
Abstract
Background
Cystic fibrosis is a rare recessive monogenic disease caused by loss-of-function mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Recently developed CFTR-modulators (i.e. drugs that rescue CFTR-function) have improved the prognosis of the disease, but are only available for certain, relatively common mutations. More than 2000 genetic variants in CFTR have been described, many of which are uncharacterized, while efficacy of CFTR-modulators can differ depending on genotype. Personalized models that can predict drug efficacy in patients with rare mutations could help to provide access to effective CFTR-modulator treatment. The forskolin induced swelling (FIS) assay in rectal organoids can robustly measure CFTR-function and CFTR-modulator efficacy, and pilot studies indicate that FIS accurately predicts treatment efficacy in individual patients. In the HIT-CF Organoid Study, rectal organoids from patients with rare CFTR-mutations are screened for CFTR-modulator efficacy to identify patients that could clinically benefit from these drugs.
Methods
In this EU-H2020 funded multi-centre study, rectal biopsies are obtained from approximately 500 subjects with rare CFTR-mutations from 17 different EU countries and sent to central laboratories. Organoids are generated, on which several novel CFTR-modulating drugs are screened for efficacy. In the process, a biobank is generated which could be used for future research. Subjects whose organoids show a response to treatment will be asked to participate in subsequent clinical trials evaluating the clinical efficacy of tested drugs.
Discussion
The HIT-CF Organoid Study applies the intestinal organoid model for personalized medicine in patients with rare variant cystic fibrosis, who are currently excluded from classical CFTR-modulator trials. Together with the subsequent clinical trials in high responders, this study aims to create a new pathway for access to CFTR-modulating drugs for patients with ultra-rare CFTR variants.
Trial registration
This trial has been registered in the Netherlands Trial Registry (NTR7520, https://www.trialregister.nl/trial/7304), a primary registry approved and recognized by WMO and ICMJE, on 2018-10-02.
Similar content being viewed by others

Background
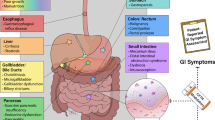
Cystic Fibrosis (CF) is a life shortening rare disease caused by loss-of-function mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene [1, 2]. Dysfunctional CFTR impairs epithelial chloride and bicarbonate transport, which leads to thickening of secretions in multiple organs, resulting in respiratory failure, endocrine dysfunction, liver cirrhosis, gastrointestinal disorders and early death [3]. CF is a monogenic recessive disease, and more than 2000 different variants in the CFTRgene have been described (genet.sickkids.on.ca) [4]. The F508del mutation is present in ~ 80% of European CF patients, but 351 other variants have been confirmed as pathogenic [5]. The characteristics and disease liability of the remaining variants remain mostly unclear. They can affect CFTR function via distinct (combinations of) mechanisms and can result in different levels of CFTR dysfunction and clinical disease [1, 6].
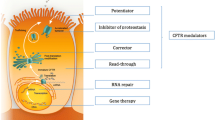
The recent introduction of so-called CFTR-modulators has caused a paradigm shift in the treatment of cystic fibrosis. Four different drug categories can increase CFTR protein availability and function, and act in a CFTR mutation specific fashion.
Ivacaftor, the first CFTR modulator on the market, was identified as CFTR potentiator for patients with so-called gating mutations that are present in ~ 5% of CF patients. In these patients ivacaftor increases the open-probability of the CFTR channel, thereby improving the anion transport [7].
A combination of ivacaftor and the corrector lumacaftor proved to be effective in patients with the most common F508del mutation [8, 9]. CFTR-correctors aim to correct impaired CFTR folding and trafficking to the cell surface [10, 11]. Typically a potentiator with one or two correctors are needed to optimally re-establish the function of the complex CFTR-channel in patients with one or two F508del alleles [12, 13], and these combination treatments can lead to effective treatment for ~ 85% of the CF population [14, 15].
