
Abstract
Intertidal macroalgae are exposed to many abiotic stress factors, and they must regularly react to changes in their environment. We used RNA-seq to describe how Porphyra umbilicalis (Rhodophyta) changes gene expression patterns to interact with different habitats. Tissue samples were taken from a typical habitat along the open-coast of the Northwest Atlantic, as well as from a rare, atypical habitat in an estuarine tidal rapid environment. Differential gene expression analyses suggest that pathogic bacteria and viruses may be a significant factor influencing the transcriptome in the human-impacted estuarine environment, but the atypical habitat does not necessarily induce more stress in Porphyra umbilicalis growing there. We found genes related to nitrogen transport are over-expressed in tissue from the open-coastal site compared to those from the estuarine site, where environmental N levels approach hypertrophic levels. Low N levels impede growth, but high levels are toxic to cells, and we use qPCR to show this species regulates expression of a putative high-affinity NH4+ transporter under low and high N conditions. Differences in expression of this transporter in these habitats appear to be inherited from parent to offspring and have general implications for adaptation to habitat in other species that are capable of asexual reproduction, as well as more specific implications for this species’ use in aquaculture.
Similar content being viewed by others
Background
Abiotic stress is a nearly universal factor affecting photosynthetic eukaryotes. Some organisms deal with stressful environmental changes better than others, but few are capable of coping with variable environmental conditions better than intertidal algae. Porphyra umbilicalis Kützing is a species of marine red algae of the North Atlantic that is known for its tolerance of desiccation (Smith and Berry 1986), rapid osmotic changes (Reed et al. 1980), and subzero temperatures (Wang et al. 2011, Green and Neefus 2014). Some of this tolerance is due to cell structure and anatomy, but the physiological mechanisms by which they respond to abiotic stress are not well understood. As a candidate for potential use in integrated multitrophic aquaculture (IMTA) to remove NH4+ produced from fish effluent (Blouin et al. 2007, Kim et al. 2007, Day et al. 2009, Green and Neefus 2016), and as a potential model organism for Rhodophyta (Gantt et al. 2010, Blouin et al. 2011), a better understanding of the effect of environment on gene expression by which it copes with stress and N uptake is necessary.
Porphyra umbilicalis typically lives on rocky substrates in the high intertidal zone, but it can also be found in estuarine tidal rapid locations on bridge pilings such as in the mouth of the Great Bay Estuary system in Dover Point, NH, USA. (West et al. 2005, Eriksen et al. 2016). Estuarine rapids are rare in the Gulf of Maine, and experience high velocity currents and changes in salinity and temperature with tidal changes. Eriksen et al. (2016) identified six Porphyra umbilicalis genotypes among populations in the Gulf of Maine based on a limited set of polymorphic microsatellite loci but showed isolation by distance is a more important factor influencing population structure than selection for habitat. This suggests the populations acclimate to different stress conditions in different habitats through physiological adjustments.
Mathieson et al. (1981b, 1981a) compared the macroalgal species composition and environmental conditions at Dover Point (DP) and a fully marine location nearby at Fort Stark (FS), located on Jaffrey Point in New Castle, NH, USA. They describe a greater range in salinities (22–31 ppt DP; 26–32 ppt FS) and water temperature (1–22 °C DP; 1–20 °C FS) at DP. Over the last 30 years, the Great Bay Estuary System and DP has experienced increasingly high N levels, largely due to discharge from sewer treatment plants in surrounding towns, and non-point sources including run-off from increasing amounts of impervious pavement and development (PREP 2013, Trowbridge et al. 2014). Nitrogen levels from 2008 to 2010 measured at nearby Cedar Point by Nettleton (2012) were classified as hypertrophic and ranged from 5.03–57.98 μM total N. In comparison, NH4+ levels at FS collected from 1973 to 1974 by Norall and Mathieson (1974) ranged from approximately 0–0.08 μM NH4+, while NO3− concentrations ranged from 0.02 to 0.23 μM. Ammonium nitrogen levels in the vicinity of DP at that time ranged from 0.06 to 0.65 μM, and NO3− concentrations ranged from 0 to 0.02 μM (Norall and Mathieson 1974).
We used RNA-seq to identify differentially expressed (DE) genes within and among populations growing in these habitats at DP and FS, NH, USA. This sampling allows us to compare gene expression between two genotypes in a single habitat (FS), but also to compare gene expression among different habitats (DP and FS) using P. umbilicalis tissue with the same genotype but also with different genotypes. Our goal was to compare gene expression patterns among libraries to describe the extent to which environment and genotype affect gene expression, to determine if one environment elicits a greater stress response, or N metabolism response than the other, and if so, to begin to describe the physiological mechanisms by which Porphyra umbilicalis copes with stress. The results shed light on how this organism interacts with its environment in atypical habitats and has implications for N uptake in IMTA systems.
Methods
Sampling
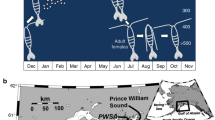
We collected tissue during high tide from two locations at Fort Stark, Jaffrey Point, New Hampshire (43° 03′ 28.3″ N 70° 42′ 41.7″ W) and from the U.S.-4/N.H.-16 bridge pilings at Dover Point, New Hampshire (43° 07′ 11.5″ N 70° 49′ 38.5″ W), mid-day in late July and early August 2012 (Fig. 1). At FS, whole thalli were removed from rocks within a 3 m radius at two locations while submersed or partially submersed. The thalli were patted dry, visible epiphytes and amphipods were carefully removed, and the tissue was flash frozen in liquid N in the field. Tissue from DP was collected during high tide from a single bridge piling where they were known to occur, inspected for preliminary species identification, cleaned of visible epiphytes, and then patted dry and frozen in liquid N in the field. We made no effort to eliminate microscopic epiphytes or endophytes, nor bacterial contaminants on or within the tissue that was demonstrated to be abundant in an earlier study by Miranda et al. (2013). A small amount of each thallus from each population was put aside while in the field in a 1.8-mL cryogenic tube for species confirmation and genotyping. Samples were returned to the lab and stored at − 80 °C until processing.

