
Abstract
Background
Pathogenic variants in the androgen receptor (AR) gene located on chromosome Xq11-12, are known to cause varying degrees of undermasculinization in 46, XY individuals. The aim of this study was to investigate the frequency of pathogenic variants in the AR gene in a cohort of 46, XY undermasculinized individuals from Indonesia who were suspected of having androgen insensitivity syndrome (AIS). All patients with 46, XY DSD referred to our center between 1994 and 2019 were collected from our clinical database. All 46, XY DSD patients without a prior molecular diagnosis with an external masculinization score (EMS) ≤ 9 were included in this study. All exons and intron–exon boundaries of AR gene were analyzed using Sanger sequencing to identify pathogenic variants of the AR gene.
Results
A cohort of 75 undermasculinized patients were selected for the study. Direct Sanger sequencing of all eight exons of the AR gene led to a genetic diagnosis in 11 patients (14.67%). All of the variants identified (p.Arg841His; p.Ile604Asn; p.Val731Met; p.Pro672Ser; p.Gln739Arg; p.Ser302Glufs*3) have been previously reported in patients with AIS.
Conclusions
This is the first study in Indonesia that highlights the significance of molecular analysis in providing a definitive diagnosis of AIS for patients with 46, XY DSD undermasculinization. This is an uncommon finding in the Indonesian population presenting with 46, XY DSD undermasculinization. A genetic diagnosis allows optimal clinical management and genetic counseling for patients and their families. As 46, XY DSD can be caused by pathogenic variants in other genes involved in gonadal development and differentiation, further genetic analysis, such as whole exome sequencing, should be carried out on those patients that did not carry an AR variant.
Similar content being viewed by others

Background
Individuals with disorders/differences of sex development (DSD) are often identified with atypical genitalia at birth or absence of secondary sex characteristics at adolescence [1,2,3]. This condition can result either from disorders in androgen synthesis or action, non-hormonal anatomical disorders, or disruption of any stages of gonadal development [4, 5]. It is important to note that disorders of androgen synthesis or action share common dysmorphic characteristics with others 46, XY DSD conditions [6]. Therefore, molecular diagnosis may help clinicians to establish a definitive diagnosis and assign clinical management plan for 46, XY DSD patients with undermasculinization in cases where hormonal data is inconclusive or where the imaging or biopsy of the gonads has not been possible.
Androgen insensitivity syndrome (AIS) is one of the most well-recognized causes of 46, XY DSD [7, 8]. AIS (OMIM# 300068) is a disorder of sexual differentiation resulting from complete or partial resistance to the biological action of androgens. Due to resistance to androgens, AIS causes defective masculinization of external genitalia in 46, XY individuals regardless of normal androgen synthesis. The clinical phenotype of AIS shows a wide spectrum of sex phenotypes, ranging from complete feminization of the external genitalia, to male undermasculinization and infertility.
The frequency of AIS is estimated to be between 1 in 20,000 and 64,000 male births [9]. AIS is caused by pathogenic variants in the androgen receptor (AR) gene which is located on the long arm of the X chromosome (Xq11-12). The AR gene consists of eight exons which encode a 919 amino acid protein comprising of four major functional domains: the N-terminal domain (NTD), the central DNA-binding domain (DBD), a C-terminal ligand-binding domain (LBD), and a hinge region connecting the LBD to the DBD [10]. The pattern of inheritance of AIS in most cases is inherited in an X-linked manner, although sporadic de novo variants have been identified [11, 12]. The most prominent molecular defects in the AR gene associated with AIS are missense variants predominantly clustered in the LBD region [7, 13]. Nevertheless, pathogenic variants of the AR gene have also been identified throughout the whole coding region [14].
Here, we performed molecular analysis of the AR gene in a cohort of 46, XY patients who presented with varying degrees of undermasculinization.
