than the RSD values of the corresponding ratios of BioGIT AUC0–0.75h values. Therefore, a simple linear regression analysis was performed with the BioGIT AUC0–0.75h ratios as the independent variable (29), for assessing the quantitative relationship between the ratios of mean plasma AUCs and the ratios of mean BioGIT AUCs. To achieve a normal and homoscedastic distribution of the points around the regression line (29), a logarithmic transformation of both the plasma AUC ratios and the BioGIT AUC ratios was applied.
RESULTS AND DISCUSSION
Solubility Classification of Model Compounds
Based on data shown in Fig. 2, dose/solubility ratios for the doses tested in the present investigation (Table I) ranged from 0.24 L (AZD2207 EQ, 8 mg dose) to 417 L (itraconazole, 200 mg dose) in Level III FaSSGF and from 0.51 L (nifedipine, 10 mg dose) to 444 L (itraconazole, 200 mg dose) in Level II FaSSIF. Therefore, all APIs at the doses tested in the present investigation were low solubility APIs, according to the Biopharmaceutics Classification System (31) and according to the Development Classification System (32).

Mean + SD (n = 3) values for apparent equilibrium solubility of Lu 35-138, fenofibrate, AZD2207, posaconazole, SB705498, cyclosporine, nifedipine, and itraconazole in Level III FaSSGF (black bars) and Level II FaSSIF (white bars). Posaconazole data were collected in Level I FaSSGF. Nifedipine data from a previous publication (30) in Level ΙI FaSSGF and Level II FaSSIF-V2 are shown. In some cases, the SD bar may not be visible, due to the small SD value
BioGIT Data for Assessing the Impact of Dose on Early Exposure
Lu 35-138C HP-β-CD Solutions
At each dose level, apparent concentrations of Lu 35-138 (non-complexed and complexed with HP-β-CD) were almost identical to the corresponding total amounts per volume, suggesting no precipitation in the duodenal compartment during the entire experiment (Fig. 3a).

Total amount per volume (black symbols, mean ± SD values), apparent concentration (white symbols, mean ± SD values) and apparent equilibrium solubility (gray symbols) in the duodenal compartment of BioGIT versus time after the initiation of the experiment using various doses of Lu 35-138C in a HP-beta-CD solution, of fenofibrate solid dispersion tablets and of AZD2207 EQ capsules in the gastric compartment. a Lu 35-138, 15 mg (circles), 40 mg (diamonds), and 75 mg (squares); b fenofibrate, 54 mg (circles) and 160 mg (diamonds); c AZD2207, 8 mg (circles), 30 mg (diamonds), and 100 mg (squares)
At the lowest dose level (15 mg), mean apparent concentrations of Lu 35-138 in the duodenal compartment were slightly higher than or similar to the apparent equilibrium solubility. Mean(SD) (n = 3) values of the equilibrium solubility in the duodenal compartment at each time point ranged from 28.26(0.69) to 22.13(0.66) μg/mL (Fig. 3a). Since mean(SD) solubility of Lu 35-138C in Level II FaSSIF was 25.17(0.11) μg/mL (Fig. 2), it could be observed that at the 15 mg dose level, HP-β-CD concentration in the duodenal compartment was too small to induce substantial solubilization effects in the duodenal compartment.
In contrast, at the two higher doses (40 and 75 mg) HP-β-CD concentrations were adequate to solubilize Lu 35-138 (Fig. 3a, gray diamonds and squares versus Fig. 1, Lu 35-138C solubility in Level II FaSSIF). In addition, contents of the duodenal compartment were apparently supersaturated, i.e., concentrations were higher than the apparent equilibrium solubility in the duodenal compartment, in all cases, except the 45 min time point with the 40 mg dose (Fig. 3a, white versus gray diamonds). Cyclodextrins, apart from solubilizing drugs through the formation of dynamic inclusion complexes (33), can also inhibit nucleation and crystal growth through non-complex based mechanisms (34); bile salts can exclude the drug from the cyclodextrin cavity and displacement can trigger supersaturation (35).