The third type of CFTR modulators are CFTR-read-through agents that promote ribosomal read-through at premature termination codons (PTCs) and thus lead to full length CFTR protein production in patients with nonsense mutations [16]. CFTR-amplifiers increase CFTR mRNA availability and might act in a genotype agnostic fashion [17]. The therapeutic targets of read-through agents and CFTR-amplifiers still need to be assessed. Another interesting CFTR-modulating drug category are CFTR-stabilizers that aim to stabilize CFTR-protein activity at the cell membrane through different molecular targets [18, 19].
CF patients with non-F508del and non-gating mutations (approximately 12,000–13,000 patients worldwide) are very heterogeneous and carry > 2000 different ultra-rare and mostly uncharacterized CFTR mutations (genet.sickkids.on.ca and cftr2.org) [4, 5]. Moreover, patients carry a combination of two CFTR variants which further complicates the prediction of drug efficacy in individual patients. The role of current CFTR-modulators in these patients is hardly described and clinical efficacy can not be tested in classical clinical trials due to extremely low patient numbers. In this population, there is a clear unmet need to match the best CFTR-modulator (combination) to each individual.
Recent stem cell technologies enable the long-term culture of patient-derived tissues in the laboratory as organoids [20, 21]. Organoids are three-dimensional cell cultures that recapitulate features of the tissue from which they originate [20]. Dekkers et al. developed a functional CFTR assay in intestinal organoids that were grown from easily obtainable rectal biopsies which provides a way to rapidly collect and biobank tissues from individual patients [22]. CFTR function can be measured upon stimulation of organoids with forskolin, which leads to CFTR phosphorylation, CFTR channel opening and subsequent transport of chloride and water into the organoid lumen. Although forskolin-induced swelling (FIS) of intestinal organoids is an indirect measurement of CFTR function as it quantifies fluid rather than ion transport, FIS is fully CFTR dependent and provides a rapid, phenotypic assessment of CFTR function. Residual CFTR function in organoids correlates with several clinical disease parameters [23, 24] and biomarkers of CFTR function [25].
Importantly, the effect of CFTR modulators on CFTR function can be robustly measured by the FIS assay through quantification of increased swelling upon drug incubation in vitro [26]. Using this assay, correlations were observed between organoid drug-induced swelling and drug responses in clinical trials at the population level [26], as well as a high-to-excellent accuracy predicting individual in vivo responses to CFTR modulators [27]. This has led to the identification of successful treatment of several patients with ultra-rare genotypes that showed high FIS to CFTR-modulating drugs in their organoids [26, 27]. Moreover, drugs that showed potential in other pre-clinical model systems but not in organoids subsequently failed to produce clinical benefit, highlighting the specificity of organoids in finding meaningful clinical changes [28]. By providing an individual assessment of CFTR modulator efficacy without a priori knowledge of the CFTR mutations, organoid CFTR function might be exceptionally suited to stratify people with ultra-rare mutations for effective treatments.
To further implement the FIS assay as a personalized medicine approach for patients with rare CFTR-mutations, we developed the HIT-CF project (www.hitcf.org) [29].
Rationale for the current study
The Human Individualized Treatment for CF (HIT-CF) project was developed to bring CFTR-modulators to CF patients with rare mutations, and has received funding through the European Union’s Horizon2020 programme (grant agreement No. 755021). The project consists of three parallel and intertwined pathways aiming to generate new treatment options and reimbursement of drugs for this patient group (Fig. 1). First, the HIT-CF Organoid Study will screen different CFTR-modulators on organoids generated from 500 patients with rare CFTR mutations across Europe. Multiple pharmaceutical companies have committed to providing their investigational drugs (currently in phase I/II clinical trials) for the screening phase. The compounds being tested comprise the full range of different CFTR-modulator categories currently in the clinical domain (i.e. read-through agents, amplifiers, correctors and potentiators). An in vitro drug screening will identify which patients are most likely to clinically respond to one of the drug(s) (combinations). Once these patients have been identified, subsequent studies will evaluate the clinical efficacy of the drugs in patients identified as being ‘in vitro responders’, with the aim to gather sufficient data for regulatory approval of the drugs in these specific patients.