Methods
Patients recruitment
Seventy-five patients with clinical and endocrinological findings consistent with 46,XY undermasculinization were included in this study, as determined by our multidisciplinary team (MDT). They were referred to our center for comprehensive evaluation of DSD. Levels of luteinizing hormone (LH), follicle stimulating hormone (FSH), and total testosterone (T) assay were performed in collaboration with a commercial laboratory. G-banded karyotyping of peripheral blood lymphocytes was performed for all patients in our laboratory. Approval was obtained by our Ethics Committee before conducting the study, and all patients and/or parents gave informed consent before they participated in the study (No. 24/EC/FK-RSDK/I/2017). Patients with a 46, XY karyotype, no prior genetic finding, and an external masculinization score (EMS) ≤ 9 were included in our genetic studies. The degree of undermasculinization was evaluated using the EMS, which summarizes clinical features in patients with ambiguous genitalia, taking into account scrotal fusion, presence of microphallus, site of urethral meatus, and presence/location and appearance of gonads [15, 16]. Where available, we included patients with increased basal T and LH and normal or slightly increased FSH levels suggestive of AIS [17].
AR gene analysis
Genomic DNA from patients was isolated from blood leukocytes using salting-out methods as described elsewhere [18]. We performed direct Sanger sequencing analysis of the AR gene in patients with an EMS ≤ 9 to confirm the molecular diagnosis of AIS. All eight exons and the intron–exon boundaries of the AR gene were amplified by PCR using specific primers pairs as reported by Listyasari et al. [12]. DNA annotations are based on NM_000044.2; protein change annotations are based on NP_000035.2; all variant annotations were confirmed using Mutalyser [19]. Nucleotide numbering starts at 1, at the A of the ATG initiation codon.
Results
A cohort of 1039 patients with clinical phenotype DSD were collected by our center between 2004 and 2019. Of these patients, 778 patients were classified as 46, XY DSD. Based on clinical diagnosis, 304 patients with 46, XY DSD with undermasculinization were identified and a total of 75 undermasculinized patients with EMS ≤ 9 were selected for this study. The clinical profiles of the patients are shown in Table 1. The mean age of the patients was 10.43 years (SD 10.54). The majority of the participant (64 individuals, 85.33%) were diagnosed in early childhood (> 1 year old) while only 11 (14.67%) of the individuals were diagnosed in infancy (< 1 year). During the initial consultation, 55 (73.33%) of the patients were being raised as males, while 19 (25.33%) of the patients were being raised as females and there was only 1 (1.33%) patient whose gender was not yet decided on. We have previously reported patients 10, 16, 18 [20], and 29 and 37 [16].
Clinical presentation
All patients presented with a wide range of atypical genitalia (Table 1). Forty out of 75 (53%) patients had bifid scrotum, 59 patients (79%) had micropenis, and most of the patients 68 (91%) had severe hypospadias. Bilateral abdominal testes were present in 17 (23%) patients; 49 (65%) patients had scrotal position, and in 9 (12%) patients were unilateral undescended testis. The EMS from this study was established in a median score of 5.5 (ranging from 1 to 9).
Hormonal analysis
In all 75 patients, basal hormonal measurements were obtained. However, 45 patients were pre-pubertal, making the hormonal assay very difficult to interpret. Among 30 patients in pubertal age, 11 had elevated or normal basal serum T levels associated with high serum LH levels followed by normal FSH levels, indicative of AIS. Seven patients had endocrine data more consistent with gonadal dysgenesis with low T levels, elevated LH and FSH levels, and 12 patients had inconclusive endocrine data.
