
Abstract
Background
Vibrio cholerae is associated with severe watery diarrheal disease among people in many parts of the world, including the coastal provinces of Southern Thailand. There are relatively few studies focusing on the genetic characterization among V. cholerae isolates in this region. Therefore, this study aimed at exploring the presence of virulence genes and DNA fingerprints among V. cholerae O1 and non-O1/non-O139 isolates obtained from clinical samples in four southern coastal provinces during the period of 2001–2009 (n = 21).
Results
All V. cholerae O1 isolates possessed ctxA, tcpA, zot, ace, hlyA, and vasH genes. However, only hlyA, vcsV2, and vasH genes were detected in the majority of the non-O1/non-O139 isolates. All O1 isolates showed indistinguishable PCR fingerprints by arbitrarily primed (AP)-PCR and enterobacterial repetitive intergenic consensus (ERIC)-PCR regardless of the geographical area and period of isolation. However, the multi-locus variable-number of tandem-repeat analysis (MLVA) could differentiate these O1 isolates (n = 11) into eight profiles. Isolates exhibiting an undistinguished MLVA profile also showed identical pulsed-field gel electrophoresis (PFGE). In addition, the O1 isolates were grouped into the same cluster by all methods used in this study.
Conclusions
This study demonstrated the presence of virulence genes and genetic diversity among different serogroups of V. cholerae isolates from clinical samples in southern Thailand. V. cholerae O1 isolated over a period of multiple years were genetically related, suggesting that they had a clonal origin, whereas non-O1/non-O139 isolates could have evolved independently.
Similar content being viewed by others
Background
Vibrio cholerae is one of the endemic pathogens causing acute diarrheal disease in several parts of the world. During the past 10 years, the highest documented outbreaks in Thailand occurred in 2001, 2004, 2007, 2010, and 2016 [1], and approximately 31% of reported cholera cases in 2007 occurred in the southern regions [2]. Many of the reported cholera cases originated from the southern coastal provinces which are located on the Malay Peninsula. The V. cholerae infections in the southern region are almost exclusively caused by consumption of contaminated seafood or poor sanitation and hygiene among fishing communities and alien laborers.
Throughout history, V. cholerae O1 serogroup has dominated the affected geographical regions. Toxigenic V. cholerae serogroups O1 and O139 have been reported to harbor the ctxA and tcpA genes, encoding the A subunit of cholera toxin (CT) and the major subunit of a toxin coregulated pilus (Tcp), respectively. Furthermore, the zot, encoding the zonula occludens toxin (Zot), and ace encoding the accessory cholera enterotoxin (Ace) were also reported to be involved in the pathogenesis of O1 and O139 serogroups. However, these genes were reported to be absent in the non-O1/non-O139 isolates [3, 4]. The stn/sto and hlyA genes, encoding a non-O1 heat-stable enterotoxin (NAG-ST) and the El Tor-like hemolysin, respectively, have also been reported to associate with non-O1/non-O139 infections [5]. In addition, the type III (T3SS) and type VI (T6SS) secretion systems are considered as additional important virulence factors of V. cholerae [6, 7].
Among the methods for studying the relatedness or differentiations among the V. cholerae isolates, pulsed-field gel electrophoresis (PFGE) is considered to be the gold standard [8,9,10]. Unfortunately, this method is time-consuming and requires specialized equipment. Polymerase chain reaction (PCR)-based typing methods, in comparison to PFGE, are easier, faster, and less expensive. These methods, including box elements PCR (BOX-PCR), arbitrarily primed PCR (AP-PCR) [3, 4, 11], enterobacterial repetitive intergenic consensus sequence PCR (ERIC-PCR) [3, 12,13,14], V. cholerae repeats-PCR (VCR-PCR) [15, 16], and multi-locus variable-number of tandem-repeat (VNTR) analysis (MLVA) [17,18,19,20] have been evaluated for typing of V. cholerae isolates obtained from various geographic regions.