Mean AUC0–0.75h values estimated from BioGIT data increased with the dose and the impact of dose was significant (Table III, p < 0.001). All post hoc pairwise comparisons resulted in statistically significant differences. In vivo only the non-complexed compound can be considered for absorption and, as indicated from relatively long Tmax values for a BCS Class II API [median Tmax values 3.5-5 h were observed (H. Lundbeck A/S, data on file)], the absorption process was extended. The complexation efficiency (36) of Lu 35-138 with HP-β-CD, i.e., the product of the association constant (kas) with the solubility of Lu 35-138 in the medium could provide an idea on whether changes in total drug concentrations directly reflect changes in free drug concentrations. Complexation efficiency = 3714 M−1 (H. Lundbeck A/S, data in file) × 6 × 10−4 (M, solubility in Level II FaSSIF, based on Fig. 2) = 0.22. This value indicates that changes in total concentrations can reflect changes in free drug concentrations (37). In line with BioGIT data, mean AUC0–1h and AUC0–2h values estimated from plasma data increased with the dose (Table III) and the impact of dose was significant (p < 0.001). All post hoc pairwise comparisons of AUC0–1h values and of AUC0–2h values resulted in statistically significant differences.
AUC0–tlast values also varied with the dose in a similar manner (H. Lundbeck A/S data on file, p < 0.001) and post hoc comparisons resulted in significant difference in all cases. These data suggest that differences in the absorption rates of Lu 35-138 with the dose from the upper small intestine, after administration as complex with HP-β-CD were maintained during the entire absorption process.
Fenofibrate Solid Dispersion Tablets
Contents of the duodenal compartment were apparently supersaturated with fenofibrate at times longer than 5 min, after the initiation of the experiment with the 54 mg tablet or with the 160 mg tablet (Fig. 3b). Mean(SD) (n = 3) apparent solubility value at each time point ranged from 12.1(3.2) to 14.91(0.60) μg/mL, in line with mean(SD) apparent solubility in Level II FaSSIF 14.42(0.15) μg/mL (Fig. 2).
At 5 min, apparent concentrations of fenofibrate were below or close to saturation, reflecting the lower concentrations in the gastric compartment during the tablet disintegration process. Mean AUC0–0.75h values calculated from BioGIT data were higher at the 160 mg dose level (Table III) and the difference from the data collected at the 54 mg dose level was statistically significant (p = 0.003). Similarly, mean AUC0–1h and AUC0–2h values calculated from previously collected plasma data were higher after administration of the 160 mg tablet. Differences of AUC0–1h and AUC0–2h values between doses were statistically significant (p < 0.001 for both comparisons) and similar with that observed in vitro (Table III).
It is interesting to note that the difference between the two doses in AUC0–tlast values estimated from plasma data was in line with BioGIT data and statistically significant; mean(SD) values were calculated to be 53(20) μg × h/mL and 116(61) μg × h/mL, after the 54 and 160 mg dose, respectively (Abbvie data on file, p < 0.001). This could be related with the fact that, based on the median Tmax values (2.0 h after both the 54 mg and the 160 mg administration), absorption was almost complete from the upper/middle small intestine, i.e., differences up to about 2 h post-dosing reflected differences in total amounts absorbed.
AZD2207 EQ Capsules
At the lowest dose level (8 mg), the apparent concentration in the duodenal compartment was close or below the apparent saturation level at all time points (Fig. 3c, white versus gray circles). Mean(SD) (n = 3) apparent equilibrium solubility at each time point ranged from 7.98(0.28) to 8.27(0.28) μg/mL, in line with mean(SD) solubility in Level II FaSSIF [8.58(0.31), Fig. 2].