Molecular analysis
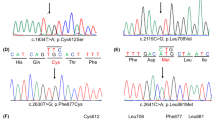
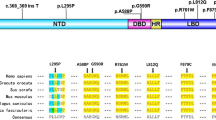
Direct Sanger sequencing of the AR gene in 75 individuals with 46,XY DSD led to the identification of 6 unique variants in 11 subjects (14.67%) (Table 2). A schematic diagram of mutations distribution of AR gene in this study is presented in Fig. 1. The majority of the variants we identified were single-nucleotide missense substitutions: c.1811T>A (p.Ile604Asn), c.2014C>T (p.Pro672Ser), c.2191G>A (p.Val731Met), c.2216A>G (p.Gln739Arg), and c.2522G>A (p.Arg841His). All of the missense variants we identified have been previously described in individuals with AIS. We also identified a single-nucleotide duplication at position c.902dup which is predicted to lead to a frame-shift and a premature stop codon at position 304 (p.Ser302Glufs*3). The identified variants did not seem to cluster to any specific protein domains: one variant was predicted to fall in the N-terminal domain (p.Ser302Glufs*3), one was in the DNA-binding domain (p.Ile604Asn), another was in the hinge region (p.Pro672Ser), while the other three variants were in the androgen-binding domain (p.Val731Met; p.Gln739Arg and p.Arg841His).

Schematic diagram showing the AR gene and the localization of the six pathogenic variants identified in the present study. The exons of the AR gene are indicated numbered boxes. AR: androgen receptor
All of the patients mentioned above (patient 7, 8, 9, 10, 13, 16, 18, 24, 29, 37, 51, and 68) carried pathogenic variants in the AR gene. Only 4 patients (7, 8, 9, 10) reached pubertal age, which allowed the hormonal assays to be interpreted. They all showed hormonal profiles with increased LH and T, and slightly increased FSH levels, consistent with AIS. All of these patients had atypical genitalia with severe hypospadias, bifid scrotal, a palpable gonad in scrotal region, and micropenis. All these patients had a low EMS ≤ 9 which is consistent with an AIS clinical profile.
Molecular analysis in a familial case of AIS
In this study, we identified the c.2522G>A;p.Arg841His pathogenic variant in an extended family with multiple affected [patients 7 (III:14), 8 (III:16), 9 (III:8), and 68 (IV:4)] across three generations (see pedigree Fig. 2), and this pathogenic variant has been reported before in patients with PAIS [14, 21,22,23,24]. Based on clinical evaluation, patients 7 (III:14), 8 (III:16), and 9 (III:8) were seen at our clinic as adults; they were raised as females but later had undergone gender reassignment. At presentation, they had severe hypospadias, bilateral palpable gonads, and presented gynecomastia at puberty then later clinically diagnosed as PAIS (Fig. 3a). While patient 68 (IV:4) was referred to our clinic when she was 1 year old, she presented with female external genitalia with bilateral palpable gonads in labial region and was diagnosed as CAIS (Fig. 3b). Her mother (III:5) and maternal grandmother (II:4) were found to be carriers of the same pathogenic variant c.2522G>A; p.R841H in the AR gene which was carried over generations in this family.

Pedigree of the family 1. The proband is indicated by an arrow. Black-filled symbols represent affected individuals; open symbols represents unaffected individuals; black spots represents carrier individuals; squares: males; circle: females

The clinical presentation of AIS within family 1. a Noted bifid scrotal, micropenis, severe hypospadias, bilateral palpable testes in scrotal region in patient 7 (III:14). b Noted minimal scrotalization, severe hypospadias and bilateral palpable testes in scrotal region in patient 68 (IV:4)
Discussion
We report here a cohort of Indonesian patients with suspected AIS and AR gene variants. The age of presentation varied among patients; however, a large proportion of the patients were referred to our center at a later age (mean:10.43 years old), while only 14.67% patients were identified in early childhood (< 1 year). Consistent with our results, Shawky et al. (2012) reported that diagnosis of patients with a DSD was often delayed, with adults as the most common presenting group in Egypt [27]. Undermasculinization can pose a diagnostic dilemma since the patients were often not aware of their condition until they experienced delayed puberty. In addition, DSD patients in Indonesia can face delays in management due to social stigmatization, cultural and religious issues, as well as inadequate laboratory facilities or a lack of health insurance to support medical treatments including hormonal and genetic examination [28, 29]. This is in contrast to developed countries, where DSD is usually identified before or shortly after birth due to atypical genitalia or sex discordance detected with prenatal testing [30, 31]. Early diagnosis can benefit patients and support parents in coping with experiences such as stigma and gender dysphoria. It can improve the quality of life, including allowing decisions about the child’s sex of rearing and surgery, rather than the shock of a late diagnosis [32, 33].