Overview of the HIT-CF Project
The second pathway is the generation of a living organoid biobank. Organoids from all 500 subjects in the HIT-CF Organoid Study will be stored for the study at Hubrecht Organoid Technology (HUB), and can be used for future drug development efforts. Currently, a biobank is set up to include materials from subjects across Europe. When patients consent to the long-term storage of their organoids, these can be used by academic laboratories worldwide, and collaborations can be explored with pharmaceutical companies to screen potential new CFTR-modulating drugs in this patient population.
The third pathway is aimed at regulatory access and reimbursement of the drugs in this patient population. Within the project, organoids are used to identify patients and data will be gathered on the predictive value of organoids. If this approach is to be implemented in regular practice, it needs the support of stakeholders such as regulatory agencies and insurance companies. Discussions with these stakeholders are ongoing to clarify what data is important, and their advice will be used to further optimize the project.
The currently ongoing HIT-CF Organoid Study (NTR7520) will be described here.
Methods
Clinical design
The aim of the HIT-CF Organoid Study is to collect rectal biopsies and culture intestinal organoids from at least 500 patients with ultra-rare CFTR mutations on both alleles. Subsequently the study aims to identify the predicted best clinical responders (based on amount of organoid swelling) to new CFTR-modulators from different pharmaceutical companies.
The study design is an international multi-center study. Rectal biopsies are collected from 500 subjects in different clinical sites across Europe, all members of the clinical trial network of the European Cystic Fibrosis Society (ECFS-CTN). Investigational study drugs will be tested on each organoid in one of three academic laboratories (University Medical Centre Utrecht (UMCU), Katholieke Universiteit Leuven (KUL) and the University of Lisboa (LIS)). A subset of responsive organoids in the academic laboratories will be further screened at HUB and ranked according to level of response to one of the lead drug combinations from each pharmaceutical partner. The subjects with the highest organoid responses will then be asked to participate in subsequent clinical trials. For more details see ‘organoid culturing and drug screening’ below.
Patient population
The target population is male and female adult (older than 16 or 18 years depending on country-specific ethical regulations) subjects diagnosed with Cystic Fibrosis with a sweat chloride concentration > 60 mmol/L, without a mutation for whom CFTR-modulating drugs are currently available (homozygous or heterozygous F508del mutations or gating mutations), and without combinations of frequently occurring class I mutations (1717-1G > A, 621 + 1G > T, 3120 + 1G > A, 1898 + 1G- > A, CFTRdele2,3 and 2183AA- > G). Patients that have a comorbidity that (to the opinion of the investigator) might pose a risk while participating in a clinical trial will be excluded from study participation. Moreover, patients that have had a lung transplantation are also ineligible.
Sample size
The study goal is to identify responders that are eligible for subsequent HIT-CF clinical trials. Our preliminary power calculations for the clinical trial with lung function (FEV1) as the primary endpoint, indicate a power of > 80% if 16 subjects are included. Therefore the study aims at recruiting 20 patients to mitigate drop-out during the study. This sample size is comparable to a previously published trial in a similar patient population and intervention [30]. As we expect that some patients will not be eligible for a clinical trial, we aim to identify 27 patients per trial eligible for recruitment. This will allow us to contact a new subject if a subject is not eligible to participate. The inclusion rate for the clinical trial needed to include 20 out of 27 patients is thus 74%. To include 16 evaluable subjects the minimum needed inclusion rate is 55%. In experiments performed so far on ultra-rare organoids, we have observed that ~ 16% of them react to the CFTR-modulating drugs ivacaftor or lumacaftor/ivacaftor, resulting in about 80 in vitro responders in the observed population of 500 subjects, which covers about 27 subjects for each of the three intended clinical trials.