In contrast, at higher doses (30 and 100 mg) contents of the duodenal compartment were apparently supersaturated (Fig. 3c, white versus gray diamonds and white versus gray squares) during the first 25 min and the first 35 min for the 30 mg dose and the 100 mg dose, respectively.
Mean AUC0–0.75h values calculated from BioGIT data increased with the dose (Table III), especially from the 8 mg dose to the 30 mg dose (Table III). The impact of dose was significant (p < 0.001) and all post hoc pairwise comparisons resulted in statistically significant differences.
Mean AUC0–1h and AUC0–2h values calculated from previously collected plasma data increased with dose (Table III). ANOVA indicated significant difference among the AUC0–2h values (p = 0.023) and post hoc pairwise comparisons of AUC0–2h values indicated statistically significant difference between the 8 mg dose and 100 mg dose. ANOVA did not indicate significant difference among the AUC0–1h values (p = 0.079); however, the statistical power for the AUC0–1h comparisons was low, 0.34.
BioGIT AUC0–0.75h, plasma AUC0–1h, and plasma AUC0–2h values normalized by the corresponding doses indicated that absorption early after administration of the highest dose was slower. However, the ratio [AUC0–tlast of the highest dose/AUC0–tlast of the lowest dose] was close to the ratio of the two doses, i.e., 11 and 12, respectively. This was in line with previous estimations on the linearity of AUC with the dose up to 100 mg (11) and implies problematic absorption rates but not extent of absorption for highest dose. Slower absorption at the highest dose could be related with increased precipitation in upper small intestine at higher doses. Such possibility was in line with the longer median Tmax values at higher doses (2, 4.5, and 2.75 h for the 8, 30, and 100 mg dose, respectively (AZ data in file).
Posaconazole Suspension
Based οn literature data, contents in the duodenal compartment were apparently supersaturated with posaconazole when using a 40 mg dose (the degree of apparent supersaturation, i.e., [apparent concentration]/[apparent equilibrium solubility], was about 3.4), but saturated when using the 400 mg dose (degree of apparent supersaturation was about 1) (4,6). Previously reported grand mean apparent solubility in the duodenal compartment (3.27 μg/ml) (6) was similar to the mean(SD) apparent solubility in Level II FaSSIF 2.370(0.080) μg/mL (Fig. 2). The difference in supersaturation between the two doses could be attributed to the more extensive precipitation for the 400 mg dose, due to the increased presence of undissolved particles coming from the gastric compartment. Such explanation was also in line with the extended Tmax value after the 400 mg dose (6 h) compared with the Tmax value observed after the 40 mg dose (3.5 h). Mean AUC0–0.75h values calculated from BioGIT data were higher at the 40 mg dose level (Table III) and the difference from the data collected at the 400 mg dose level was statistically significant (p < 0.001). Mean AUC0–1h values were also higher at the 40 mg dose level. The ratio [AUC0–tlast of the 400 mg dose/AUC0–tlast of the 40 mg dose] (4.8) was much lower than the ratio of the two doses (10), implying that the increased precipitation in upper small intestine, after the 400 mg dose, decreased, also, the total % absorbed.
BioGIT Data for Assessing the Impact of Formulation on Early Exposure
SB705498 Immediate Release Products
Contents of the duodenal compartment were apparently supersaturated in all cases, apart from 5 min when the capsule was used (Fig. 4a). At 5 min, the capsule shell was not dissolved in the gastric compartment. Mean(SD) (n = 3) apparent equilibrium solubility at each time point during the experiment with capsules ranged from 2.39(0.21) to 2.58(0.15) μg/mL. Mean (n = 2) apparent equilibrium solubility at each time point during the experiment with tablets ranged from 2.77 to 3.39 μg/mL. These values were similar to the mean(SD) (n = 3) values estimated in Level II FaSSIF [2.728 (0.012) μg/mL (Fig. 1)].