We identified numerous AR gene variants in our cohort. In our previous study, we reported pathogenic variants in the AR gene in patient 10, 16, and 18 [20]. The AR c.1811T>A; p.(Ile604Asn) pathogenic variant in patient 10 is located in the DBD region of the AR, close to the D box in the second zinc cluster which is involved in AR dimerization and hormone-responsive element. The p.Ile604Asn has been shown to display normal translocation to the nucleus upon hormone stimulation but has completely lost its DNA-binding capacity as shown by live cell imaging, which explains its total lack of transcriptional activity from two distinct promoters [20]. The c.2014C>T; p.(Pro672Ser) in patient 16 is located in the helix 1 of the LBD and encoded by exon 4 of the AR gene. The c.2216A>G; p.(Gln739Arg) pathogenic variant in patient 18 caused substitution of the polar yet uncharged glutamine residue by a charged arginine residue caused a PAIS phenotype [20].
The c.2191G>A; p.(Val731Met) in patient 13, 24, and 51 has been reported before in two patients with prostate cancer [25, 26]. It has been proposed that the replacement of p.Val731Met allows for cellular proliferation with low-androgen concentrations [26]. Newmark et al. identified codon 730 as a highly conserved region among all steroid receptors therefore this pathogenic variant may impair its transcriptional activity by preventing binding to the target genes [25]. However, the biologic pathway for undermasculinization caused by this pathogenic variant is still not fully understood.
In our previous study, patient 37 has been reported as a male with undermasculinization [16]. The frameshift mutation c.902dup; p.(Ser302Glufs*3) in patient 37 is located in in the first exon of the AR gene. The frameshift mutation results in a truncated protein of three amino acids. The pathogenic variant was located in the NTD domain containing the transcriptional regulatory region; thus, it potentially causes disruption of transcriptional factor binding sites of AR gene that would induce partial phenotype in this patients [10, 34].
A familial case of AIS with diverse phenotypic expressivity is presented in this study. DNA analysis has shown a missense mutation in exon 7 of AR gene (c.2522G>A) leading to a change of arginine into histidine at position 841 (p.Arg841His) in the ligand binding domain of the AR gene. This particular codon is reported as a mutational ‘hotspot’ [13]. Imasaki et al. and Mazen et al. reported phenotypic diversity in families carrying the same variant, in which the index patient was female but the other family members were reared as boys [23, 35].
Phenotypic diversity among siblings with PAIS who share the same pathogenic variant has been well documented [23, 24]. This intrafamilial variance may be due to factors other than abnormalities in the AR gene, such as co-regulators affecting the functional characteristics of androgen receptor, including ligand selectivity and DNA-binding ability [35]. In some patients, somatic mosaicism for an AR gene mutation could also be responsible [36].
The finding of EMS in this study of undermasculinized male patients showed similar results to that observed previously with this subset of AIS patients [9, 37]. The EMS itself cannot influence gender assignment, but could be used to determine when to inquire or request advice from a specialist. Based on a study by Ahmed et al., it would seem reasonable to seek a specialist’s opinion in cases of undermasculinized newborns with a score of 10 or less [9]. An additional benefit of the EMS is the ability to objectively analyze biochemical, genetic, and clinical features of AIS [9].