Collection map of Porphyra umbilicalis used in study
We extracted DNA from the small samples set aside for genotyping in a 96-well plate format using a modification of the method by Page (2010) described in Eriksen et al. (2016). Species identification was confirmed using restriction fragment length polymorphisms (RFLP) as described by Teasdale et al. (2002).
RNA-seq library preparation
Tissue from each population was genotyped at three polymorphic microsatellite loci according to Eriksen et al. (2016). The thallus samples were then pooled into libraries based on genotype as genotype 3 (g3) or genotype 6 (g6) (Table 1); no individuals from DP were assayed as g3. The number of individuals collected was relatively low due to small population size. The pools of tissue were ground to a fine powder in a mortar and pestle with liquid N. We extracted the total nucleic acids from the tissue as described above and in Eriksen et al. (2016). Following extraction, the total nucleic acids were treated with RNase-free DNase (1500 Kunitz units, Qiagen, Germantown, MD, USA) per the manufacturer’s instructions at the maximum volume. DNase was removed with equal volumes of 24:1 chloroform to isoamyl alcohol and centrifuged at 2440×g for 5 min at 4 °C. The supernatant was pipetted off, and 1/10 volumes of 3 M sodium acetate was added. The RNA was then precipitated with two volumes of cold 100% ethanol at − 20 °C for 1 h. The tubes were subsequently centrifuged at 12,000×g for 10 min to pellet the RNA, which was then washed with 500 μL cold 70% ethanol twice before being re-pelleted by centrifugation at 12,000×g for 2 min. The quality and quantity of RNA was assessed by evaluating concentration and A260/A280 and A260/A230 ratios on a NanoDrop 2000c spectrophotometer (ThermoFisher Scientific, Grand Island, NY, USA). Integrity of the RNA samples was measured on an Agilent 2100 Bioanalyzer at the Hubbard Center for Genomic Studies at the University of New Hampshire, Durham, USA; RNA integrity numbers (RIN) were high and ranged from 8.8 for the DP pool, to 9.2 for the FS g6 pool.
Library preparation by polyA cDNA isolation and sequencing on a HiSeq2000 platform was performed by Otogenetics Corporation (Norcross, GA, USA). Reads with adaptor contamination and low support bases were filtered out by the bioinformatics core at Otogenetics. The sequences were then mapped to the reference library Porphyra umbilicalis non-redundant EST contigs (assembly B, available at http://porphyra.rutgers.edu/ downloaded February, 2013; Chan et al. 2012a) using CLC Workbench; all sequences that did not map to the reference library were treated as contaminant sequence and eliminated from the dataset. Therefore, the results represent expression of Porphyra genes rather than expression of the meta-transcriptome to the extent that the EST database was culled of contaminant sequences (Chan et al. 2012a). Differential expression analysis was conducted in CLC Workbench, using RPKM (reads per kb per million reads) values to evaluate gene expression levels, and Kal’s Z test to evaluate significance. Contigs with at least a twofold difference in expression and a significance level less than a Bonferroni-adjusted value of α were considered DE among the libraries.
The DE sequences were submitted to the KEGG Automated Annotation Server (KAAS) under default parameters for annotation. We used BLAST2GO to conduct BLASTX searches with a less stringent evalue cutoff of 1e−3 against the NCBI non-redundant database for sequence and pathway annotation (Conesa et al. 2005; Conesa and Götz 2008; Götz et al. 2008). Mapping and Interpro Scan analyses were conducted with BLAST2GO default values. Analysis of Gene Ontology (GO) annotations was done in WEGO (Ye et al. 2006). GO enrichment analysis was conducted in R v. 3.2.3 using the package topGO (Alexa and Rahnenfuhrer 2010). Heat maps for expression levels were created using the R package gplots (Warnes et al. 2016).
Tissue cultures, tissue acclimation, and quantitative PCR
The expression levels of one DE contig (P_umbilicalis_esContig4469) that was annotated as an amt was evaluated by qPCR under low and high N conditions in tissue from multiple populations maintained in culture under common garden conditions. Tissue was sampled from the field at FS and DP and assayed as g3. Neutral spores were harvested from multiple individuals and grown to size in culture from spores according to Redmond et al. (2014). Cultures were maintained in 1–2-L Erlenmeyer flasks containing modified Von Stosch enriched (VSE) seawater (Ott 1966) with NH4Cl serving as the N source at 5 mM concentration. Salinity was measured weekly and adjusted to 30 ppt. Constant aeration was applied, ambient temperatures were maintained at 15 °C, and tissue was kept under a constant day neutral photoperiod (12:12 light to dark) at a 30-s average value of 30 μmol photons m−2 s−1.
Pieces of tissue between 0.2–0.3 g (fresh weight, patted dry) from three different thalli from each population were placed in 250-mL flasks containing either sterile seawater for low N conditions (< 5 μM NH4+), and an equal amount of tissue from the same three thalli was placed in Von Stosch enriched (VSE) seawater under high N conditions (5 mM NH4+). Different pieces of tissue from the same thallus were thus evaluated under both low and high N conditions. The samples were left to acclimate to the N conditions for 10–14 days under the culture conditions described above. Von Stosch enriched water was replaced in high N experimental treatments every 2 days to ensure constant high N conditions; however, the unenriched sterile seawater was not replaced to allow the N to become depleted.