Αpparent concentration [white symbols; mean ± SD values (continuous lines) or individual values (dotted lines] and apparent equilibrium solubility (gray symbols) in the duodenal compartment of BioGIT versus time after the initiation of the experiment using various formulations of SB705498, cyclosporine, and nifedipine in the gastric compartment. a SB705498 tablets (circles, dose 400 mg) and capsules (diamonds, dose 400 mg); b Sandimmun® (circles, dose 300 mg) and Sandimmun Neoral® (diamonds, dose 175 mg); c Adalat® capsules (diamonds, dose 10 mg) and Macorel® tablets (circles, dose 10 mg)
Mean AUC0–0.75h values estimated from BioGIT data were higher for the tablets (Table IV). The difference between the two formulations in the AUC0–1h values and in the AUC0–2h values estimated from previously collected plasma data (Table IV) were statistically significant (p = 0.025 and p = 0.002, respectively). The difference in total exposure was also significant (p = 0.004); [dose tablet/dose capsule = 1 whereas [AUC0–tlast tablet/AUC0–tlast capsule] = 1.7 (GSK data on file). This may be related to the short duration of absorption and its completion in the upper/middle small intestine (median Tmax values were 2 and 1.5 h after tablets and capsules, respectively).
Cyclosporine Lipid-Based Products
The concentrations in the duodenal compartment were below the apparent saturation level at all times for both products (Fig. 4b). During the experiments with Sandimmun® capsules, mean (n = 2) values of the apparent equilibrium solubility of cyclosporine ranged from 36.4 to 42.5 μg/mL (Fig. 4b), similar to the mean (SD) (n = 3) values estimated in Level II FaSSIF [43.76(0.34) μg/mL (Fig. 1)]. In contrast, during the experiment with Sandimmun Neoral® capsules, a much higher apparent equilibrium solubility of cyclosporine was measured at most time points ranging from 38.6 to 3172 μg/mL (n = 5, median 1747 μg/mL) and reflecting the increased solubilizing capacity of excipients in the Sandimmun Neoral® formulation. However, it should also be considered that the efficiency of separation of lipid particles from the micellar phase of contents in the duodenal compartment during the filtration (cutoff size 0.2 μm) may be a potential issue with self-emulsifying drug delivery systems.
Mean AUC0.75h values calculated from the BioGIT data were higher for Sandimmun Neoral® capsules, in line with both the mean AUC0–1h and the mean AUC0–2h values calculated from previously collected plasma data (Table IV). Differences between the two tested products in AUC0–0.75h in AUC0–1h values and in AUC0–2h values were significant (p < 0.001 for each comparison). However, differences between the two products in early exposure in vivo, i.e., between AUC0–1h or between AUC0–2h, were overestimated by BioGIT data (Table IV), most likely, due to the inefficient separation of lipid particles from the micellar phase of contents of duodenal compartment of BioGIT.
Nifedipine Products
Contents of the duodenal compartment of BioGIT were apparently unsaturated with nifedipine in all cases, except in one case: 15 min after the initiation of the experiment with Adalat® capsule contents were apparently saturated (Fig. 4c). Mean(SD) (n = 3) apparent solubility value at each time point ranged from 23.8(3.5) to 24.8(3.0) μg/mL, slightly higher than the apparent solubility in Level II FaSSIF-V2 previously reported (19.77 μg/mL (Fig. 2). Mean AUC0–0.75h values calculated from BioGIT data were higher when Adalat® capsule was used (Table IV) and the difference from the data collected with Macorel® tablet was statistically significant (p < 0.001). The difference was in line with the mean differences between Macorel® tablet and Adalat® capsule for AUC0–1h values (Table IV), AUC0–2h values (Table IV), Tmax values (1.33 h Macorel® tablet vs. 0.16 h Adalat® capsule), and Cmax values (44.2 μg/mL Macorel® tablet vs. 110 μg/mL Adalat® capsule) (26,27). Due to the low dose (based on the apparent solubility values), differences in early exposure were not reflected in total exposure for this highly permeable API; [dose Adalat® capsule/dose Macorel® tablet = 1 whereas [AUC0–tlast Adalat® capsule/AUC0–tlast Macorel® tablet] = 0.96.