There are also complexities regarding gender assignments for infants with DSD. As mentioned in this study, during genetic counseling in a familial case of AIS, it was revealed that the grandmother (II:4) had not disclosed this fact to the mother (III:5) until she was giving birth to patient 68. She came to our clinic for further evaluation after knowing the baby presented with atypical genitalia which was similar to that seen other family members. When informed about the pathogenic variant inherited in this family, the parents changed their daughter’s gender to male without consent and discussion with our MDT. In the genetic counseling session, the gender identity of the affected individual must be considered with care to avoid assumption that chromosomal sex or gene variants will determine gender identity. In this patient, surgery should be postponed until the affected child can consent.
We often see 46,XY DSD patients with PAIS, who have been assigned a female gender at birth, but undergo gender change in their adolescence [38]. Therefore, the cytogenetic, hormonal, and genetic analysis should precede the gender assignment of infants with DSD. However, medical management in Indonesia faces health resource issues, socio-cultural, and legal obstacles that hinder medical options for the affected individuals [28]. Patients often seek medical consultation at a later age, when they have reached puberty, and often our team is confronted by patients with emotional distress, gender confusion, and social stigmatization [29, 39]. It is critical that patients with DSD are assigned to a multidisciplinary team who can accurately care for their needs immediately after identification.
Due to the heterogeneous nature of DSD, determining a genetic diagnosis can be challenging. In many developing countries with limited resources, genetic testing is only available for some DSD-associated genes through Sanger sequencing and is only useful when the phenotype is highly suggestive of a single gene [12]. However, single-gene sequencing can be time-consuming and costly and only yields a low-molecular diagnosis [40].
The rapidly falling costs of genomic sequencing have recently led to the use of next generation sequencing (NGS) for the genetic diagnosis of DSD [41]. A study using a targeted gene panel sequencing in a large international patient cohort with 46, XY DSD reported likely genetic diagnosis in 43% of patients [42]. NGS-based methods may in future provide a molecular diagnosis to our undiagnosed patients [43, 44].
Conclusions
In summary, our molecular diagnosis identified six unique pathogenic variants in the AR gene in a cohort of undermasculinized 46, XY DSD patients. Suspected AIS patients can share overlapping features with patients that carry variants in other genes involved in gonadal development, and a genetic finding in AR rules out the latter. Further studies using next-generation sequencing such as targeted gene panels for DSD or whole exome sequencing to identify pathogenic variants in other DSD genes should be considered. This is the first report of Indonesian patients with undermasculinized 46, XY DSD and an EMS classification who were diagnosed using molecular diagnostic approaches.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AIS:
-
Androgen insensitivity syndrome
- AR:
-
Androgen receptor
- CAIS:
-
Complete androgen insensitivity syndrome
- DBD:
-
DNA-binding domain
- DSD:
-
Disorders/differences of sex development
- EMS:
-
External masculinization score
- FSH:
-
Follicle stimulating hormone
- LBD:
-
Ligand-binding domain
- LH:
-
Luteinizing hormone
- MAIS:
-
Mild androgen insensitivity syndrome
- MDT:
-
Multidisciplinary team
- NGS:
-
Next generation sequencing
- NTD:
-
N-terminal domain
- PAIS:
-
Partial androgen insensitivity syndrome