Study procedures
The study procedure performed is collection of rectal biopsies by either forceps during sigmoidoscopy or using a rectal suction device. These are not part of regular medical treatment. Each site has the possibility to choose the procedure they are most experienced in. The success rate of generating organoids out of two forceps biopsies is > 92%. Success rates for rectal suction device is > 95% if four biopsies are obtained, and ~ 80% if two biopsies are obtained (unpublished data from UMCU/HUB). Therefore, either two forceps or four rectal suction biopsies will be obtained per subject. If no organoids can be generated, the patient will be re-contacted and asked whether they are willing to undergo another biopsy. The biopsies will have no influence on regular medical care. If intestinal organoids have already been generated for a subject in a previous study or for routine medical care and can be made available for testing, subjects do not need to undergo the study procedure again.
Rectal suction biopsy
Biopsies can be collected using a rectal suction device. This device is used to facilitate diagnosis of the disease Morbus Hirschsprung, and to obtain material for Intestinal Current Measurement for the diagnosis of Cystic Fibrosis. Therefore, many CF-centres have experience with rectal suction biopsies. A working instruction is provided to the sites with detailed instructions how to perform the procedure.
To obtain the best possible biopsies, patients will be treated with an enema (sodiumphosphate), to clean the most distal part of the colon. If this is contraindicated, biopsies will be collected without a preceding enema. During the procedure, patients will lie on their side, with knees pulled upwards towards the chest. Next, the rectal suction device is introduced rectally, and inserted ~ 6 cm. The rectal suction device is connected by a rubber tube to a vacuum meter, and the vacuum meter is attached by a rubber tube to a 50 cc plastic syringe. The assisting nurse sucks air into the suction device by pulling on the plunger of the syringe. This creates a vacuum, and intestinal tissue is sucked into the suction device through an opening with a diameter of 2.0 mm. This tissue is subsequently cut by the rectal suction device, and the tissue is collected. The diameter of the collected tissue is around 2 mm, and less than 1 mm thick. The biopsies are taken on the dorsolateral sides of the rectum, because no pain receptors are present there and no major blood vessels are present. It is important that the device opening has passed the anal sphincter to avoid pain. Collecting organoids using a rectal suction device has a very low risk of complications. Friedmacher et al. have recently published a systematic review, in which they found that the risk of any complication is 0.63%. Persistent rectal bleeding was the most common complication with an incidence of 0.5% [31]. The article states that in very young children (< 1 year of age), the risk is relatively higher. We will perform this procedure in adult subjects, and subjects are instructed to call the hospital if bleeding is persistent or if they have any other complaints. To detect any prolonged bleeding and intervene as early as possible, patients will stay in the hospital for 1 hour after the procedure.
Forceps biopsy
Biopsies can be collected by forceps during rectoscopy. This device is used by trained gastro-enterologists to obtain material for a wide spectrum of diseases. The procedure is done endoscopically with a flexible endoscope. The endoscope is introduced rectally. The biopsies are obtained on sight, thereby avoiding any arteries or veins. In a recently published article, the safety of this procedure was evaluated by studying 353 forceps biopsies. No major complications were reported. Only one patient complained of abdominal pain following the procedure, but fully recovered after 4 h observation [32]. Patients are observed after the procedure to quickly notice any possible complications.
Generating organoids from rectal biopsies
At the HUB organoids will be generated from the rectal biopsies using the protocol as previously published [22, 33, 34]. If organoids are received from study sites, these will be further cultured to ensure viability of material. From HUB, organoids will be transferred to the Cystic Fibrosis Research Laboratory of University Medical Center Utrecht, the Organ Systems Laboratory of KU Leuven and the Cystic Fibrosis Research Laboratory of the University of Lisbon. Samples will be transported by a specialist courier service.
Organoid culturing and drug screening
UMCU protocols for CFTR function measurements have been established under ISO-9001 conditions and HUB has ISO-270001 compliant protocols. The most updated protocols of UMC and HUB have been combined and shared with the laboratories in Leuven and Lisbon for preparation of organoid culture media, organoid growing, and CFTR function measurement by forskolin-induced swelling (FIS) [33]. From these, one standard lab manual has been generated for all the labs to make sure the procedures are standardised. Additionally, identical lot numbers and preferably batches of reagents will be obtained by all labs to ensure the most optimal standardisation.