Southern coastal provinces of Thailand are considered to be the endemic regions for cholera. In this study, we examined V. cholerae isolates obtained from diarrheal patients in these areas for their virulence gene profiles and also elucidate their genetic relationships using well established PCR-based fingerprinting methods. Moreover, the discriminatory ability of each method was also determined.
Methods
Bacterial strain
A total of 21 V. cholerae isolates (VC1-VC21) from clinical samples in four southern coastal provinces were included in this study (Table 1). All isolates were obtained between 2001 and 2009 from sporadic cholera cases as part of the routine microbiological diagnosis, and were kindly provided by Hat Yai Hospital, Songkhla, Thailand. These isolates were confirmed as V. cholerae by ompW-based PCR [21]. Serogrouping was performed by slide agglutination using O1 and O139 antisera. Bacterial genomic DNA was extracted using a Genomic DNA extraction kit (Geneaid, Taiwan).
PCR detection of virulence genes
V. cholerae isolates were screened for the presence of eight virulence genes, including ctxA, tcpA, zot, ace, stn/sto, hlyA, vcsV2 (T3SS), and vasH (T6SS) genes. All primers, amplicon sizes, and PCR conditions are listed in Table 2. PCR products were visualized after electrophoresis in 1.8% agarose gel.
Molecular typing by AP-PCR, ERIC-PCR, and VCR-PCR
DNA from pure cultures was extracted using the Genomic DNA Mini Kit (Geneaid, Taiwan) according to the manufacturer’s instructions. AP-PCR was performed using a single nucleotide primer, Primer 2 (5′-GTTTCGCTCC) [22]. ERIC-PCR was performed as previously described using the primer pair ERIC1R (5′- ATGTAAGCTCCTGGGGATTCAC-3′) and ERIC2 (5′-AAGTAAGTGACTGGGGTGAGCG-3′) [14]. For AP-PCR and ERIC-PCR, ExTaq DNA polymerase (TaKaRa, Japan) was used instead of conventional Taq DNA polymerase. For VCR-PCR, the primers VCR-5′ (5′-TCCCTCTTGAGGCGTTTGTTAC-3′) and VCR-3′ (5′-AGCCCCTTAGGCGGGCGTTAA-3′) were used [16], and the amplification was carried out as described by Teh et al. [15]. The amplification products were analyzed by electrophoresis using a 1.5% agarose gel. DNA patterns were compared by the unweighted pair group method with arithmetic mean (UPGMA) using the Dice coefficient. A dendrogram was constructed using Bionumeric software v.7.6 (Applied Maths, Belgium).
MLVA and data analysis
MLVA was conducted using primers and protocols described previously [17]. Three selected loci (VC0436-7, VC1650, and VCA0171) were amplified by PCR using 5′ fluorescent-labeled forward primers and analyzed by capillary electrophoresis using an ABI 3730xl Genetic Analyzer (Applied BioSystems, USA). Fragment sizes from each VNTR locus were determined using the GeneMapper software (v.4.1) (Applied BioSystems, USA) and were converted into copy numbers using the following equation: Number of repeats (bp) = [fragment size (bp) − flanking regions (bp)]/repeat size (bp) [23]. The repeat copy numbers were analyzed, and a dendrogram was constructed by Bionumeric software v.7.6 (Applied Maths, Belgium).
PFGE
Isolates with undistinguished DNA profile by the above methods were further determined by PFGE using the PulseNet standardized protocol for V. cholerae [9]. Chromosomal DNA of V. cholerae was digested with restriction enzyme Not I (NEB, USA), and the digested DNA fragments were separated on 1% Pulse-Field Certified agarose gel by CHEF-DRIII system (Bio-Rad Laboratories, California, USA). The gel was stained with ethidium bromide, and the DNA patterns were analyzed using Bionumeric software v.7.6 (Applied Maths, Belgium).
Discriminatory index and typeability
The discriminatory powers were calculated using previously published formula [24]. The polymorphism information index or Nei’s diversity index (DI) of each VNTR locus was calculated individually using the formula: D = 1 − Σ (allele frequency)2 [25]. Typeability was calculated from the formula recommended previously [26].