- T:
-
Testosterone
References
Warne GL, Raza J (2008) Disorders of sex development (DSDs), their presentation and management in different cultures. Rev Endocr Metab Disord 9(3):227–236. https://doi.org/10.1007/s11154-008-9084-2
Mendonca BB, Domenice S, Arnhold IJ, Costa EM (2009) 46,XY disorders of sex development (DSD). Clin Endocrinol (Oxf) 70(2):173–187. https://doi.org/10.1111/j.1365-2265.2008.03392.x
Massanyi EZ, Dicarlo HN, Migeon CJ, Gearhart JP (2013) Review and management of 46,XY disorders of sex development. J Pediatr Urol 9(3):368–379. https://doi.org/10.1016/j.jpurol.2012.12.002
Hughes IA (2008) Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab 22(1):119–134. https://doi.org/10.1016/j.beem.2007.11.001
Wisniewski AB, Batista RL, Costa EMF, Finlayson C, Sircili MHP, Denes FT et al (2019) Management of 46,XY differences/disorders of sex development (DSD) throughout life. Endocr Rev 40(6):1547–1572. https://doi.org/10.1210/er.2019-00049
Nagaraja MR, Rastogi A, Raman R, Gupta DK, Singh SK (2010) Molecular diagnosis of 46,XY DSD and identification of a novel 8 nucleotide deletion in exon 1 of SRD5A2 gene. J Pediatr Endocrinol 23:379–385. https://doi.org/10.1515/jpem.2010.059
Jaaskelainen J (2012) Molecular biology of androgen insensitivity. Mol Cell Endocrinol 352(1-2):4–12. https://doi.org/10.1016/j.mce.2011.08.006
Berglund A, Johannsen TH, Stochholm K, Viuff MH, Fedder J, Main KM et al (2016) Incidence, prevalence, diagnostic delay, and clinical presentation of female 46,XY disorders of sex development. J Clin Endocrinol Metab 101(12):4532–4540. https://doi.org/10.1210/jc.2016-2248
Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, Rownland J et al (2000) Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 85(2):658–665. https://doi.org/10.1210/jcem.85.2.6337
Eisermann K, Wang D, Jing Y, Pascal LE, Wang Z (2013) Androgen receptor gene mutation, rearrangement, polymorphism. Transl Androl Urol 2(3):137–147. https://doi.org/10.3978/j.issn.2223-4683.2013.09.15
Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A (2008) Androgen insensitivity syndrome: clinical features and molecular defects. Hormones. 7(3):217–229. https://doi.org/10.14310/horm.2002.1201
Listyasari NA, Robevska G, Santosa A, Bouty A, Juniarto AZ, van den Bergen J et al (2019) Genetic analysis reveals complete androgen insensitivity syndrome in female children surgically treated for inguinal hernia. J Invest Surg:1–7. https://doi.org/10.1080/08941939.2019.1602690
Gottlieb B, Beitel LK, Trifiro MA (2001) Variable expressivity and mutation databases: the androgen receptor gene mutations database. Hum Mutat 17:382–388. https://doi.org/10.1002/humu.1113
Audi L, Fernández-Cancio M, Carrascosa A, Andaluz P, Torán N, Piró C et al (2010) Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J Clin Endocrinol Metab 95(4):1876–1888. https://doi.org/10.1210/jc.2009-2146
Su R, P. Adam M, Ramsdell L, Y. Fechner P, Shnorhavorian M (2015) Can the external masculinization score predict the success of genetic testing in 46,XY DSD? AIMS Genet 2(2):163–172. https://doi.org/10.3934/genet.2015.2.163
Juniarto AZ, van der Zwan YG, Santosa A, Hersmus R, de Jong FH, Olmer R et al (2012) Application of the new classification on patients with a disorder of sex development in indonesia. Int J Endocrinol 2012:237084. https://doi.org/10.1155/2012/237084
Ahmed SF, Achermann JC, Arlt W, Balen A, Conway G, Edwards Z et al (2016) Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol (Oxf) 84(5):771–788. https://doi.org/10.1111/cen.12857
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16(3):1215. https://doi.org/10.1093/nar/16.3.1215
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE (2008) Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat 29(1):6–13. https://doi.org/10.1002/humu.20654