Drugs to be tested
The screening of the organoids will be divided up based on which drugs are being tested.
Flatley Discovery Labs (FDL) is developing CFTR correctors and potentiators, and combinations of these drugs will be tested in the screen. Proteostasis Therapeutics (PTI) is developing CFTR correctors, potentiators and an amplifier, which will all be tested in the drug screen.
Drugs from FDL and PTI will be tested within the same screen because of their comparable modes-of-action. Eloxx Pharmaceuticals is developing a CFTR read-through drug that will be screened separately, since the distinct mode-of-action necessitates different assay conditions. Moreover, the drugs developed by FDL and PTI are not expected to work on premature termination codons. Therefore, only organoids with maximum one PTC will be screened for responsiveness to FDL and PTI drugs, and organoids with at least one PTC will be screened for efficacy of ELOXX drugs.
In both screens, organoids propagated in 24-wells plates will be transferred to 96-well plates for drug testing. Each experimental 96-wells plate will first go through several quality control checks to ensure that no pipetting errors have been made and the organoid cultures are of good quality by using reference organoid lines. These reference organoid lines are well characterized and the response of these reference organoids to the drugs from FDL, PTI and Eloxx should be within the range of what we have detected previously.
CFTR function and response to the different drugs is measured by time-lapse confocal microscopy as the relative increase in surface area (as area under the curve, AUC) of calcein-green labelled organoids at 10 min intervals for 60 min after stimulation with forskolin. Technological duplicates are measured for each condition, and individual drug responses are corrected for residual CFTR function upon forskolin stimulation.
For FDL and PTI compounds, F508del homozygous organoids will be included in each experiment for comparison. For ELOXX compounds, G542X homozygous organoids will be included in each experiment for comparison.
The best responding 27 organoids per lead product will be eligible for the clinical trials.
SNP fingerprinting analysis and CFTR mutation sequencing
As part of HUB’s quality control procedures, to confirm that no swap of material occurred during the whole screening process, genetic material from the biopsy tissue and the generated organoids will be isolated and submitted to SNP fingerprinting analysis. The obtained genetic material will be also used to confirm the CFTR mutation by PCR-DNA sequencing of the patient derived organoids for the subsequent clinical trial.
Organoid biobanking for future use
The organoids generated will be stored by HUB for the duration of the study. During the HIT-CF project, a new biobank is being created to store the organoids for future use and make them available to researchers worldwide. Ethical experts will interview stakeholders and CF patients in order to explore the optimal governance of the biobank, and to generate an informed consent form that covers all aspects of this biobank.
Once this new biobank has been set up, patients will be asked to sign informed consent for permission to use their organoids in this biobank. This will only occur if the local study site has ethical approval for the separate biobank. If patients refuse, the organoids will be destroyed after the HIT-CF Organoid Study. If patients sign the informed consent, their organoids will enter the organoid biobank.
Discussion
The HIT-CF Organoid Study applies intestinal organoids for personalized medicine in cystic fibrosis. Rare genetic variants of cystic fibrosis are currently excluded in CFTR-modulator trials, while it is expected that some of these variants are very responsive to treatment. By generating patient-specific organoids for these rare genotypes, a functional assessment of drug response per individual can be generated and treatment can be tailored to the individual. With the accompanying subsequent clinical trials, an innovative opportunity is created for CF-patients with rare mutations to get access to new CFTR-modulator drugs.
Personalized medicine approaches using organoids are ongoing in several diseases, but are still limited in scale. Several oncological trials are being conducted, such as in lung (clinicaltrials.gov identifier NCT03979170), breast (NCT03544047) and pancreatic cancer (NCT03544255). However, these studies are single-centre and aim to retrospectively compare in vivo and in vitro treatment responses. In the field of cystic fibrosis, pilot studies found that organoids can identify individuals that clinically benefit from the CFTR-modulating drugs ivacaftor and lumacaftor/ivacaftor with high accuracy, but these studies had a limited sample size [26, 27]. By including up to 500 subjects in sites across Europe and performing confirmatory clinical trials, our project will be the first to prospectively assess the feasibility of using organoids for personalized medicine on such a large scale.