Results
Virulence gene profiles
A total of six virulence gene profiles (A to F) were observed (Table 3). The most prevalent virulence genes detected were hlyA (95%) and vasH (95%), and the distribution of virulence genes was related to serogroups. All of the O1 isolates were positive for the ctxA, tcpA, zot, ace, hlyA, and vasH virulence genes. None of the O1 isolates were positive for the stn/sto and vcsV2 (T3SS) genes. All non-O1/non-O139 isolates were negative for tcpA gene. Interestingly, ctxA, zot, and/or ace genes were detected in two isolates of V. cholerae non-O1/non-O139 (VC12 and VC13). In this study, one non-O1/non-O139 isolate (VC15) was negative for all virulence gene tested (Table 3).
Molecular typing of V. cholerae isolates by AP-PCR, ERIC-PCR, and VCR-PCR
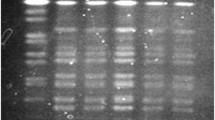
The 21 V. cholerae isolates were differentiated into 10, 5, and 9 profiles based on DNA patterns obtained by AP-PCR, ERIC-PCR, and VCR-PCR, respectively (Table 1). Good correlation was observed between the fingerprinting results of AP-PCR, ERIC-PCR, and VCR-PCR. All of the O1 isolates (n = 11) yielded identical fingerprints by AP-PCR and ERIC-PCR, and highly similar patterns by VCR-PCR (Fig. 1). In addition, only AP-PCR and VCR-PCR could distinguish serogroup O1 from non-O1/non-O139 isolates. The fingerprints of non-O1/non-O139 isolates tended to be more diverse, but were not correlated with geographical areas and period of isolations. In this study, AP-PCR and VCR-PCR of non-O1/non-O139 isolates resulted in amplification of more DNA fragments than ERIC-PCR, although no VCR-PCR product was observed from two isolates (VC12 and VC15). The dendrograms generated for all V. cholerae isolates based on AP-PCR method are shown in Fig. 1. These methods showed low discriminatory power in typing, especially among the O1 isolates.

Dendrogram and PCR fingerprints for comparison of V. cholerae isolates. The dendrogram is based on the PCR fingerprints generated by AP-PCR. The PCR fingerprints of ERIC-PCR and VCR-PCR are provided for direct comparison
MLVA and phylogenetic analysis
All V. cholerae isolates were distinguished into 18 MLVA profiles (Table 1). All profiles were represented by a single isolate except for MLVA type 1 that was represented by four isolates (VC1, VC2, VC3, and VC4). The PFGE profiles of these four isolates showed identical restriction patterns (data not shown). The copy numbers of VNTR alleles in VC0436-7, VC1650, and VCA0171 loci ranged from 4–12, 3–13, and 7–24 repeats, respectively (Table 1). In total, 9, 9, and 11 alleles were observed in VC0436-7, VC1650, and VCA0171, respectively.
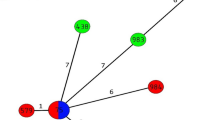
To demonstrate the relationships among V. cholerae isolates, a minimum spanning tree (MST) was constructed based on the MLVA profiles (Fig. 2). All O1 isolates were grouped into the same cluster (cluster I). In this cluster, four of the O1 isolates (VC1, VC2, VC3, and VC4) obtained during 2001 from different regions (Songkhla, Nakhon Si Thammarat, and Phuket) shared the same profile (9-13-21). Interestingly, a non-O1/non-O139 isolate (VC12) obtained during the same period was in this cluster. Cluster II composed of non-O1/non-O139 isolates which were genetically diverse (Fig. 2).