Following the acclimation period, the tissue was removed from the culture medium, patted dry, and immediately frozen in liquid nitrogen. The RNA was extracted via the methods described above and assessed for quality using a NanoDrop 2000c spectrophotometer. We used the GoTaq® Probe 2-step RT-qPCR system (Promega, Madison, WI, USA) to prepare the cDNA.
The transcription elongation factor 1 (ElF1) is among the optimal reference genes tested for comparative gene expression analysis related to stress tolerance and development in Pyropia yezoensis (Kong et al. 2015). We identified contig P_umbilicalis_esisotig06418 as a putative ElF1, containing a putative zinc-binding domain with four conserved cysteine residues and an elongation transcription factor 1-like domain (pfam05129). For our reference gene, we designed primers to amplify a region of this contig (5′-CGT CGA CCC AGT TGC TAT AC-3′, and 5′-CTG CGA ACA CGA GCA GT-3′). Sequences acquired from tissue using these primers were a reciprocal best BLASTx hit with 97% identity and an evalue of 7e−35 to a transcription elongation factor from P. yezoensis (Uji et al. 2010).
The primers for the putative Porphyra umbilicalis amt target gene were designed to amplify a 128-bp region of P_umbilicalis_esContig4469. The forward primer (5′-GCG TAC TAC TTG TTC GGG TAT G-3′) was designed to span an exon-exon boundary, over an intron that was discovered in contig P_umbilicalis_esContig961 between base 210 and 360 (Eriksen 2014). Quantitative PCR was conducted in triplicate for each sample using a Roche LightCycler 2.0 and 20 μL reactions containing 1X GoTaq® Probe qPCR Master Mix (Promega), 100 nM reference gene primers (above), and 250 nM reference gene probe (5′-/56-FAM/CAG GGC GGT/ZEN/GAT GGT CAT CTC ATG/3IABkFQ/-3′; Integrated DNA Technologies), 900 nM of the target gene forward primer and reverse primer (5′-GCA AAC GTC CAC TGA AAG AAC-3′), 250 nM of the probe (5′-/5HEX/TCG GAC GAC/ZEN/ACG CAA GAG TTT CAT/3IABkFQ/-3′), and 16.5–39.5 ng prepared cDNA. Thermal cycling conditions included a one-time, 2 min hot start at 95 °C, then a cycle consisting of a denaturation step of 15 s at 95 °C, an annealing step at 55 °C for 10 s, and an extension step at 60 °C for 30 s which was repeated for 55 times, and followed by a single cooling step at 40 °C for 30 s.
Relative differential expression of the target amt gene was assessed using the calculations described by Pfaffl (2012) against expression of ElF1 as the reference gene. The normalizer sample was cDNA from total RNA extracted from cultured Schoodic Point tissue growing under standard culture conditions as described above. We also sampled tissue under high and low N conditions for cultured tissue from Schoodic Point (g1) and Quoddy Head (g4), ME as outgroup samples (Fig. 1).
Statistical analyses of expression were done in IBM SPSS v. 21. We used a two-way ANOVA to determine the effects of population and N level on relative expression. The full dataset was log transformed according to Zar (1999) in order to meet the homogeneity of variance and normality assumptions of the two-way ANOVA. Simple main effects with a Bonferroni adjustment were used to determine which populations had significant differences in gene expression.
Results
Differential gene expression: same environment, different genotype (FSg3 × FSg6)
There were 299 contigs with at least a twofold significant difference in expression when the two FS libraries were compared (FSg3 × FSg6) (Table 2). The KEGG Automated Annotation Server (KAAS) annotated relatively few of the contigs that were DE among the two genotypes from the same environment, and assigned them to 59 different pathways (Table 3). BLAST2GO found significant hits for more contigs (Table 4) and assigned GO annotations to about 92% of the DE contigs.
Differential gene expression: different environment, same genotype (FSg3 × DPg3)
There were more contigs DE among the FS g3 (FSg3) and DP (DPg3) libraries than among the two FS libraries (Table 2). More of the DE contigs were annotated by KAAS (Table 3) and BLAST2GO (Table 4); contigs were assigned to nearly three times as many pathways. Compared to the FSg3 × FSg6 comparison, there was also a greater abundance of DE putative Porphyra contigs associated with biosynthesis of secondary metabolites, plant-microbe-interaction pathways such as “infection by Vibrio cholerae,” and various plant antibiotic biosynthesis pathways (Table 5).
Differential gene expression: different environment, different genotype (FSg6 × DPg3)
The greatest number of DE contigs was found in the comparison between the FS g6 (FSg6) and DPg3 libraries (Table 2). However, fewer of the DE contigs were annotated by KAAS (Table 3) than in the comparison between the FSg3 × DPg3 libraries. Similar to the comparison of FSg3 × DPg3, there was also an abundance of DE contigs associated with plant-microbe-interaction pathways (Table 5). BLAST2GO found significant hits for only about 60% of DE contigs (Table 4).
Differentially expressed candidate genes related to salinity and temperature stress.
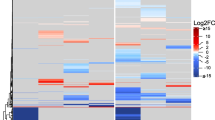
Among contigs annotated by the BLAST2GO, 499 were identified as putatively coding for enzymes related to oxidation-reduction process (GO:0055114). Hierarchical clustering of expression patterns breaks these contigs into two groups (Fig. 2). In one group, contigs are highly upregulated in FSg6 and significantly downregulated in DPg3 libraries; in the other group, contigs are downregulated in both FS libraries and upregulated in DPg3. Genes involved in ROS control such as l-ascorbate peroxidase (APX) and catalase (CAT) were found in both groups, and expression levels had no clear correlation to habitat. Two contigs were identified as putative manganese superoxide dismutases (Mn-SOD), and both were over-expressed in the DPg3 library (Fig. 2). Fifteen contigs were annotated to GO:0009408, response to heat, but there is no clear upregulation of all heat shock proteins in any library (Fig. 3). Only one homolog of the kinases involved in the mitogen-activated protein kinase (MAPK) signaling cascade involved in stress signaling in green plants was found among the libraries, and there was no clear upregulation of these contigs among the libraries (Additional files 1 and 2). Calmodulins, Ca2+-dependent protein kinases, and other genes involved in calcium signaling in response to osmotic stress in green plants are not among the contigs found in these libraries.