Itraconazole Products
Based on literature data, contents of the duodenal compartment were significantly supersaturated with itraconazole during the entire duration of the experiment with both formulations, with the solution resulting to higher degrees of supersaturation (42–102) compared with the capsule (3.3–18) (6). Mean apparent solubility in the duodenal compartment during the experiments were 0.47 and 1.19 μg/mL with the capsule and the solution, respectively (6). These values were in line with the mean(SD) apparent solubility of itraconazole in Level II FaSSIF 0.45(0.12) μg/mL (Fig. 2) and the solubilizing effect of HP-β-CD. Mean AUC0–0.75h values calculated from BioGIT data were higher when using Sporanox® solution (Table IV) and the difference from the data collected with Sporanox® capsules was significant (p < 0.001). Mean AUC0–1h and mean AUC0–2h values were also higher with Sporanox® solution (Table IV), however, not statistically different from the corresponding Sporanox® capsule values (p = 0.063 and p = 0.063, respectively). Based on the association constant of itraconazole with HP-beta-cyclodextrin (approximately 2000 M−1 at pH 7 (38) and the solubility in Level II FaSSIF (0.45 μg/mL) (Fig. 2), complexation efficiency (estimated as described above for Lu 35-138) is very low (0.0013) and changes in total drug concentrations should directly reflect changes in free drug concentrations (37). Therefore, the lack of significant differences between the two formulations in early exposure in vivo was most likely due to the low power of the in vivo comparisons (0.110 and 0.223 for the AUC0–1h and the AUC0–2h comparison, respectively).
Statistical Evaluation of the Usefulness of BioGIT Data for the Prediction of Differences in Early Exposure
Scatter plots of the ratios of mean plasma AUC0–1h and AUC0–2h values versus ratios of mean BioGIT AUC0–0.75h values were constructed, after excluding cyclosporine data (Supplemental 1). Cyclosporine data, although trending to the correct direction, they were outliers (in vivo ratios are 2.3 and 1.7 whereas in vitro ratio is 40), due to inefficient separation of lipid particles from the micellar phase of contents of duodenal compartment and, thus, substantial overestimation of difference in early exposure by the BioGIT data. Spearman rank order correlations was significant for both the AUC0–1h vs. AUC0–0.75h ratios (n = 9; p < 0.0001; R = 0.87) and for the AUC0–2h versus AUC0–0.75h ratios (n = 9; p < 0.0001; R = 0.87), suggesting that BioGIT data were useful for qualitative evaluation of differences up to 2 h after administration.
The linear regression of the log-transformed ratios of mean plasma AUC0–1h values with the log-transformed ratios of mean BioGIT AUC0–0.75h values was significant [p = 0.001; power = 0.935; slope 0.91(0.18); intercept 0.28(0.10)]. The Pearson correlation coefficient was 0.89. Best fitted line and associated 95% confidence limits are presented graphically in Fig. 5. Based on this correlation, BioGIT AUC0–0.75h ratios of two doses or two different products between 0.3 and 10 directly reflect corresponding plasma AUC0–1h ratios.

The best fitted line and the associated 95% confidence limits for the regression of log-transformed ratios of mean plasma AUC0–1h values vs. the log-transformed ratios of mean BioGIT AUC0–0.75h values. Ratios were estimated by using the lowest dose or the formulation with the lower AUC0–0.75h in BioGIT as the denominator
The linear regression of the ratios of log-transformed ratios of mean plasma AUC0–2h values with the log-transformed ratios of mean BioGIT AUC0–0.75h values was also significant (p = 0.022). However, the less favorable statistical characteristics (power = 0.650; R = 0.74) suggest that despite the higher accuracy in estimating early exposure, raw BioGIT data may not be used for quantitative evaluation of differences for periods longer than about 1 h after administration.