Minimum spanning tree of the 21 V. cholerae isolates based on MLVA profiles. Each circle represents a unique MLVA profile. The color of the circles corresponds to serogroups. The circle size is proportional to the number of isolates. A distance of one locus between two MLVA profiles is indicated by a thick line, a distance of two loci is indicated by a gray line, and a distance of three loci is indicated by a dot line. The dotted circle indicates the MLVA clusters (I and II)
Discriminatory index and typeability
In this study, the comparison of Simpson’s index of diversity revealed that MLVA exhibited the highest discrimination value (0.97) (Table 4). VCA0171 was identified to be the most polymorphic loci with a much higher DI (0.89) than the others. VC0436-7 and VC1650 had the DI value of 0.79 and 0.83, respectively. All typing methods, except VCR-PCR, were able to type 100% of the isolates examined (Table 4).
Discussion
In this study, V. cholerae O1 and non-O1/non-O139 obtained from clinical samples in southern Thailand were molecularly characterized to obtain an understanding of their virulence and genetic relationship. The distribution of ctxA, tcpA, zot, and ace genes was strongly correlated with O1 serogroup. Although these virulence genes are rarely detected from the non-O1/non-O139 V. cholerae isolates, ctxA, zot, and/or ace genes were detected in two of the non-O1/non-O139 isolates (VC12 and VC13) examined in this study. The occurrence of ctxA-positive non-O1/non-O139 isolates was found in India and Brazil [27, 28].
In this study, vcsV2 (T3SS) was not detected in all clinical isolates of V. cholerae O1. This supports the previous studies from Rahman et al. (2008) which demonstrated that most clinical isolates of V. cholerae O1 and O139 were negative for T3SS [29]. The presence of T3SS in majority of non-O1/non-O139 isolates was also reported in Bangladesh, China, Nigeria, Germany, and Austria [29,30,31,32]. However, the incidence of T3SS among V. cholerae non-O1/non-O139 isolated in this study was 73% (8/11) which is higher than that reported in India (31.5%) [6] and Bangladesh (38.9%) [33]. The role of T3SS in human infections by non-O1/non-O139 was reported to correlate with increased hemolytic titers and motility [6]. Thus, the presence of T3SS might be essential not only for environmental fitness but also for the pathogenesis of non-O1/non-O139 in human hosts. In this study, vasH (T6SS) gene was detected in almost all isolates, regardless of serogroups. This supports the previous report which demonstrated that T6SS are conserved between V. cholerae O1, O139, and non-O1/non-O139 [34]. However, T6SS expression differs between strains and is strictly regulated in O1 and O139 serogroups. The role of T6SS was reported to play a role in V. cholerae fitness in the aquatic environments, and it is repressed at initial infection in human hosts [34].
Several established DNA banding pattern-based genotyping methods, including AP-PCR, ERIC-PCR, and VCR-PCR, were used to analyze the genetic relatedness of V. cholerae O1 and non-O1/non-O139 isolates. The O1 isolates (VC1-VC11) in this study yield DNA fingerprints that were identical by AP-PCR or highly similar by VCR-PCR but distinct from those of non-O1/non-O139 isolates (VC12-VC21). However, ERIC-PCR could not differentiate some of non-O1/non-O139 (VC16-VC21) from O1 isolates. Low discrimination of these methods in V. cholerae O1 typing was also reported among the strains isolated from India, Malaysia, Taiwan, and Iran [11, 35,36,37]. This may be due to the highly conserved genomes among the V. cholerae O1 serogroup. Thus, these results confirmed the clonality among isolates of V. cholerae O1.
The MLVA method was shown to be more effective for studying the genetic relationships among V. cholerae isolates than other PCR-based typing methods [38] because this typing method relies on the detection of multiple tandem repeats on bacterium genomes that evolve rapidly [17]. To increase the accuracy of this method, capillary DNA sequencing of PCR fragments from each VNTR locus is used instead of agarose gel electrophoresis. In previous reports, the VNTR locus VCA0171 (located on chromosome II) of V. choleare Indian and Haiti isolates had the greatest diversity compared with VC0436-7 and VC1650 loci [18, 39]. The discriminatory ability of VcA VNTR based on VCA0171 locus for typing of V. cholerae strains was better than PFGE [40]. The results from this study support these findings.