Heat map showing expression levels of a select number of contigs within the RNA-seq libraries annotated as GO:0055114, oxidation-reduction process. The gray dotted center line represents mean expression. Yellow to white shades represent expression above the mean; orange to red shades represent expression below the mean. A heat map showing the expression levels of all contigs annotated as GO:0055114 is provided as Additional file 1

Heat map showing expression of all contigs within the RNA-seq libraries annotated as GO:0009408, response to heat. The gray dotted center line represents mean expression. Yellow to white shades represent expression above the mean; orange to red shades represent expression below the mean
Differentially expressed candidate genes related to nitrogen metabolism
There are 16 contigs found in all libraries that were annotated as GO:0006807, nitrogen compound metabolic process. Three contigs are annotated as NH4+ transporters (AMT), one is annotated as a nitrate transporter, and three are annotated as nitrate reductase (NR) (Fig. 4).

Heat map showing expression of all contigs within the RNA-seq libraries annotated as GO:0006807, nitrogen metabolism. The gray dotted center line represents mean expression. Yellow to white shades represent expression above the mean; orange to red shades represent expression below the mean
The transporters are consistently over-expressed in the FSg6 library and under-expressed in the DP library. Two contigs annotated as putative NH4+ transporters are over-expressed in FS tissue compared to DP tissue in comparisons of FSg3 × DPg3 and FSg6 × DPg3 libraries. The contig P_umbilicalis_esContig4469 annotated also as a putative ammonium transporter and was over-expressed in both FS libraries by over eightfold compared to the DPg3 library. Of the contigs annotated as putative nitrate reductases, two are over-expressed in FSg6, and the third is over-expressed in the DP library.
Quantitative PCR of a putative ammonium transporter
All populations experienced significantly higher relative expression of a putative NH4+ transporter P_umbilicalis_esContig4469 (hereafter NH4-4469) gene under low N conditions than high N conditions (Fig. 5). Individuals from FS experienced the greatest difference in relative expression under high and low N conditions (mean difference in relative expression = 0.65 ± 0.21 SD; F(1, 70) = 98.72, P < 0.001); individuals from DP also experienced a difference in relative expression to a lesser extent that was still significant (mean difference relative expression = 0.2 ± 0.21 SD; F(1,70) = 6.139, P = 0.003).

Relative expression patterns of contig NH4-4469 annotated as an AMT under low (< 5 μM NH4+) and high (5 mM NH4+) N conditions for three pieces of tissue cultured neutral spores from algal populations collected at FS, DP, Quoddy Head, and Schoodic Point. Error bars represent the standard error of the mean. Letters denote significance at the Bonferroni-adjusted alpha = 0.017. There was no significant difference in the mean relative expression among populations under high N treatments, and therefore, no letters are given to denote significance
Under low N conditions, there was a significant difference among populations in relative expression of this gene (F(3, 70) = 16.307, P < 0.001, partial η2 = 0.411). FS tissue had significantly higher relative expression rates than DP and Quoddy Head (P < 0.001 for both). The difference in relative expression under low N conditions for tissue from FS and Schoodic Point was not significant at a Bonferroni-adjusted alpha = 0.017 (mean difference relative expression 0.24, SE = 0.08, P = 0.02).
Under high N conditions, relative expression of this putative NH4+ transporter was low in all populations. There were no significant differences in relative expression patterns among populations under high N conditions (F(3, 70) = 1.856, P = 0.145, partial η2 = 0.074).
Discussion
We looked at differential expression patterns in Porphyra umbilicalis growing in a typical open-coastal habitat and a rare, atypical habitat in an estuarine tidal rapid habitat. Our goal was to understand how this species adjusts gene expression to acclimate to these different environments, and to understand N metabolism in this species that is a candidate for NH4+ removal in IMTA systems. We find evidence that environment has a greater effect on gene expression differences than genotype alone, though there is a cumulative effect of environment and genotype on gene expression. There is no evidence that one environment elicits a higher stress response than another, but the high N load at DP has a significant effect on N transporter activity that appears to be trans-generationally inherited.
Differential expression analysis
The comparison of differential expression among libraries represents a breakdown of some of the factors affecting gene regulation. The comparison of expression patterns among FSg3 and FSg6 libraries represents differences due to genotype within the same environment, if we assume the micro-habitats of the two collection locations are roughly similar. Relative to the other comparisons among libraries, this comparison yielded the fewest DE contigs and the fewest annotations. The list of DE genes were enriched for innocuous terms related to cellular parts, and do not include terms that may be related to cellular stress such as oxidation-reduction process, phosphorylation, or protein ubiquitination (Table 3). Genotypic differences among these samples may thus be interpreted as a minor factor influencing expression levels compared to the effects of environment as illustrated in the other library comparisons (Table 2), and a factor that largely influences the expression of genes of unknown function, as only 21 and 41% of the DE contigs were annotated by KAAS and BLAST2GO, respectively (Tables 3 and 4).