One of the strengths of this study is the collaboration with the European Cystic Fibrosis Society – Clinical Trials Network (ECFS-CTN). This network consists of 58 sites in 17 countries that share standardized working procedures and training, and have been involved in the study design. The study will be run only in ECFS-CTN sites, which will enable a quick start-up and ensures smooth communication between the sites and sponsor.
A risk of this project is that the drugs screened have yet to be clinically approved. It is therefore unclear whether all of these drugs will eventually reach the market and become publicly available. By involving three different pharmaceutical companies with several drugs in the pipeline and including ivacaftor and tezacaftor/ivacaftor in the drug screening, this risk is minimized. Another uncertainty is the number of responsive subjects that we expect to find. The current prediction of ~ 16% responsive subjects is based on a small dataset, and these numbers could differ depending on geographical distribution and efficacy of the experimental compounds. However, since drugs with innovative modes-of-action are investigated, the responsive population could be larger than previously anticipated. Moreover, efficacy of the experimental compounds is expected to be better than currently available drugs, which could result in a higher power in our clinical studies and therefore a reduced number of responsive subjects could be sufficient.
An important outcome of this study will be the large dataset created by combining all the individual results. This dataset will generate extensive knowledge on how different genotypes respond to drugs, and will become publicly available, which can be of great use to the scientific community. Furthermore, if the subjects consent, organoids that have been generated will be made available for future research. This biobank can be accessed by other scientists, and newly developed drugs can be tested on those organoids. Around 14 companies are currently developing drugs that aim to restore CFTR function (https://www.cff.org/Trials/pipeline) [35]. The impact of this study is therefore amplified, since many of these new drugs or experimental compounds could be screened.
The HIT-CF Organoid Study creates the opportunity of CFTR-modulating treatment in a CF patient population that has previously been largely ignored. Several drugs will be tested in vitro to stratify patients according to the best possible drug, thereby tailoring treatment to the individual patient. Moreover, extensive knowledge will be generated about the effects of drugs on a spectrum of genotypes, and the subsequently available biobank will be very useful for future drug development and research into Cystic Fibrosis.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed for this manuscript.
Abbreviations
- CF:
-
Cystic Fibrosis
- CFTR:
-
Cystic Fibrosis Transmembrane Conductance Regulator
- ECFS-CTN:
-
European Cystic Fibrosis Society – Clinical Trial Network
- ELOXX:
-
Eloxx Pharmaceuticals
- FDL:
-
Flatley Discovery Labs
- FIS:
-
Forskolin induced swelling
- HIT-CF:
-
Human Individualized Treatment for CF
- HUB:
-
Hubrecht Organoid Technology
- KUL:
-
Katholieke Universiteit Leuven
- LIS:
-
LIS
- PTC:
-
Premature termination codon
- PTI:
-
Proteostasis Therapeutics
- UMCU:
-
University Medical Centre Utrecht
References
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–80.
De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;2600(16):1–13. https://doi.org/10.1016/S2213-2600(16)00023-0.
Elborn JS. Cystic fibrosis. Lancet. 2016; [cited 2016 May 4]; Available from: http://www.sciencedirect.com/science/article/pii/S0140673616005766.
Cystic Fibrosis Mutation Database. [cited 2020 Apr 15]. Available from: http://www.genet.sickkids.on.ca/.
The Clinical and Functional TRanslation of CFTR (CFTR2); Available at http://cftr2.org.
Kerem E, Corey M, Kerem B-S, Rommens JM, Markiewicz D, Levison H, et al. The relation between genotype and phenotype in cystic fibrosis - analysis of the most common mutation (F508del). N Engl J Med. 1990;323(22):1517–22 Available from: http://www.ncbi.nlm.nih.gov/pubmed/2278545.
Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci. 2009;106(44):18825–30 Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0904709106.
Boyle MP, Bell SC, Konstan MW, SA MC, Rowe SM, Rietschel E, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–38. https://doi.org/10.1016/S2213-2600(14)70132-8.