In this study, MLVA showed an agreement with epidemiological data. Four O1 isolates that shared the same PFGE pattern and MLVA profile (VC1, VC2, VC3, and VC4) were obtained during the same period. The association between V. cholerae serogroups, year and site of isolation, and the genotypic clusters was previously reported by other studies [18, 19]. In this study, it is possible that these areas were probably contaminated with the same clone of V. cholerae and this strain may be responsible for the occurrence of cholera in the study regions during that period.
An isolate of non-O1/non-O139 (VC12) was grouped together with O1 isolates in MLVA cluster I. This isolate harbored all O1-specific virulence genes, except tcpA which may be due to the variation in the primer binding site. This isolate may has evolved from O1 serogroup strain by O-antigen shifting during the evolutionary step as previously occurred in V. cholerae serogroups O139, O26, and O37 [41,42,43]. It is possible that new toxigenic non-O1/non-O139 V. cholerae isolate with epidemic potential may emerge in the future. In contrast to VC12, the isolate VC13 possessed non-O1/non-O139 background (MLVA cluster II) (Fig. 2). This isolate may acquire the O1-specific virulence genes, ctxA and/or zot, by horizontal gene transfer. The presence of ctxA and/or zot genes among non-O1/non-O139 isolates demonstrated the potential of natural genetic exchange in this organism which may have occurred in aquatic ecosystems or inside the human host [27, 44]. Interestingly, the repeat numbers of VC0436-7 locus in one of the non-O1/non-O139 isolates (VC14) were related to those of the O1 isolates. Thus, isolate VC14 was excluded from cluster II. Clusters may change if more VNTR loci were included in the analysis. Further study is needed to support the relationship among these isolates.
Conclusion
The genetic relationship within serogroups of clinical V. cholerae isolates was demonstrated in this study. However, most of the non-O1/non-O139 isolates were very heterogeneous regarding their virulence gene patterns and DNA fingerprints. The fingerprinting methods should be applied according to their serogroups and sources of isolation. AP-PCR may be enough for typing among non-O1/non-O139 isolates. However, MLVA which exhibited higher discriminatory power than those methods should be used for typing of V. cholerae serogroup O1. Understanding the association between bacterial virulence characteristics, genotypes, year, and source of isolation is necessary for epidemiological surveillance of V. cholerae infections.
Abbreviations
- Ace:
-
Accessory cholera enterotoxin
- AP-PCR:
-
Arbitrarily primed-PCR
- CT:
-
Cholera toxin
- DI:
-
Diversity index
- ERIC-PCR:
-
Enterobacterial repetitive intergenic consensus-PCR
- MLVA:
-
Multi-locus variable-number of tandem-repeat analysis
- NAG:
-
Non-agglutinating V. cholerae serogroup non-O1/non-O139
- NAG-ST:
-
Non-O1 heat-stable enterotoxin
- PCR:
-
Polymerase chain reaction
- PFGE:
-
Pulsed-field gel electrophoresis
- T3SS:
-
Type III secretion systems
- T6SS:
-
Type VI secretion systems
- Tcp:
-
Toxin coregulated pilus
- UPGMA:
-
Unweighted pair group method with arithmetic mean
- VCR-PCR:
-
V. cholerae repeats-PCR
- VNTR:
-
Variable number of tandem repeat
- Zot:
-
Zonula occludens toxin
References
AESR, Annual Epidemiological Surveillance Report: Cholera 2015 Thailand, Bureau of Epidemiology. http://www.boe.moph.go.th/Annual/AESR2015/aesr2558/Part%201/07/cholera.pdf (2016). Accessed 20 May 2016.
AESR, Annual Epidemiological Surveillance Report: Cholera 2007 Thailand: Bureau of Epidemiology. http://www.boe.moph.go.th/Annual/ANNUAL2550/Part2/Table/Table9_1_4.html (2008). Accessed 20 May 2016.