The comparison of expression patterns among FSg3 and DPg3 libraries may represent differences due to environment only, because the tissue collected from DP was assayed as the same genotype as the FS material. These genotypes are only the same to the limited extent which the three polymorphic EST-SSR loci described in Eriksen et al. (2016) can identify genotypes, but these markers represented the most accurate way to identify genotypes available at the time of study. This comparison yielded many more DE contigs than the FSg3 × FSg6 comparison (Table 2), and more with known functions (Tables 3 and 4). There was a higher proportion of DE genes with GO terms associated with immune system processes and viral reproduction. The difference in expression levels of immune system processes and viral reproduction among comparisons of environment is also reflected in KAAS data, which annotated more contigs attributed to plant-microbe-interactions, such as viral infection, Vibrio cholerae infection, and plant-pathogen interactions (Table 5). This suggests that the heavily human-impacted estuarine environment harbors more pathogens that macroalgae populations must contend with when living there. Seaweed has been shown to act as a reservoir for various pathogenic Vibrio populations (Mahmud et al. 2007, Mahmud et al. 2008, Gonzalez et al. 2014, Martinez and Padilla 2016).
The comparison of patterns among FSg6 and DPg3 libraries represents the interactive effect of genotype and environment on expression. It contains the most DE contigs (Table 2), but fewer were of known function (Tables 3 and 4). It more closely resembles the FSg3 × DPg3 comparison in terms of the abundance of pathogen and infection-related contigs (Table 5). For some contigs (such as those associated with GO terms related to the establishment of localization, molecular transducer activity, and transporter activity), there appears to be a cumulative effect of environment and genotype on expression of the pathway.
The environment thus appears to be the largest factor influencing differential expression among the libraries, and environment-by-genotype interactions also appear to be significant. The effects of pathogens and microbes appear to have a significant effect on expression levels of putative Porphyra genes via upregulation of genes involved in infection pathways, plant-pathogen interaction pathways, and various antibiotic synthesis pathways in the DPg3 library.
Stress response genes
Temperature and salinity conditions are two environmental factors that populations in these habitats may experience differently, possibly leading to a stress response. Populations under temperature and salinity stress should have elevated levels of expression of genes encoding antioxidant proteins (Dring 2005). Sampath-Wiley et al. (2008) found elevated levels of stress-related antioxidant enzymes such as glutathione reductase (GTR) and CAT in emersed Porphyra umbilicalis tissue found in a typical open-coastal habitat under high temperature stress. Using microarrays to evaluate expression patterns of Chondrus crispus, Collén et al. (2007) found that genes for antioxidant proteins such as APX, CAT, dehydroascorbate reductase, glutathione peroxidase, GTR, methionine sulfoxide reductase, manganese superoxide dismutase, and thioredoxin were downregulated in hypo-osmotic conditions compared to the non-stress control. A different study found upregulation of APX in Gracilaria changii exposed to hypo-osmotic conditions compared to the control exposed to 30 ppt salinity (Teo et al. 2009).
In the RNA-seq libraries analyzed in this study, there was no consistent pattern in expression of ROS contigs; some were upregulated in FS libraries, and others were upregulated in DP libraries (Fig. 2, Additional files 1 and 2). Contigs that are annotated as phosphorylation genes that are involved in stress signaling pathways in green plants, such as the mitogen-activated kinases (MAPK/MAPKK/MAPKKK, though only MAPK appears to exist in P. umbilicalis), also show no consistent pattern (Additional file 2). Heat shock proteins show no consistent pattern (Fig. 3). Based on these data, there is thus no clear evidence to suggest one or the other population is under more stress during submersed conditions in these habitats. Porphyra umbilicalis has non-rigid cell walls made of mannan and xylan (Lobban and Harrison 1997) and porphyran (Morrice et al. 1984) that allow the cell wall to expand and contract along with the plasma membrane under different osmotic conditions. The difference in salinity at the time of sampling was small and likely not outside of the range which P. umbilicalis is capable of coping with anatomical adjustments. Further work will be necessary to investigate specific hypotheses about the physiological response of this species to abiotic stress.
Nitrogen metabolism
Nitrogen (N) is an essential and often a limiting nutrient for macroalgae; it plays an important role in construction of amino acids and pigments such as chlorophyll and phycobilins (Reed 1990). Low nutrient stress may exacerbate damage to the photosystems via other abiotic stress factors and lead to ROS production by slowing the electron transport chain, but high levels of N cause the intermediaries of N assimilation (NO2− and NH4+) to accumulate to toxic levels if the N assimilation process is not properly regulated within the cell (Taiz and Zeiger 2010; Raven 1980).
Nitrogen metabolism is thus tightly regulated by the cell, and the first step of regulation is intake. In barley, high-affinity NH4+ transport systems are active at low concentrations of NH4+, but they are downregulated when internal N concentrations reach sufficient levels (Glass et al. 2001). In the Porphyra umbilicalis EST database, Chan et al. (2012a) found seven contigs that code for putative NH4+ transporters (Chan et al. 2012b); thus, it seems reasonable to assume that transporters with different affinities for NH4+ may be present within the Porphyra genome as well.
Nitrogen uptake appears to be downregulated in DP tissue based on RNA-seq data (Fig. 4). Two contigs annotated as putative ammonium transporters (amts) and another annotated as a nitrate transporter were under-expressed in DP tissue. The contig P_umbilicalis_esContig4469 (NH4-4469) is identified by BLAST searches as a high-affinity transporter, and qPCR confirmed decreased expression of this gene under high N conditions, and increased expression under low N which is consistent with high-affinity transporters (Fig. 5).