One issue with the validation of novel methodologies in regard to their biorelevance is the existence of in vivo data of formulations which are appropriate for the specific setup and for which dose units are still available to be tested in the in vitro model. The OrBiTo project provided this opportunity but, still, for some of the formulations, only few data points could be used (based on the existing plasma data). This was the main reason for testing AUC values estimated up to 1 h and up to 2 h post-dosing.
CONCLUDING REMARKS
The present study demonstrated that the BioGIT system is useful for qualitative detection of the impact of dose and of formulation on early exposure. Importantly, BioGIT AUC0–0.75h ratios of two doses or two different products between 0.3 and 10 directly reflect corresponding plasma AUC0–1h ratios.
Data of this study confirm earlier observations based on which the BioGIT system is useful for estimating drug concentrations in the upper small intestine during the first hour after administration in the fasted state, especially when supersaturation of contents and/or drug precipitation are involved. Relevant information could be very useful in physiologically based pharmacokinetic (PBPK) modeling. Based on a series of recent publications from the OrBiTo project (39,40,41), prospective PBPK modeling of plasma profiles, after oral administration, using a bottom-up approaches is limited by the reliability of estimation of clearance and volume of distribution, quality of intestinal permeability data, quality of solubility data, and characterization of intestinal metabolism or transporter involvement. The development of methodologies for implementing BioGIT data into PBPK models and methodologies for the evaluation of processes taking place in the middle/lower small intestine using the BioGIT system as a basis would greatly improve PBPK modeling approaches and facilitate the development of orally administered drug products.
References
Selen A, Dickinson PA, Müllertz A, Crison JR, Mistry HB, Cruañes MT, et al. The biopharmaceutics risk assessment roadmap for optimizing clinical drug product performance. J Pharm Sci. 2014;103:3377–97.
Carino SR, Sperry DC, Hawley M. Relative bioavailability estimation of carbamazepine crystal forms using an artificial stomach-duodenum model. J Pharm Sci. 2006;95:116–25.
Tsume G, Amidon L, Takeuchi S. Dissolution effect of gastric and intestinal pH for BCS class II drug, pioglitazone: new in vitro dissolution system to predict in vivo dissolution. J Bioequiv Availab. 2013;5:224–7.
Kourentas A, Vertzoni M, Stavrinoudakis N, Symillidis A, Brouwers J, Augustijns P, et al. An in vitro biorelevant gastrointestinal transfer (BioGIT) system for forecasting concentrations in the fasted upper small intestine: design, implementation, and evaluation. Eur J Pharm Sci. 2016;80:106–14.
Ruff A, Holm R, Kostewicz ES. Evaluating the predictability of the in vitro transfer model and in vivo rat studies as a surrogate to investigate the supersaturation and precipitation behaviour of different albendazole formulations for humans. Eur J Pharm Sci. 2017;105:108–18.
Kourentas A, Vertzoni M, Symillides M, Hens B, Brouwers J, Augustijns P, et al. In vitro evaluation of the impact of gastrointestinal transfer on luminal performance of commercially available products of posaconazole and itraconazole using BioGIT. Int J Pharm. 2016;515:352–8.
Kourentas A, Vertzoni M, Khadra I, Symillides M, Clark H, Halbert G, et al. Evaluation of the impact of excipients and an albendazole salt on albendazole concentrations in upper small intestine using an in vitro biorelevant gastrointestinal transfer (BioGIT) system. J Pharm Sci. 2016;105:2896–903.
Holm R, Andresen L, Strange C. Oral bioavailability of a poorly aqueous drug from three different SBE7-β-cyclodextrin based formulations in beagle dogs. Results Pharma Sci. 2011;1:57–9.
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing Fenofibrate. AAPS J. 2007;9:E344–52.