Elfferich P, Juniarto AZ, Dubbink HJ, van Royen ME, Molier M, Hoogerbrugge J et al (2009) Functional analysis of novel androgen receptor mutations in a unique cohort of indonesian patients with a disorder of sex development. Sex Dev 3(5):237–244. https://doi.org/10.1159/000252814
Hiort O, Huang Q, Sinnecker GHG, Sadeghi-Nejad A, Kruse K, Wolfe HJ et al (1993) Single strand conformation polymorphism analysis of androgen receptor gene mutations in patients with androgen insensitivity syndromes: application for diagnosis, genetic counseling, and therapy. J Clin Endocrinol Metab 77(1):262–266. https://doi.org/10.1210/jcem.77.1.8325950
Beitel LK, Kazemi-Esfarjani P, Kaufman M, Lumbroso R, DiGeorge AM, Killinger DW et al (1994) Substitution of arginine-839 by cysteine or histidine in the androgen receptor causes different receptor phenotypes in cultured cells and coordinate degrees of clinical androgen resistance. J Clin Invest 94(2):546–554. https://doi.org/10.1172/jci117368
Imasaki K, Hasegawa T, Okabe T, Sakai Y, Haji M, Takayanagi R et al (1994) Single amino acid substitution in the hormone-binding domain of the androgen receptor leads to incomplete androgen insensitivity syndrome associated with a thermolabile androgen receptor. Eur J Endocrinol 130:569–574. https://doi.org/10.1530/eje.0.1300569
Evans BAJ, Hughes IA, Bevan CL, Patterson MN, Gregory JW (1997) Phenotypic diversity in siblings with partial androgen insensitivity syndrome. Arch Dis Child 76:529–531. https://doi.org/10.1136/adc.76.6.529
Newmark JR, Hardy DO, Tonb DC, Carter BS, Epstein JI, Isaacs WB et al (1992) Androgen receptor gene mutations in human prostate cancer. Proc Natl Acad Sci U S A 89:6319–6323. https://doi.org/10.1073/pnas.89.14.6319
Sanchez D, Rosell D, Honorato B, Lopez J, Arocena J, Sanz G (2006) Androgen receptor mutations are associated with Gleason score in localized prostate cancer. BJU Int 98(6):1320–1325. https://doi.org/10.1111/j.1464-410X.2006.06438.x
Shawky RM, Nour El-Din SM (2012) Profile of disorders of sexual differentiation in the Northeast region of Cairo, Egypt. Egypt J Med Hum 13(2):197–205. https://doi.org/10.1016/j.ejmhg.2012.03.003
Ediati A, Faradz SM, Juniarto AZ, van der Ende J, Drop SL, Dessens AB (2015) Emotional and behavioral problems in late-identified Indonesian patients with disorders of sex development. J Psychosom Res 79(1):76–84. https://doi.org/10.1016/j.jpsychores.2014.12.007
Ediati A, Juniarto AZ, Birnie E, Okkerse J, Wisniewski A, Drop S et al (2017) Social stigmatisation in late identified patients with disorders of sex development in Indonesia. BMJ Paediatr Open 1(e000130):1–9. https://doi.org/10.1136/bmjpo-2017-000130
Lee PA, Nordenstrom A, Houk CP, Ahmed SF, Auchus R, Baratz A et al (2016) Global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr 85(3):158–180. https://doi.org/10.1159/000442975
Delot EC, Papp JC, Workgroup D-TG, Sandberg DE, Vilain E (2017) Genetics of disorders of sex development: the DSD-TRN experience. Endocrinol Metab Clin North Am 46(2):519–537. https://doi.org/10.1016/j.ecl.2017.01.015
Raza J, Zaidi SZ, Warne GL (2019) Management of disorders of sex development - With a focus on development of the child and adolescent through the pubertal years. Best Pract Res Clin Endocrinol Metab 33(3):101297. https://doi.org/10.1016/j.beem.2019.101297
Ediati A, Verrips GHW, Juniarto AZ, Faradz SMH, Drop SLS, Dessens AB (2018) Quality of life in late-treated patients with disorders of sex development: insights for patient-centered care. Front Pediatr 6:434. https://doi.org/10.3389/fped.2018.00434
Kamanu FK, Medvedeva YA, Schaefer U, Jankovic BR, Archer JA, Bajic VB (2012) Mutations and binding sites of human transcription factors. Front Genet 3:100. https://doi.org/10.3389/fgene.2012.00100