The difference in expression patterns from the RNA-seq data of amt NH4-4469 (high expression in FS tissue and lower expression in DP tissue) is predicted based on environmental N levels; however, the difference persisted in tissue grown under similar N conditions in culture. Tissue grown from neutral spores from field collected tissue from FS and DP and maintained in culture under the same high NH4+ conditions (5 mM) maintained significant differences in expression patterns of this amt when later exposed to low N conditions (Fig. 5). Tissue grown from neutral spores from parental tissue collected at FS had significantly higher expression of this putative high-affinity amt under low N conditions than tissue grown from spores from DP. Similarly, tissue grown from FS spores had significantly higher expression levels of this amt gene than outgroup tissue grown from Quoddy Head spores, which is also exposed to atypically high environmental N levels (Garside and Garside 2004) similar to DP.
The finding suggests tissue from FS is genetically predisposed to thrive under lower N conditions by upregulating expression of a gene necessary to take in more NH4+ when it is scarce; or vice versa, that tissue from DP is genetically predisposed to thrive under potentially toxic loads of N. No sequence polymorphism among genotypes was found within a 465-bp region containing both coding and intron sequence of this gene using Sanger sequencing (Eriksen 2014), though upstream sequence data were not available. Heritability of expression patterns from parent to offspring encompasses the field of epigenetics, but the mechanism by which that information is passed through generations when the offspring are generated through sexual reproduction is complicated by erasure of genomic imprints during gametogenesis and embryogenesis (Gibson and Weir 2005); however, the asexual life cycle of the populations of Porphyra umbilicalis in this study theoretically could allow for simple transmission of environmentally induced epigenetic signals. Environmentally induced methylation changes are heritable in apomictic dandelions (Verhoeven et al. 2010) and has been shown to be an important mechanism of acclimation and adaptation in dandelion range expansion (Verhoeven and Preite 2014, Preite et al. 2015). Adaptation to environment by epigenetic signals may explain why previous studies using EST-SSR markers found no evidence of genetic structure in populations in open-coastal and estuarine environments in the Gulf of Maine (Eriksen et al. 2016). While the data are merely suggestive at this point, further exploration of polymorphism within the regulatory regions, or epigenetic signals in the regulatory region of this amt gene may be warranted.
Conclusions
Our results have implications for use of Porphyra umbilicalis in integrated multitrophic aquaculture (IMTA). The species has been suggested as a possible partner in IMTA systems (Blouin et al. 2007); however, the levels at which fish experience nitrogen toxicity in aquaculture systems is relatively low (Randall and Tsui 2002) compared to the NH4+ concentrations at which P. umbilicalis thrives in culture. Populations of P. umbilicalis representing different asexual lineages may have different genetically controlled abilities to take up NH4+ from fish effluent at lower environmental concentrations, depending on the expression of high-affinity NH4+ transporters such as NH4-4469. Researchers seeking to identify lineages with an increased ability to take up NH4+ from aquaculture systems might look for populations growing in low-nitrogen habitats such as open-coastal sites away from human influences.
Abbreviations
- amt :
-
Ammonium (NH4+) transporter
- APX:
-
Ascorbate peroxidase
- CAT:
-
Catalase
- DE:
-
Differentially expressed
- DP:
-
Collection location, Dover Point, NH, USA
- EST:
-
Expressed sequence tag
- FS:
-
Collection location, Fort Stark, Jaffrey Point, NH, USA
- GTR:
-
Glutathione reductase
- NH4 :
-
Ammonium
- NO2 :
-
Nitrate
- qPCR:
-
Quantitative polymerase chain reaction
- RFLP:
-
Restriction fragment length polymorphism
- RIN:
-
RNA integrity number
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
References
Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for Gene Ontology. 2010;R package version 2.22.0.
Blouin N, Xiugeng F, Peng J, Yarish C, Brawley SH. Seeding nets with neutral spores of the red alga Porphyra umbilicalis (L.) Kützing for use in integrated multi-trophic aquaculture (IMTA). Aquaculture. 2007;270(1):77–91.
Blouin NA, Brodie JA, Grossman AC, Xu P, Brawley SH. Porphyra: a marine crop shaped by stress. Trends Pl Sci. 2011;16(1):29–37.
Chan CX, Blouin NA, Zhuang Y, Zäuner S, Prochnik SE, Lindquist E, Lin S, Benning C, Lohr M, Yarish C. Porphyra (Bangiophyceae) transcriptomes provide insights into red algal development and metabolism. J Phycol. 2012a;48(6):1328–42.
Chan CX, Zäuner S, Wheeler G, Grossman AR, Prochnik SE, Blouin NA, Zhuang Y, Benning C, Berg GM, Yarish C. Analysis of Porphyra membrane transporters demonstrates gene transfer among photosynthetic eukaryotes and numerous sodium-coupled transport systems. Plant Physiol. 2012b;158(4):2001–12.
Collén J, Guisle-Marsollier I, Léger JJ, Boyen C. Response of the transcriptome of the intertidal red seaweed Chondrus crispus to controlled and natural stresses. New Phytol. 2007;176(1):45–55.
Conesa A, Götz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008; https://doi.org/10.1155/2008/619832.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Day JP, Neefus CD, Yarish C. Development of a modular integrated recirculating aquaculture system using Porphyra for bioremediation of marine finfish effluent. J Phycol. 2009;45:31–2.
Dring MJ. Stress resistance and disease resistance in seaweeds: the role of reactive oxygen metabolism. Adv Bot Res. 2005;43:175–207.
Eriksen RL. Population genetics and organism-environment interactions of Porphyra umbilicalis Kützing in the Gulf of Maine Doctoral thesis University of New Hampshire 2014.
Eriksen RL, Green LA, Klein AS. Genetic variation within and among asexual populations of Porphyra umbilicalis Kützing (Bangiales, Rhodophyta) in the Gulf of Maine, USA. Bot Mar. 2016;59(1):1–12.
