
Abstract
Background
Ethnomedicinally, the family Polygonaceae is famous for the management of cancer. Various species of this family have been reported with anticancer potentials. This study was designed to isolate anticancer compounds from ethnomedicinally important species Polygonum barbatum.
Methods
The column chromatography was used for the isolation of compounds from the solvent fraction of P. barbatum. The characterization of isolated compounds was performed by various spectroscopic techniques like UV, IR, mass spectrometry and 1D-2D NMR spectroscopy. Keeping in view the ethnomedicinal importance of the family, genus and species of P. barbatum, the isolated compounds (1–3) were screened for anticancer potentials against oral cancer (CAL-27) and lungs cancer (NCI H460) cell lines using MTT assay. Active compound was further investigated for apoptosis by using morphological changes and flow cytometry analysis. In vivo anti-angiogenic study of the isolated compounds was also carried using chorioallantoic membrane assay. Docking studies were carried out to explore the mechanism of anticancer activity.
Results
Three dihydrobenzofuran derivatives (1–3) have been isolated from the ethyl acetate fraction of P. barbatum. The structures of isolated compounds were elucidated as methyl (2S,3S)-2-(3,4-dimethoxyphenyl)-4-((E)-3-ethoxy-3-oxoprop-1-en-1-yl)-7-methoxy-2,3-dihydrobenzo-furan-3-carboxylate (1), (E)-3-((2S,3S)-2-(3,4-dimethoxyphenyl)-7-methoxy-3-(methoxy carbonyl)-2,3-dihydrobenzofuran-4-yl)acrylic acid (2) and (2S,3S)-4-((E)-2-carboxyvinyl)-2-(3,4-dimethoxyphenyl)-7-hydroxy-2,3-dihydrobenzofuran-3-carboxylic acid (3). The compound 1 was found to be more potent with IC50 of 48.52 ± 0.95 and 53.24 ± 1.49 against oral cancer cells as compared to standard drug (IC50 = 97.76 ± 3.44 μM). Both compound also inhibited lung cancer cells but at higher concentrations. Morphological and flow cytometry analysis further confirms that compound 1 induces apoptosis after 24 to 48 h treatment. In antiangiogenesis assay, compounds 1, 2 and 3 exhibited IC50 values of 8.2 ± 1.1, 13.4 ± 1.1 and 57.7 ± 0.3 μM respectively. The docking studies revealed that the compounds under study have the potential to target the DNA and thymidylate synthase (TS).
Conclusion
Based on its overwhelming potency against the tested cell lines and in angiogenesis assay, compound 1 can be further evaluated mechanistically and can be developed as anticancer drug candidate.
Similar content being viewed by others

Introduction
Polygonum barbatum is a perennial herb, mostly found in shady and marshy places near river banks, ponds and other aquatic areas in different countries of South-East Asia [25, 26]. The species belongs to the family Polygonaceae represented by 19 genera and 103 species in Pakistan out of total 55 genera and approximately 1200 species worldwide [21]. Traditionally, various species of Polygonaceae family are used for the management of cardiovascular diseases, hypertension and cancer [14, 29]. Based on the ethnomedicinal background, some species have been verified scientifically for the management of cancer [1, 2, 4, 17]. The leaf extract of P. barbatum has been used in Chinese traditional medicine for the treatment of ulcer, and root extract as astringent and as carminative agent [10, 20, 30].
Anti-inflammatory, antinociceptive and diuretic activities of P. barbatum has been previously reported [10, 25]. Similarly, P. barbatum also possess potential analgesic, brine shrimps, cholinergic, as well as spasmolytic potentials [10, 11, 24]. In indigenous system of medicine, it has been used as potential anticancer agent against oral cancer [18]. P. barbatum has also been reported to have antimicrobial activity, suggesting it to be a source of potential antibiotic agents [18].
Additionally, the therapeutic and biological potentials of some species of the genus Polygonum have also been previously reported [5,6,7,8, 19, 21, 22]. Phytochemical investigations of various species of this genus showed the presence of secondary metabolites, particularly anthraquinones, flavonoids, as well as phenylpropanoid [9, 16]. Furthermore, secondary metabolites such as acetophenone, viscozulenic acid and sitosterone have been previously isolated from P. barbatum [26]. Similarly, Farooq et al. reported sesquiterpenes from P. barbatum possessing potential anti-proliferative activity and inhibitory effect on cancer cell migration [13].