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015:1–12 Available from: http://www.ncbi.nlm.nih.gov/pubmed/25981758.
Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci. 2011;108(46):18843–8 Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1105787108.
Cuyx S, De Boeck K. Treating the underlying cystic fibrosis transmembrane conductance regulator defect in patients with cystic fibrosis. Semin Respir Crit Care Med. 2019;40(6):762–74. https://doi.org/10.1055/s-0039-1696664. Epub 2019 Oct 28.
Veit G, Xu H, Dreano E, Avramescu RG, Bagdany M, Beitel LK, et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat Med. 2018;24(11):1732–42 Available from: http://www.nature.com/articles/s41591-018-0200-x.
Veit G, Da Fonte DF, Avramescu RG, Premchandar A, Bagdany M, Xu H, et al. Mutation-specific dual potentiators maximize rescue of CFTR gating mutants. J Cyst Fibros. 2019; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1569199319309245.
Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet (London, England). 2019;6736(19):6–14 Available from: http://www.ncbi.nlm.nih.gov/pubmed/31679946.
Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor–Tezacaftor–Ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–19 Available from: http://www.nejm.org/doi/10.1056/NEJMoa1908639.
Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91.
Dukovski D, Villella A, Bastos C, King R, Finley D, Kelly JW, et al. Amplifiers co-translationally enhance CFTR biosynthesis via PCBP1-mediated regulation of CFTR mRNA. J Cyst Fibros. 2020; In Press. Available from: https://www.sciencedirect.com/science/article/pii/S1569199320300527.
Marozkina NV, Yemen S, Borowitz M, Liu L, Plapp M, Sun F, et al. Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc Natl Acad Sci U S A. 2010;107(25):11393–8 Available from: http://www.ncbi.nlm.nih.gov/pubmed/20534503.
Sigoillot M, Overtus M, Grodecka M, Scholl D, Garcia-Pino A, Laeremans T, et al. Domain-interface dynamics of CFTR revealed by stabilizing nanobodies. Nat Commun. 2019;10(1). https://doi.org/10.1038/s41467-019-10714-y.
Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190–4 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23744940.
Clevers H. Modeling development and disease with organoids. Cell. 2016;165(7):1586–97. https://doi.org/10.1016/j.cell.2016.05.082.
Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–45. https://doi.org/10.1038/nm.3201.
de Winter-de Groot KM, Janssens HM, van Uum RT, Dekkers JF, Berkers G, Vonk A, et al. Stratifying infants with cystic fibrosis for disease severity using intestinal organoid swelling as a biomarker of CFTR function. Eur Respir J. 2018;52(3):1702529 Available from: http://erj.ersjournals.com/lookup/doi/10.1183/13993003.02529-2017.
de Winter de Groot KM, Berkers G, Marck van der Wilt REP, van der Meer R, Vonk A, Dekkers JF, et al. Forskolin-induced swelling of intestinal organoids correlates with disease severity in adults with cystic fibrosis and homozygous F508del mutations. J Cyst Fibros. 2019;(xxxx):1–6. https://doi.org/10.1016/j.jcf.2019.10.022.
Zomer-van Ommen DD, de Poel E, Kruisselbrink E, Oppelaar H, Vonk AM, Janssens HM, et al. Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J Cyst Fibros. 2018;17(3):316–24. https://doi.org/10.1016/j.jcf.2018.02.007.
Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med. 2016;8(344):344ra84 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27334259.
Berkers G, van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, de Winter-de Groot KM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019;26(7):1701–1708.e3. https://doi.org/10.1016/j.celrep.2019.01.068.
Zomer-van Ommen DD, Vijftigschild LAW, Kruisselbrink E, Vonk AM, Dekkers JF, Janssens HM, et al. Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J Cyst Fibros. 2016;15(2):158–62. https://doi.org/10.1016/j.jcf.2015.07.007.
HIT-CF Europe. [cited 2020 Apr 15]. Available from: https://www.hitcf.org/.