Clarysse S, Brouwers J, Tack J, Annaert P, Augustijns P. Intestinal drug solubility estimation based on simulated intestinal fluids: comparison with solubility in human intestinal fluids. Eur J Pharm Sci. 2011;43:260–9.
Tannergren C, Karlsson E, Sigfridsson K, Lindfors L, Ku A, Polentarutti B, et al. Biopharmaceutic profiling of salts to improve absorption of poorly soluble basic drugs. J Pharm Sci. 2016;105:3314–23.
Courtney R, Wexler D, Radwanski E, Lim J, Laughlin M. Effect of food on the relative bioavailability of two oral formulations of posaconazole in healthy adults. Br J Clin Pharmacol. 2004;57:218–22.
Czogalla A. Oral cyclosporine A—the current picture of its liposomal and other delivery systems. Cell Mol Biol Lett. 2009;14:139–52.
el Tayar N, Mark AE, Vallat P, Brunne RM, Testa B, van Gunsteren WF. Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: evidence from partition coefficients and molecular dynamics simulations. J Med Chem. 1993;36:3757–64.
Wagner C, Thelen K, Willmann S, Selen A, Dressman JB. Utilizing in vitro and PBPK tools to link ADME characteristics to plasma profiles: case example nifedipine immediate release formulation. J Pharm Sci. 2013;102:3205–19.
Augustijns P, Wuyts B, Hens B, Annaert P, Butler J, Brouwers J. A review of drug solubility in human intestinal fluids: implications for the prediction of oral absorption. Eur J Pharm Sci. 2014;57:322–32.
Markopoulos C, Andreas CJ, Vertzoni M, Dressman J, Reppas C. In-vitro simulation of luminal conditions for evaluation of performance of oral drug products: choosing the appropriate test media. Eur J Pharm Biopharm. 2015;93:173–82.
Griffin BT, Kuentz M, Vertzoni M, Kostewicz E, Fei Y, Faisal W, et al. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: case studies with fenofibrate. Eur J Pharm Biopharm. 2014;86:427–37.
Psachoulias D, Vertzoni M, Butler J, Busby D, Symillides M, Dressman J, et al. An in vitro methodology for forecasting luminal concentrations and precipitation of highly permeable lipophilic weak bases in the fasted upper small intestine. Pharm Res. 2012;29:3486–98.
Ran Υ, Zhao L, Xu Q, Yalkowsky S. Solubilization of cyclosporin A. AAPS Pharm Sci Tech. 2001;2:23–6.
Vertzoni M, Reppas C, Archontaki H. Sensitive and simple liquid chromatographic method with ultraviolet detection for the determination of nifedipine in canine plasma. Anal Chim Acta. 2006;28:298–304.
Endrenyi L, Csizmadia F, Tothfalusi L, Balch AH, Chen M-L. The duration of measuring partial AUCs for the assessment of bioequivalence. Pharm Res. 1998;5:399–404.
Hens B, Brouwers J, Corsetti M, Augustijns P. Supersaturation and precipitation of posaconazole upon entry in the upper small intestine in humans. J Pharm Sci. 2016 Sep;105(9):2677–84.
Walravens J, Brouwers J, Spriet I, Tack J, Annaert P, Augustijns P. Effect of pH and comedication on gastrointestinal absorption of posaconazole. Clin Pharmacokinet. 2011;50:725–34.
Kovarik JM, Mueller EA, van Bree JB, Tetzloff W, Kutz K. Reduced inter- and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J Pharm Sci. 1994;83:444–6.
Rashid TJ, Martin U, Clarke H, Waller DG, Renwick AG, George CF. Factors affecting the absolute bioavailability of nifedipine. Br J Clin Pharmacol. 1995 Jul;40(1):51–8.
Niopas I, Daftsios AC, Xanthakis I, Nikolaidis N. Relative bioavailability study of two nifedipine tablet formulations in healthy male volunteers. Int J Clin Pharmacol Ther. 2000 Jun;38(6):309–14.