Cancer, a second leading cause of death, resulted in 8.8 million deaths in 2015 globally [32]. In Pakistan, the three deadliestof them are breast cancer, oral cancer and lungs cancer with death rate of 26.76, 8.71 and 5.53 per 100,000 population, respectively [33]. Despite of the current advances in therapeutic approaches, there is an immense need of new potential compounds that can be used as safe anticancer drugs. Keeping in view the ethnomedicinal importance of Polygonaceae and potential therapeutic efficiency of P. barbatum, we were able to isolate three dihydrobenzofuran derivatives from its ethyl acetate fraction. To study the anticancer effect, we targeted oral squamous cell carcinoma and large cell lung carcinoma. Furthermore, we also assessed the potential of these isolated dihydrobenzofuran derivatives on normal vascular development in chick embryos using a multifaceted and low-cost chicken chorioallantoic membrane (CAM) assay.
Methods
The Polygonum barbatum (whole plant) was collected from different areas of Khyber Pakhtunkhwa in October 2015 and specimen having voucher # 66130 has been deposited in herbarium at Department of Botany, postgraduate college Abbottabad, Pakistan.
The shade dried plant (5.4 kg) was ground into fine powder, extracted with methanol and filtered twice. The crude extract (245 g) was obtained from filtrate by using vacuum rotary evaporator and partitioned into n-hexane (85 g), ethyl acetate (48 g) and n-butanol (94 g) fraction. Further investigation and purification of compounds was done through column chromatography of ethyl acetate fraction selected based on TLC.
Column chromatography
The column chromatography of ethyl acetate fraction was carried out using n-hexane/ethyl acetate as solvent with increasing polarity of ethyl acetate up to 100%, later followed by methanol. Various sub-fractions (1–12) were obtained as a result of column chromatography and sub-fractions (4–12) were re-selected on the basis of TLC and subjected to re-column chromatography to purify compounds (1–3). The compound 1 (7 mg) was obtained at polarity n-hexane:ethyl acetate (65:35) from sub-fractions 4–10 while compound 2 and 3 were obtained from re-column chromatography of sub-fractions (11–12) at polarity of n-hexane:ethyl acetate (58:42) and (55:45) respectively.
Instrumentation
The NMR spectra were recorded by using Bruker AMX-400 spectrometer (1H-NMR at 400 MHz and 13C-NMR at 100 MHz). The values of chemical shift and coupling constant were recorded as ppm and Hertz (Hz) respectively. The Varian MAT-312 spectrometer was used to record HR-EI-MS and EI-MS spectra. The E. Merck 230–400 mesh silica gel was used to pack columns while pre-coated silica gel plates were employed for TLC analysis. In addition, Ceric sulphate solution in 10% sulphuric acid (H2SO4) has been used for detection of UV active compounds.
Methyl (2S,3S)-2-(3,4-dimethoxyphenyl)-4-((E)-3-ethoxy-3-oxoprop-1-en-1-yl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylate (1)
A yellow gum; UV λmax: 306 (2.4), 220 (3.4) nm; IR (KBr) ʋmax: 3036, 2810, 1734, 1440, 1204 cm−1; HR-EI-MS (m/z): Calcd. 442.1628, mol. formula C24H26O8; Obs. 442.1621; EI-MS (m/z): 442, 414, 396, 370, 338, 306, 264, 180, 140, 126, 96; [∝] 25 D + 60 (c 0.39, CHCl3); 1H NMR (400 MHz, CD3OD) δH (ppm): 6.90 (d, J = 9.1 Hz, H-5, 1H), 7.30 (d, J = 9.1 Hz, H-6, 1H), 7.80 (d, J = 16.5 Hz, H-7, 1H), 6.54 (d, J = 16.5 Hz, H-8, 1H), 4.30 (q, J = 8.1 Hz, H-10, 2H), 1.10 (t, J = 8.1 Hz, H-11, 3H), 6.80 (d, J = 1.8 Hz, H-2′, 1H), 6.61 (d, J = 8.2 Hz, H-5′, 1H), 6.63 (dd, J = 8.2, 1.8 Hz, H-6′, 1H), 5.92 (d, J = 5.2 Hz, H-7′, 1H), 4.68 (d, J = 5.2 Hz, H-8′, 1H), 3.60 (s, 4-OCH3, 3H), 3.64 (s, 3′-OCH3, 3H), 3.66 (s, 4′-OCH3, 3H), 3.55 (s, 9′-OCH3, 3H); 13C NMR (100 MHz, CD3OD) δC (ppm): 126.6 (C-1), 129.7 (C-2), 151.9 (C-3), 146.3 (C-4), 121.3 (C-5), 122.4 (C-6), 143.7 (C-7), 120.7 (C-8), 170.1 (C-9), 66.2 (C-10), 19.4 (C-11), 135.9 (C-1′), 116.3 (C-2′), 153.2 (C-3′), 154.5 (C-4′), 118.6 (C-5′), 124.8 (C-6′), 82.4 (C-7′), 58.1 (C-8′), 174.6 (C-9′), 57.8 (4-OCH3), 60.1 (3′-OCH3), 60.5 (4′-OCH3), 54.8 (9′-OCH3).