Kumar P, Jain M, Goel A, Bhadauria S, Sharma S, Kamboj D, et al. A large cholera outbreak due to a new cholera toxin variant of the Vibrio cholerae O1 El Tor biotype in Orissa, Eastern India. J Med Microbiol. 2009;58:234–8.
Singh D, Matte MH, Matte G, Jiang S, Sabeena F, Shukla B, et al. Molecular analysis of Vibrio cholerae O1, O139, non-O1, and non-O139 strains: clonal relationships between clinical and environmental isolates. Appl Environ Microbiol. 2001;67:910–21.
Ceccarelli D, Chen A, Hasan NA, Rashed SM, Huq A, Colwell RR. Vibrio cholerae non-O1/non-O139 carrying multiple virulence factors and V. cholerae O1 in the Chesapeake Bay, Maryland. Appl Environ Microbiol. 2015;81:1909–18.
Chatterjee S, Ghosh K, Raychoudhuri A, Chowdhury G, Bhattacharya M, Mukhopadhyay A, et al. Incidence, virulence factors, and clonality among clinical strains of non-O1, non-O139 Vibrio cholerae isolates from hospitalized diarrheal patients in Kolkata, India. J Clin Microbiol. 2009;47:1087–95.
Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host Microbe. 2009;5:234–43.
Cameron DN, Khambaty FM, Wachsmuth IK, Tauxe RV, Barrett TJ. Molecular characterization of Vibrio cholerae O1 strains by pulsed-field gel electrophoresis. J Clin Microbiol. 1994;32:1685–90.
Cooper K, Luey C, Bird M, Terajima J, Nair G, Kam K, et al. Development and validation of a PulseNet standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio cholerae. Foodborne Pathog Dis. 2006;3:51–8.
Kam KM, Luey CKY, Tsang YM, Law CP, Chu MY, Cheung TL, et al. Molecular subtyping of Vibrio cholerae O1 and O139 by pulsed-field gel electrophoresis in Hong Kong: correlation with epidemiological events from 1994 to 2002. J Clin Microbiol. 2003;41:4502–11.
Ranjbar R, Pourshafie M, Sadeghifard N, Karami A, Hamidian M, Izadi M, et al. Molecular characterization of epidemic isolates of Vibrio cholerae O1 by arbitrarily primed PCR (AP-PCR). Iran J Public Health. 2008;37:83–7.
Chokesajjawatee N, Zo Y-G, Colwell RR. Determination of clonality and relatedness of Vibrio cholerae isolates by genomic fingerprinting, using long-range repetitive element sequence-based PCR. Appl Environ Microbiol. 2008;74:5392–401.
Preeprem S, Mittraparp-arthorn P, Bhoopong P, Vuddhakul V. Isolation and characterization of Vibrio cholerae isolates from seafood in Hat Yai City, Songkhla, Thailand. Foodborne Pathog Dis. 2014;11:881–6.
Rivera I, Chowdhury M, Huq A, Jacobs D, Martins M, Colwell RR. Enterobacterial repetitive intergenic consensus sequences and the PCR to generate fingerprints of genomic DNAs from Vibrio cholerae O1, O139, and non-O1 strains. Appl Environ Microbiol. 1995;61:2898–904.
Teh CSJ, Thong KL, Osawa R, Chua KH. Comparative PCR-based fingerprinting of Vibrio cholerae isolated in Malaysia. J Gen Appl Microbiol. 2011;57:19–26.
Tokunaga A, Yamaguchi H, Morita M, Arakawa E, Izumiya H, Watanabe H, et al. Novel PCR-based genotyping method, using genomic variability between repetitive sequences of toxigenic Vibrio cholerae O1 El Tor and O139. Mol Cell Probes. 2010;24:99–103.
Danin-Poleg Y, Cohen LA, Gancz H, Broza YY, Goldshmidt H, Malul E, et al. Vibrio cholerae strain typing and phylogeny study based on simple sequence repeats. J Clin Microbiol. 2007;45:736–46.