Brouwers J, Geboers S, Mols R, Tack J, Augustijns P. Gastointestinal behavior of itraconazole in humans—part 1: supersaturation from a solid dispersion and a cyclodextrin-based solution. Int J Pharm. 2017;525:211–7.
Sokal R.R. and Rohlf F.J. in «Biometry», pp540–549, 2nd edition, 1981, W.H. Freeman and Co, New York.
Andreas CJ, Tomaszewska I, Muenster U, van der Mey D, Mueck W, Dressman JB. Can dosage form-dependent food effects be predicted using biorelevant dissolution tests? Case example extended release nifedipine. Eur J Pharm Biopharm. 2016;105:193–202.
Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20.
Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J Pharm Sci. 2010;99:4940–54.
Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–35.
Brewster M, Neeskens P, Peeters J. Solubilization of itraconazole as a function of cyclodextrin structural space. J Incl Phenom Macro Chem. 2007;57:561–6.
Stappaerts J, Augustijns P. Displacement of itraconazole from cyclodextrin complexes in biorelevant media: in vitro evaluation of supersaturation and precipitation behavior. Int J Pharm. 2016;511:680–7.
Loftsson T, Brewster ME. Cyclodextrins as functional excipients: methods to enhance complexation efficiency. J Pharm Sci. 2012;101:3019–32.
Olesen NE, Westh P, Holm R. Displacement of drugs from cyclodextrin complexes by bile salts: a suggestion of an intestinal drug-solubilizing capacity from an in vitro model. J Pharm Sci. 2016;105:2640–7.
Peeters J, Neeskens P, Tollenaere JP, Van Remoortere P, Brewster ME. Characterization of the interaction of 2-hydroxypropyl-beta-cyclodextrin with itraconazole at pH 2, 4, and 7. J Pharm Sci. 2002 Jun;91(6):1414–22.
Margolskee A, Darwich AS, Pepin X, Pathak SM, Bolger MB, Aarons L, et al. IMI-oral biopharmaceutics tools project—evaluation of bottom-up PBPK prediction success part 1: characterisation of the OrBiTo database of compounds. Eur J Pharm Sci. 2017;96:598–609.
Darwich AS, Margolskee A, Pepin X, Aarons L, Galetin A, Rostami-Hodjegan A, et al. IMI - Oral biopharmaceutics tools project—evaluation of bottom-up PBPK prediction success part 3: identifying gaps in system parameters by analysing in silico performance across different compound classes. Eur J Pharm Sci. 2017;96:626–42.
Margolskee A, Darwich AS, Pepin X, Aarons L, Galetin A, Rostami-Hodjegan A, et al. IMI - Oral biopharmaceutics tools project—evaluation of bottom-up PBPK prediction success part 2: an introduction to the simulation exercise and overview of results. Eur J Pharm Sci. 2017;96:610–25.
Acknowledgements
Most of the work was performed within the OrBiTo project which is funded by the Innovative Medicines Initiative Joint Undertaking under Grant Agreement No 115369. Nifedipine data were collected outside the OrBiTo project at Astellas Pharma Europe B.V., Leiden, during the internship of V. Barmpatsalou. The authors would like to thank N. Stavrinoudakis (National and Kapodistrian University of Athens) for collecting the itraconazole solubility data in Level III FaSSGF and in Level II FaSSIF.
Furthermore, this article is based upon work from COST Action UNGAP, supported by COST (European Cooperation in Science and Technology).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(DOCX 45 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kourentas, A., Vertzoni, M., Barmpatsalou, V. et al. The BioGIT System: a Valuable In Vitro Tool to Assess the Impact of Dose and Formulation on Early Exposure to Low Solubility Drugs After Oral Administration. AAPS J 20, 71 (2018). https://doi.org/10.1208/s12248-018-0231-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-018-0231-8