(E)-3-((2S,3S)-2-(3,4-dimethoxyphenyl)-7-methoxy-3-(methoxycarbonyl)-2,3-dihydrobenzofuran-4-yl) acrylic acid (2)
A yellowish-brown gummy solid; UV λmax: 308 (1.8), 218 (2.6), 202 (3.2) nm; IR (KBr) ʋmax: 3020, 2822, 1743, 1710, 1610, 1540, 1201 cm−1; HR-EI-MS (m/z): Calcd. 414.1315, mol. formula C22H22O8; Obs. 414.1304; EI-MS (m/z): 414, 396, 368, 336, 304, 182, 140, 126, 96; [∝] 25 D +46 (c 0.190, CHCl3);1H NMR (400 MHz, CD3OD) δH (ppm): 6.88 (d, J = 9.2 Hz, H-5, 1H), 7.28 (d, J = 9.2 Hz, H-6, 1H), 7.70 (d, J = 15.4 Hz, H-7, 1H), 6.50 (d, J = 15.4 Hz, H-8, 1H), 6.80 (d, J = 1.9 Hz, H-2′, 1H), 6.65 (d, J = 8.8 Hz, H-5′, 1H), 6.72 (dd, J = 8.8, 1.9 Hz, H-6′, 1H), 5.90 (d, J = 4.8 Hz, H-7′, 1H), 4.70 (d, J = 4.8 Hz, H-8′, 1H), 3.60 (s, 4-OCH3, 3H), 3.62 (s, 3′-OCH3, 3H), 3.65 (s, 4′-OCH3, 3H), 3.53 (s, 9′-OCH3, 3H); 13C NMR (100 MHz, CD3OD) δC (ppm): 124.5 (C-1), 128.2 (C-2), 150.6 (C-3), 147.8 (C-4), 120.2 (C-5), 125.1 (C-6), 142.4 (C-7), 119.6 (C-8), 172.1 (C-9), 134.7 (C-1′), 114.3 (C-2′), 152.7 (C-3′), 153.1 (C-4′), 117.6 (C-5′), 123.1 (C-6′), 84.1 (C-7′), 59.2 (C-8′), 174.1 (C-9′), 58.6 (4-OCH3), 61.3 (3′-OCH3), 61.6 (4′-OCH3), 56.4 (9′-OCH3).
(2S,3S)-4-((E)-2-carboxyvinyl)-2-(3,4-dimethoxyphenyl)-7-hydroxy-2,3-dihydrobenzofuran-3-carboxylic acid (3)
A brown gummy solid; UV λmax: 320 (2.1), 270 (3.1), 210 (1.9) nm; IR (KBr) ʋmax: 3370, 2950, 2820, 1710, 1640, 1480, 1240 cm−1; HR-EI-MS (m/z): Calcd. 386.1002, mol. formula C20H18O8; Obs. 386.997; EI-MS (m/z): 386, 368, 350, 342, 338, 306, 184, 140, 126, 96; [∝] 25 D +80 (c 0.172, CHCl3);1H NMR (400 MHz, CD3OD) δH (ppm): 6.89 (d, J = 8.8 Hz, H-5, 1H), 7.27 (d, J = 8.8 Hz, H-6, 1H), 7.78 (d, J = 17.1 Hz, H-7, 1H), 6.48 (d, J = 17.1 Hz, H-8, 1H), 6.74 (d, J = 2.1 Hz, H-2′, 1H), 6.64 (d, J = 7.9 Hz, H-5′, 1H), 6.68 (dd, J = 7.9, 2.1 Hz, H-6′, 1H), 5.91 (d, J = 5.5 Hz, H-7′, 1H), 4.74 (d, J = 5.5 Hz, H-8′, 1H), 3.58 (s, 3′-OCH3, 3H), 3.60 (s, 4′-OCH3, 3H); 13C NMR (100 MHz, CD3OD) δC (ppm): 124.2 (C-1), 128.1 (C-2), 140.4 (C-3), 146.6 (C-4), 119.9 (C-5), 123.1 (C-6), 141.4 (C-7), 120.8 (C-8), 173.1 (C-9), 135.7 (C-1′), 118.6 (C-2′), 153.1 (C-3′), 154.6 (C-4′), 116.7 (C-5′), 125.3 (C-6′), 84.4 (C-7′), 57.1 (C-8′), 175.6 (C-9′), 59.1 (3′-OCH3), 59.8 (4′-OCH3).