Ghosh R, Nair GB, Tang L, Morris JG, Sharma NC, Ballal M, et al. Epidemiological study of Vibrio cholerae using variable number of tandem repeats. FEMS Microbiol Lett. 2008;288:196–201.
Okada K, Roobthaisong A, Nakagawa I, Hamada S, Chantaroj S. Genotypic and PFGE/MLVA analyses of Vibrio cholerae O1: geographical spread and temporal changes during the 2007–2010 cholera outbreaks in Thailand. PLoS One. 2012;7:e30863.
Olsen JS, Aarskaug T, Skogan G, Fykse EM, Ellingsen AB, Blatny JM. Evaluation of a highly discriminating multiplex multi-locus variable-number of tandem-repeats (MLVA) analysis for Vibrio cholerae. J Microbiol Methods. 2009;78:271–85.
Nandi B, Nandy RK, Mukhopadhyay S, Nair GB, Shimada T, Ghose AC. Rapid method for species-specific identification of Vibrio cholerae using primers targeted to the gene of outer membrane protein OmpW. J Clin Microbiol. 2000;38:4145–51.
Williams JG, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990;18:6531–5.
Slack AT, Dohnt MF, Symonds ML, Smythe LD. Development of a multiple-locus variable number of tandem repeat analysis (MLVA) for Leptospira interrogans and its application to Leptospira interrogans serovar Australis isolates from Far North Queensland, Australia. Ann Clin Microbiol Antimicrob. 2005;4:10.
Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol. 1988;26:2465–6.
Weir BS. Genetic data analysis. Methods for discrete population genetic data. Sunderland, MA: Sinauer Associates, Inc. Publishers; 1990.
Burucoa C, Lhomme V, Fauchere JL. Performance criteria of DNA fingerprinting methods for typing of Helicobacter pylori isolates: experimental results and meta-analysis. J Clin Microbiol. 1999;37:4071–80.
Madhusudana RB, Surendran P. Detection of ctx gene positive non-O1/non-O139 V. cholerae in shrimp aquaculture environments. J Food Sci Technol. 2013;50:496–504.
Rivera IN, Chun J, Huq A, Sack RB, Colwell RR. Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl Environ Microbiol. 2001;67:2421–9.
Rahman MH, Biswas K, Hossain MA, Sack RB, Mekalanos JJ, Faruque SM. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol. 2008;27:347–55.
Luo Y, Ye J, Jin D, Ding G, Zhang Z, Mei L, et al. Molecular analysis of non-O1/non-O139 Vibrio cholerae isolated from hospitalised patients in China. BMC Microbiol. 2013;13:52.
Marin MA, Thompson CC, Freitas FS, Fonseca EL, Aboderin AO, Zailani SB, et al. Cholera outbreaks in Nigeria are associated with multidrug resistant atypical El Tor and non-O1/non-O139 Vibrio cholerae. PLoS Negl Trop Dis. 2013;7:e2049.
Schirmeister F, Dieckmann R, Bechlars S, Bier N, Faruque S, Strauch E. Genetic and phenotypic analysis of Vibrio cholerae non-O1, non-O139 isolated from German and Austrian patients. Eur J Clin Microbiol Infect Dis. 2014;33:767–78.
Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci. 2002;99:1556–61.
Unterweger D, Kitaoka M, Miyata ST, Bachmann V, Brooks TM, Moloney J, et al. Constitutive type VI secretion system expression gives Vibrio cholerae intra- and interspecific competitive advantages. PLoS One. 2012;7:e48320.
Chen C-H, Shimada T, Elhadi N, Radu S, Nishibuchi M. Phenotypic and genotypic characteristics and epidemiological significance of ctx + strains of Vibrio cholerae isolated from seafood in Malaysia. Appl Environ Microbiol. 2004;70:1964–72.