Mazen I, Lumbroso S, Ghaffar SA, Salah N, Sultan C (2004) Mutation of the androgen receptor (R840S) in an Egyptian patient with partial androgen insensitivity syndrome: Review of the literature on the clinical expression of different R840 substitutions. J Endocrinol Invest 27:57–60. https://doi.org/10.1007/BF03350912
Boehmer ALM, Bruggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MCT, Niermeijer MF et al (2001) Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab 86(9):4151–4160. https://doi.org/10.1210/jcem.86.9.7825
Ahmed SF, Khwaja O, Hughes IA (2000) The role of a clinical score in the assessment of ambiguous genitalia. BJU Int 85(1):120–124. https://doi.org/10.1046/j.1464-410x.2000.00354.x
Mazur T (2005) Gender dysphoria and gender change in androgen insensitivity or micropenis. Arch Sex Behav 34(4):411–421. https://doi.org/10.1007/s10508-005-4341-x
Ediati A, Juniarto AZ, Birnie E, Drop SLS, Faradz SMH, Dessens AB (2013) Body image and sexuality in Indonesian adults with a disorder of sex development (DSD). J Sex Res 52(1):15–29. https://doi.org/10.1080/00224499.2013.816260
Ahmed SF, Bashamboo A, Lucas-Herald A, McElreavey K (2013) Understanding the genetic aetiology in patients with XY DSD. Br Med Bull 106:67–89. https://doi.org/10.1093/bmb/ldt008
Hughes LA, McKay Bounford K, Webb E, Dasani P, Clokie S, Chandran H et al (2019) Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr Connect. https://doi.org/10.1530/EC-18-0376
Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J et al (2016) Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol 17(1):243. https://doi.org/10.1186/s13059-016-1105-y
Barseghyan H, Delot EC, Vilain E (2018) New technologies to uncover the molecular basis of disorders of sex development. Mol Cell Endocrinol 468:60–69. https://doi.org/10.1016/j.mce.2018.04.003
Bever YV, Bruggenwirth HT, Wolffenbuttel KP, Dessens AB, Groenenberg IAL, Knapen M et al (2020) Under-reported aspects of diagnosis and treatment addressed in the Dutch-Flemish guideline for comprehensive diagnostics in disorders/differences of sex development. J Med Genet 57(9):581–589. https://doi.org/10.1136/jmedgenet-2019-106354
Acknowledgements
MDT members of Dr. Kariadi Hospital/National Diponegoro University Hospital.
Ethics committee
Prof. Dr. dr. Tjahjono, Sp.PA (K) FIAC: the chairman of ethics committee, Faculty of Medicine Diponegoro University.
Dr. Soejoto, PAK, Sp.KK (K): the Dean Faculty of Medicine Diponegoro University.
Funding
This study was funded by Diponegoro University Research Grant No. 474-73/UN7.P4.3/PP/2018.
Author information
Authors and Affiliations
Contributions
SMHF devised the project and the main conceptual ideas of the study. NAL performed the experiments under supervision of GR and KLA. NAL processed the experimental data, performed the analysis and interpretation of data, drafted the manuscript, and designed the figures and tables. NAL, AZJ, GR, KLA, AHS, and SMHF revised the manuscript critically for important intellectual content. All authors gave final approval of the submitted version and any substantially modified version that involves the authors’ contributions to the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of Faculty of Medicine Diponegoro University with approval number 92/EC/FK-RSDK/2008. The patient provided written consent.
Consent for publication
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient/parent/guardian/relative of the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Listyasari, N.A., Juniarto, A.Z., Robevska, G. et al. Analysis of the androgen receptor (AR) gene in a cohort of Indonesian undermasculinized 46, XY DSD patients. Egypt J Med Hum Genet 22, 14 (2021). https://doi.org/10.1186/s43042-021-00134-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00134-3