Anticancer activity
The isolated compounds (1–3) were investigated for anticancer activity against human oral squamous cell carcinoma cell line (CAL 27) and human large cell lung carcinoma cell line (NCI H460) as described recently [13]. The cell lines were purchased from ATCC and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 100 µg/mL streptomycin and 100 units/mL penicillin at 37 °C in a humidified 5% CO2 incubator. The CAL 27 and NCI H460 cells were seeded in 96 well plates with the density of 1.5 × 104 and 1 × 104 cells per well, respectively and incubated for 24 h. The cells were then treated with different concentrations of compound 1, 2 and 3 for 48 h. 1% DMSO was used as vehicle control and empty wells were used as negative control. The standard anticancer drugs namely 5-Fluorouracil and Cisplatin were used against CAL 27 and NCI H460, respectively. Quantification of metabolically active cells were determined using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) dye which reduces to insoluble Formazan crystals by the action of mitochondrial succinate dehydrogenase. The soluble MTT dye was aspirated off and insoluble crystals were dissolved using DMSO. The optical density of each well was determined by SpectraMax microplate reader at 570 nm. IC50 values were calculated using software EZ-Fit Enzyme Kinetics by Perrella Scientific. For morphological changes cells treated at IC50 and IC70 were observed under phase contrast microscopy (Nikon, Japan) at 24 and 48 h respectively.
YO-PRO-1 apoptotic assay
Oral cancer (CAL-27) cells (5 × 105) were seeded in a 6-well plate and incubated for 24 h in presence of 5% CO2. After incubation compound 1 was added at IC50 and IC70 concentration and further incubated for 48 h. Following incubation cells were harvested by using trypsin (0.05%) and pallet out after washing three times at 400×g for 5 min to remove media and trypsin. Cell pallets were suspended in 1 mL PBS and stained with Vybrant Apoptosis Assay kit # 4 (Invitrogen, USA) containing YO-PRO-1 and Propidium Iodide (PI) dye according to manufacturer protocol. Briefly, cells were incubated with 1 μL YO-PRO-1 stock solution (Component A) and 1 μL PI stock solution (Component B) for 30 min on ice. Later, cells were analyzed on FACSCalibur (Becton–Dickinson, USA). YO-PRO-1 and PI were excited at 488 nm, and fluorescence was measured at 530 and 620 nm, respectively. A total of 10,000 events were acquired from each sample. The percentages of live, apoptotic and dead cells were determined using CellquestPro software.
Chorioallantoic membrane (CAM) assay
Chorioallantoic membrane (CAM) assay was performed according to our previously reported procedure [15]. The experiments were carried out as per the approval of the ethical committee, Department of Pharmacy, University of Malakand, Pakistan according to the animals Bye-Laws 2008 (Scientific Procedure Issue-1). The fertilized domestic chicken eggs were purchased from poultry form Chakdara, Pakistan. The fertilized eggs were incubated for 5–6 days at 37 °C with slowly shaking for at least two to three times a day. After the incubation period, the seven-day old eggs were examined under the flash light for detection of the embryo head. After that a small hole was bored at the narrow end of the eggs and 0.5–1 mL of albumin was sucked with the help of eighteen-gauge hypodermic needle so that yolk sacs fell down from the shell membrane. The shell around the embryo air sac was removed through forceps and the shell membrane at the base of air sac was peel away. On 8th day, a thermanox cover slip was carefully placed on the surface of CAM loaded with different samples for each concentration (10 µL) and were placed in incubator. After 24 to 48 h, the numbers of vessels on the surface of CAM were observed. The numbers of blood vessels in each egg was counted by figuring out the actual number of blood vessels in 1 cm2 area of the CAM adjacent to the sample application site. Vessels radially converging in the direction of the center were counted under a microscope. At least twenty eggs were used for each sample dose. The % of increase and inhibition were calculated using formula;
Median inhibitory concentrations (IC50) for anti-angiogenic assay were calculated via regression analysis of dose response curve by adding all the concentration and percent activity.