Chhotray G, Pal B, Khuntia H, Chowdhury N, Chakraborty S, Yamasaki S, et al. Incidence and molecular analysis of Vibrio cholerae associated with cholera outbreak subsequent to the super cyclone in Orissa, India. Epidemiol Infect. 2002;128:131–8.
Shangkuan Y-H, Tsao C-M, Lin H-C. Comparison of Vibrio cholerae O1 isolates by polymerase chain reaction fingerprinting and ribotyping. J Med Microbiol. 1997;46:941–8.
Li W, Raoult D, Fournier PE. Bacterial strain typing in the genomic era. FEMS Microbiol Rev. 2009;33:892–916.
Ali A, Chen Y, Johnson JA, Redden E, Mayette Y, Rashid MH, et al. Recent clonal origin of cholera in Haiti. Emerg Infect Dis. 2011;17:699–701.
Bhowmick T, Das M, Sarkar B. Evaluation of VcA VNTR as a strain-typing and phylogeny study method of Vibrio cholerae strains. Epidemiol Infect. 2010;138:1637–49.
Berche P, Poyart C, Abachin E, Lelievre H, Vandepitte J, Dodin A, et al. The novel epidemic strain O139 is closely related to the pandemic strain O1 of Vibrio cholerae. J Infect Dis. 1994;170:701–4.
Cariri FAMO, Costa A, Melo C, Theophilo GND, Hofer E, de Melo Neto O, et al. Characterization of potentially virulent non-O1/non-O139 Vibrio cholerae strains isolated from human patients. Clin Microbiol Infect. 2010;16:62–7.
Li M, Shimada T, Morris JG, Sulakvelidze A, Sozhamannan S. Evidence for the emergence of non-O1 and non-O139 Vibrio cholerae strains with pathogenic potential by exchange of O-antigen biosynthesis regions. Infect Immun. 2002;70:2441–53.
Jiang S, Chu W, Fu W. Prevalence of cholera toxin genes (ctxA and zot) among non-O1/O139 Vibrio cholerae strains from Newport Bay, California. Appl Environ Microbiol. 2003;69:7541–4.
Fields P, Popovic T, Wachsmuth K, Olsvik Ø. Use of polymerase chain reaction for detection of toxigenic Vibrio cholerae O1 strains from the Latin American cholera epidemic. J Clin Microbiol. 1992;30:2118–21.
Singh D, Isac SR, Colwell R. Development of a hexaplex PCR assay for rapid detection of virulence and regulatory genes in Vibrio cholerae and Vibrio mimicus. J Clin Microbiol. 2002;40:4321–4.
Shi L, Miyoshi S-i, Hiura M, Tomochika K-i, Shimada T, Shinoda S. Detection of genes encoding cholera toxin (CT), zonula occludens toxin (ZOT), accessory cholera enterotoxin (ACE) and heat-stable enterotoxin (ST) in Vibrio mimicus clinical strains. Microbiol Immunol. 1998;42:823.
Acknowledgements
We extend our gratitude to Dr. Kamonnut Singkhamanan, Faculty of Medicine, Prince of Songkla University.
Funding
This work was supported by the government budget of Prince of Songkla University (grant number SCI580246b), the Faculty of Science Research Fund (contract no. 2-2556-02-020), and the Graduate School of Prince of Songkla University.
Availability of data and materials
The dataset and materials of the current study are available through the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
PM and VV developed the research plan. ST performed the research work and analyzed and interpreted the data with help of SP and PM. ST and SP prepared the manuscript. PM and VV edited the manuscripts. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. All isolates were kindly provided by Hat Yai Hospital, Songkhla, Thailand. Laboratory records and patient information were not obtained. Thus, this study does not require ethics approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tulatorn, S., Preeprem, S., Vuddhakul, V. et al. Comparison of virulence gene profiles and genomic fingerprints of Vibrio cholerae O1 and non-O1/non-O139 isolates from diarrheal patients in southern Thailand. Trop Med Health 46, 31 (2018). https://doi.org/10.1186/s41182-018-0113-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41182-018-0113-x