Gantt E, Berg GM, Bhattacharya D, Blouin NA, Brodie JA, Chan CX, Collén J, Cunningham FX Jr, Gross J, Grossman AR, Karpowicz S. Porphyra: complex life histories in a harsh environment: P. umbilicalis, an intertidal red alga for genomic analysis. In: Red algae in the genomic age. Netherlands: Springer; 2010. p. 129–48.
Garside C, Garside JC. Nutrient sources and distributions in Cobscook Bay. Northeast Nat. 2004;11(sp2):75–86.
Gibson G, Weir B. The quantitative genetics of transcription. Trends Genet. 2005;21(11):616–23.
Glass ADM, Britto DT, Kaiser BN, Kronzucker HJ, Kumar A, Okamoto M, Rawat SR, Siddiqi MY, Silim SM, Vidmar JJ, Zhuo D. Nitrogen transport in plants, with an emphasis on the regulation of fluxes to match plant demand. J Plant Nutr Soil Sci. 2001;164(2):199–207.
Gonzalez DJ, Gonzalez RA, Froelich BA, Oliver JD, Noble RT, McGlathery KJ. Non-native macroalga may increase concentrations of Vibrio bacteria on intertidal mudflats. Marine Ecol Prog Ser. 2014;505:29–36.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nuc Acids Res. 2008;36(10):3420–35.
Green LA, Neefus CD. The effects of short-and long-term freezing on Porphyra umbilicalis Kützing (Bangiales, Rhodophyta) blade viability. J Exp Mar Biol Ecol. 2014;461:499–503.
Green LA, Neefus CD. Effects of temperature, light level, and photoperiod on the physiology of Porphyra umbilicalis Kützing from the Northwest Atlantic, a candidate for aquaculture. J Appl Phycol. 2016;28(3):1815–26.
Kim JK, Kraemer GP, Neefus CD, Chung IK, Yarish C. Effects of temperature and ammonium on growth, pigment production and nitrogen uptake by four species of Porphyra (Bangiales, Rhodophyta) native to the New England coast. J Appl Phycol. 2007;19(5):431–40.
Kong F, Cao M, Sun P, Liu W, Mao Y. Selection of reference genes for gene expression normalization in Pyropia yezoensis using quantitative real-time PCR. J Appl Phycol. 2015;27:1003–10.
Lobban CS, Harrison PJ. Seaweed ecology and physiology. New York, USA: Cambridge University Press; 1997.
Mahmud ZH, Neogi SB, Kassu A, Mai Huong BT, Jahid IK, Islam MS, Ota F. Occurrence, seasonality and genetic diversity of Vibrio vulnificus in coastal seaweeds and water along the Kii Channel, Japan. FEMS Microbiol Ecol. 2008;64(2):209–18.
Mahmud ZH, Neogi SB, Kassu A, Wada T, Islam MS, Nair GB, Ota F. Seaweeds as a reservoir for diverse Vibrio parahaemolyticus populations in Japan. Int J Food Microbiol. 2007;118(1):92–6.
Martinez JN, Padilla PI. Isolation and characterization of agar-digesting Vibrio species from the rotten thallus of Gracilariopsis heteroclada Zhang et Xia. Marine Environ Res. 2016;119:156–60.
Mathieson AC, Hehre EJ, Reynolds NB. Investigations of New England marine algae I: a floristic and descriptive ecological study of the marine algae at Jaffrey Point, New Hampshire, USA. Bot Mar. 1981a;24:521–32.
Mathieson AC, Reynolds NB, Hehre EJ. Investigations of New England marine algae II: the species composition, distribution and zonation of seaweeds in the Great Bay Estuary System and the adjacent open coast of New Hampshire. Bot Mar. 1981b;24(10):533–44.
Miranda LN, Hutchison K, Grossman AR, Brawley SH. Diversity and abundance of the bacterial community of the red macroalga Porphyra umbilicalis: did bacterial farmers produce macroalgae? PLoS One. 2013;8(3):e58269.
Morrice LM, McLean MW, Long WF, Williamson FB. Porphyran primary structure. In: Bird C, Ragan M, editors. Eleventh International Seaweed Symposium, vol. 22: Developments in Hydrobiology. Springer Netherlands; 1984. p. 572–5. https://doi.org/10.1007/978-94-009-6560-7_118.
Nettleton J. Tracking environmental trends in the Great Bay Estuarine System: an examination of water quality and nuisance macroalgal blooms. Durham: Doctoral thesis University of New Hampshire; 2012.
Norall TL, Mathieson AC. Nutrient studies of the Great Bay Estuary System Jackson Estuarine Laboratory. Durham, New Hampshire: University of New Hampshire; 1974.
Ott FD. A selected listing of xenic algal cultures, vol. 72. Woods Hole: Systematics-Ecology Program, Marine Biological Laboratory; 1966.
Page AF. Detection and avoidance of polysaccharides in plant nucleic acid extractions Thermo Fisher Scientific, Wilmington, DE, USA. 2010.
Pfaffl MW. Quantification strategies in real-time polymerase chain reaction. Quantitative real-time PCR. Appl Microbiol. 2012;53–62.
Preite V, Snoek LB, Oplaat C, Biere A, Putten WH, Verhoeven KJF. The epigenetic footprint of poleward range-expanding plants in apomictic dandelions. Mol Ecol. 2015;24(17):4406–18.
PREP State of Our Estuaries Piscataqua Region Estuaries Partnership, Durham, New Hampshire, USA. 2013.
Randall DJ, Tsui TKN. Ammonia toxicity in fish. Mar Pollut Bull. 2002;45(1):17–23.
Raven JA. Nutrient transport in microalgae. Adv Microbial Physiol. 1980;21:47–226.
Redmond S, Green LA, Yarish C, Kim J, Neefus C. New England seaweed culture handbook. 2014.