Docking studies
Docking studies were carried out using Molecular Operating Environment [27] software. Crystal structures of DNA and Human Thymidylate Synthase (TS) was retrieved from Protein Data Bank with code numbers 3LPV and 1JU6 respectively. Validation of docking procedure was carried out by using re-dock method. Ligand and DNA/enzyme preparation, active site determination and docking procedure was carried out by using our previously reported procedures [3, 15]. The lowest energy minimized pose was used for further analysis. Ligand-interaction module of MOE was used to calculate the 2D ligand–enzyme interactions. The view of the docking results and analysis of their surface with graphical representations were done using MOE and discovery studio visualizer [12].
Results and discussion
Isolation and Characterization of benzofuran derivatives
The ethyl acetate fraction of P. barbatum upon column chromatography gave three benzofuran derivatives (1–3). Using various spectroscopic analyses and published literature, the structures of isolated compounds were determined.
Compound 1
Compound 1, a white gummy solid having molecular formula C24H26O8 as suggested by molecular ion peak of 442.1621 in HR-EI-MS was obtained by repeated chromatography. Other m/z peaks were obtained at 442, 414, 396, 370, 338, 306, 264, 180, 140, 126, and 96. The UV spectrum showed absorption bands at λmax 306 (2.4) and 220 (3.4) nm while IR spectrum showed absorption at 3036, 2810, 1734, 1440, 1204 cm−1 suggesting the presence of alkene, ester, and alkyl aryl ether moiety.
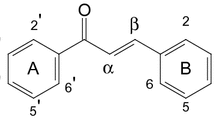
The 1H-NMR spectrum showed two ortho coupled proton with chemical shift value of δH 6.90 (d, J = 9.1 Hz, H-5, 1H) and 7.30 (d, J = 9.1 Hz, H-6, 1H). Similarly, two doublets for other aromatic protons were also revealed in 1H-NMR spectrum at δH 6.80 (d, J = 1.8 Hz, H-2′, 1H), δH 6.61 (d, J = 8.2 Hz, H-5′, 1H,) while doublet of doublet was observed for proton at δH 6.63 (dd, J = 8.2, 1.8 Hz, H-6′, 1H). The coupling observed between two aliphatic protons at δH 5.92 (d, J = 5.2 Hz, H-7′, 1H) and δH 4.68 (d, J = 5.2 Hz, H-8′, 1H) suggested the presence of dihydrobenzofuran ring [34]. The protons at position H-7′ and H-8′ were suggested at trans position to each other as indicated by value of coupling constant, i.e. 5.2 Hz. The 1H-NMR also revealed the presence of two olefinic protons at δH 7.80 (d, J = 16.5 Hz, H-7, 1H) and δH 6.54 (d, J = 16.5 Hz, H-8, 1H) evident from trans-olefinic protons with extended conjugation with carbonyl group. A quartet was observed for methylene protons at position H-10 (δH 4.30, q, J = 8.1 Hz) while the terminal methyl group at position H-11 (δH 1.10, t, J = 8.1 Hz) was directly connected with methylene group. In addition, four singlets for methoxy group (–OCH3) were observed in 1H-NMR spectrum at chemical shift values of δH 3.60 (3H, s, H-4), δH 3.64 (3H, s, H-3′), δH 3.66 (3H, s, H-4′), and δH 3.55 (3H, s, H-9′) (Table S1 in Additional file 1).
The 13C-NMR along with DEPT spectrum showed signals for 24 carbon atoms comprising of one methyl, one methylene, nine methine, seven quaternary and four methoxy groups. In addition, two carbonyl carbon atoms were observed at δC 170.1 (C-9) and δC 174.6 (C-9′) while the olefinic carbons at position H-7 and H-8 resonated at δC 143.7 and 120.7 respectively (as shown in Fig. 1). The only downfield methylene appeared at δC 66.2, while the methyl group resonated at δC 19.4 ppm. The two methine carbons of dihydrobenzofuran centered at δC 82.4 for C-7′ and δC 58.1 for C-8′. The chemical shift values of aromatic carbon and methoxy groups have been shown in Additional file 1: Table S2.