De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–80. https://doi.org/10.1016/j.jcf.2014.09.005.
Friedmacher F, Puri P. Rectal suction biopsy for the diagnosis of Hirschsprung’s disease: a systematic review of diagnostic accuracy and complications. Pediatr Surg Int. 2015;31(9):821–30 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26156878.
Servidoni MF, Sousa M, Vinagre AM, Cardoso SR, Ribeiro MA, Meirelles LR, et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol. 2013;13:91 Available from: http://www.ncbi.nlm.nih.gov/pubmed/23688510.
Vonk AM, Van P, Ramalho AS, De Boeck K, Van CK ED, et al. Protocol for application , standardization and validation of the Forskolin-induced swelling assay in cystic fibrosis human colon organoids protocol for application , standardization and validation of the Forskolin-induced swelling assay in cystic fibrosis. STAR Protoc. 2020;100019 Available from: https://star-protocols.cell.com/protocols/72.
Boj SF, Vonk AM, Statia M, Su J, Vries RRG, Beekman JM, et al. Forskolin-induced swelling in intestinal organoids: an in vitro assay for assessing drug response in cystic fibrosis patients. J Vis Exp. 2017;(120):1–12 Available from: http://www.jove.com/video/55159/forskolin-induced-swelling-intestinal-organoids-an-vitro-assay-for.
Cystic Fibrosis Foundation Drug Development Pipeline. [cited 2020 Apr 15]. Available from: https://www.cff.org/Trials/pipeline.
Acknowledgements
We would like to thank the following people in particular for contributing to the design of the HIT-CF Organoid Study: Hilde De Keyser, Elise Lammertyn (CF Europe), Dr. Silke van Koningsbruggen-Rietschel (European Cystic Fibrosis Society – Clinical Trial Network), Dr. Lutz Naehrlich (European Cystic Fibrosis Society - Patient Registry), Judith Pool (Julius Clinical Research), Marc van de Craen (Patergrus), Begoña Aguilera, Johanna Pott, Rob G. J. Vries, Sylvia F. Boj (Hubrecht Organoid Technology), Kris De Boeck, François Vermeulen, Anabela S. Ramalho (Katholieke Universiteit Leuven, Katholieke Universiteit Leuven, Dept. of Development and Regeneration), Iris A.L. Silva, Margarida D. Amaral (University of Lisboa, Faculty of Sciences, BioISI- Biosystems & Integrative Sciences Institute), Matthew Goddeeris (Eloxx Pharmaceuticals), and Geoffrey Gilmartin (Proteostasis Therapeutics).
Funding
This study was funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 755021.
Author information
Authors and Affiliations
Consortia
Contributions
PM was involved in the conception and design of the protocol, and has drafted the manuscript. SM was involved in the conception and design of the protocol. AV was involved in the conception and design of the protocol. JMB was involved in the conception and design of the protocol, and has revised the manuscript. CKE was involved in the conception and design of the protocol and has revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Medical Research Ethics Committee (METC) of the University Medical Center Utrecht. The study will be performed according to Good Clinical Practice guidelines, and participants will have to sign an informed consent form before they can be included in this study.
Consent for publication
Not applicable.
Competing interests
CKE reports grants from GSK, Nutricia, TEVA, Gilead, Vertex, ProQR, Proteostasis, Galapagos NV, and grants from Eloxx outside the submitted work and has a patent 10006904 with royalties paid. JMB is inventor on a patent related to CFTR function measurements in organoids and has received financial royalties. He also participates in industry-sponsored clinical trials (Galapagos NV, Vertex Pharmaceuticals, Proteostasis, Eloxx Pharmaceuticals) and received travel reimbursements and speaker’s fees. The other authors have nothing to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van Mourik, P., Michel, S., Vonk, A.M. et al. Rationale and design of the HIT-CF organoid study: stratifying cystic fibrosis patients based on intestinal organoid response to different CFTR-modulators. transl med commun 5, 9 (2020). https://doi.org/10.1186/s41231-020-00060-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41231-020-00060-3