Reed RH. Solute accumulation and osmotic adjustment. In: Cole KM, Sheath RG, editors. Biology of the red algae. New York: Cambridge University Press; 1990. p. 147–70.
Reed RH, Collins JC, Russell G. The effects of salinity upon cellular volume of the marine red alga Porphyra purpurea (Roth) C Ag. J Exp Bot. 1980;31(125):1521–37.
Sampath-Wiley P, Neefus CD, Jahnke LS. Seasonal effects of sun exposure and emersion on intertidal seaweed physiology: fluctuations in antioxidant contents, photosynthetic pigments and photosynthetic efficiency in the red alga Porphyra umbilicalis Kützing (Rhodophyta, Bangiales). J Exp Mar Biol Ecol. 2008;361(2):83–91.
Smith CM, Berry JA. Recovery of photosynthesis after exposure of intertidal algae to osmotic and temperature stresses: comparative studies of species with differing distributional limits. Oecologia. 1986;7:6–12.
Taiz L, Zeiger E. Plant Physiology. 5th ed. Sunderland: Sinauer Associates, Inc Publishers; 2010.
Teasdale B, West A, Taylor H, Klein A. A simple restriction fragment length polymorphism (RFLP) assay to discriminate common Porphyra (Bangiophyceae, Rhodophyta) taxa from the Northwest Atlantic. J Appl Phycol. 2002;14(4):293–8.
Teo SS, Ho CL, Teoh S, Rahim RA, Phang SM. Transcriptomic analysis of Gracilaria changii (Rhodophyta) in response to hyper- and hypo-osmotic stresses. J Phycol. 2009;45(5):1093–9.
Trowbridge P, Wood PE, Wood MA, Underhill JT, Healy DS. New Hampshire Department of Environmental Services. Great Bay nitrogen non-point source study R-WD-13-10. 2014.
Uji T, Takahashi M, Saga N, Mikami K. Visualization of nuclear localization of transcription factors with cyan and green fluorescent proteins in the red alga Porphyra yezoensis. Mar Biotechnol. 2010;12(2):150–9.
Verhoeven KJ, Jansen JJ, van Dijk PJ, Biere A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010;185(4):1108–18.
Verhoeven KJ, Preite V. Epigenetic variation in asexually reproducing organisms. Evolution. 2014;68(3):644–55.
Wang W-J, Wang F-J, Zhu J-Y, Sun X-T, Yao C-Y, Xu P. Freezing tolerance of Porphyra yezoensis (Bangiales, Rhodophyta) gametophyte assessed by chlorophyll fluorescence. J Appl Phycol. 2011;23(6):1017–22. https://doi.org/10.1007/s10811-010-9634-3.
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. gplots: various R programming tools for plotting data R package version 301. 2016. https://cran.r-project.org/web/packages/gplots/index.html.
West AL, Mathieson AC, Klein AS, Neefus CD, Bray TL. Molecular ecological studies of New England species of Porphyra (Rhodophyta, Bangiales). Nova Hedwigia. 2005;80(1–2):1–24.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. WEGO: a web tool for plotting GO annotations. Nucleic acids research. 2006;34(suppl_2):W293–W297.
Zar JH. Biostatistical analysis. 4th ed. New Jersey: Prentice Hall; 1999. p. 663.
Acknowledgements
Drs. W. Kelley Thomas, Leland Jahnke, Arthur Mathieson, and Chris Neefus provided invaluable input and advice. Greg Smith and Anna helped with collecting, and Dr. Andrew Page and Shay Polsky provided technical assistance with a ThermoFisher Scientific NanoDrop 2000c. Jobriah Anderson at the HCGS performed all genotyping and Sanger sequencing, as well as helped with the Agilent Bioanalyzer. Dr. Kazu Okamoto of the HCGS performed the differential expression analysis. We also thank Drs. Kirk Broders and Franz Lichtner for the technical assistance with the Roche Light Cycler 2.0.
Funding
This work was supported by the Leslie S. Hubbard Marine Program Endowment to Support Marine Research Development/Equipment. Partial funding was provided by the New Hampshire Agricultural Experiment Station. This is Scientific Contribution Number 2784. This work was supported by the USDA National Institute of Food and Agriculture Hatch Project 0222799.
Availability of data and materials
The dataset supporting the conclusions of this article as well as Additional files are available in the figshare.com repository, https://doi.org/10.6084/m9.figshare.5467027 and https://figshare.com/s/2c5b2cf2155b4562f469.
Author information
Authors and Affiliations
Contributions
RLE designed and implemented all experiments, analyzed the data, and wrote the manuscript. ASK contributed to the experimental design and provided funding and manuscript comments. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The samples at Schoodic Point, Acadia National Park, were collected under permit number #ACAD-2012-SCI-0013 issued to Dr. Jessica Muhlin from April 16, 2012, to April 25, 2012.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Heat map showing expression levels of all contigs within the RNA-seq libraries annotated as GO:0055114, oxidation-reduction process. The gray dotted center line represents mean expression. Yellow to white shades represent expression above the mean; orange to red shades represent expression below the mean. (TIFF 450 kb)
Additional file 2:
Heat map showing expression levels of all contigs within the RNA-seq libraries annotated as part of the mitogen-associated protein kinase (MAPK/MAPKK/MAPKKK) pathway. The gray dotted center line represents mean expression. Yellow to white shades represent expression above the mean; orange to red shades represent expression below the mean. (TIFF 79 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Eriksen, R.L., Klein, A.S. Organism-environment interactions and differential gene expression patterns among open-coastal and estuarine populations of Porphyra umbilicalis Kützing (Rhodophyta) in the Northwest Atlantic. Fish Aquatic Sci 21, 28 (2018). https://doi.org/10.1186/s41240-018-0103-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41240-018-0103-2