Structure of compounds 1–3
The HMBC spectrum revealed strong interaction of protons at H-5 and H-6 with C-4 and C-1 while protons of methoxy group substituent at this position (C-4) also showed strong correlation with C-4. The olefinic proton at position H-7 showed strong HMBC correlation with C-1 and C-8 while other olefinic proton at H-8 showed HMBC correlation with C-1, C-7 and C-9 (carbonyl carbon of ethyl ester moiety). The placement of methyl ester as substituent on dihydrobenzofuran ring was confirmed by HMBC correlation of methoxy protons were connected with carbonyl carbon at C-9′ as evident from Fig. 2. This placement was further supported by H-7′ and H-8′ protons which showed strong correlations with C-9′ carbon. The position of two methoxy group at position 3′ and 4′ was also confirmed by their HMBC correlation as depicted in Fig. 2. The aromatic protons at position 2′ and 6′ showed strong correlation with C-1′ and C-7′ confirming the placement of this aromatic ring at dihydrobenzofuran ring.

Important HMBC correlation of compound 1
The relative configuration of aliphatic protons at position H-7′ and H-8′ was suggested trans to each other by Nuclear Overhauser Effect (NOE) as well as the coupling constant value (i.e., 5.2 Hz) suggested them to be at Trans position to each other. All the spectral data of 1H-NMR, 13C-NMR, HR-EI-MS, HMBC, and COSY along with comparison of previous literature the structure of compound 1 was confirmed as methyl (2S,3S)-2-(3,4-dimethoxyphenyl)-4-((E)-3-ethoxy-3-oxoprop-1-en-1-yl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylate.
Compound 2
Compound 2 was isolated as yellowish gummy solid and was assigned molecular formula C22H22O8 as suggested by molecular ion peak of 414.1304 in HR-EI-MS while other peaks with m/z values were obtained at 414, 396, 368, 336, 304, 182, 140, 126 and 96. The UV absorption bands were observed at 308 (1.8), 218 (2.6) and 202 (3.2) nm suggesting the presence of alkene, carboxylic acid, ester and ether moieties which were further supported by IR absorption bands at 3020, 2822, 1743, 1710, 1610, 1540, 1201 cm−1.
The 1H-NMR spectrum showed doublet for proton at δH 6.65 (d, J = 8.8 Hz, H-5′, 1H) having ortho coupling with δH 6.72 (dd, J = 8.8, 1.9 Hz, H-6′, 1H) which was further coupled with δH 6.80 (d, J = 1.9 Hz, H-2′, 1H). Similarly, two ortho coupled aromatic protons of dihydrobenzofuran with chemical shift value of δH 6.88 (d, J = 9.2 Hz, H-5, 1H) and 7.28 (d, J = 9.2 Hz, H-6, 1H) were also observed in 1H-NMR quite similar to compound 1. The two trans olefinic protons centered at δH 7.70 (d, J = 15.4 Hz, H-7, 1H) and 6.50 (d, J = 15.4 Hz, H-8, 1H) were also revealed by 1H-NMR spectrum. The two aliphatic protons at δH 5.90 (d, J = 4.8 Hz, H-7′, 1H) and 4.70 (d, J = 4.8 Hz, H-8′, 1H) were trans oriented to each other as suggested by their coupling constant value of 4.8 Hz [34]. The trans orientation of H-7′ and H-8′ was further supported by NOE experiment. When H-7′ was irradiated, H-8′ did not show any increase in intensity of H-8′ confirmed the trans orientation. In addition, singlets for methoxy groups were appeared at δH 3.60 (3H, s, 4-OMe), 3.62 (3H, s, 3′-OMe) and 3.65 (3H, s, 4′-OMe), while the methoxylgroup of ester moiety appeared at δH 3.53 as singlet in 1H-NMR spectrum.
The 13C-NMR spectra of compound 2 were quite identical to compound 1 that showed the presence of 22 carbon atoms including one methoxyl group of ester moiety, nine methine, seven quaternary and three methoxy groupson aromatic rings along with two carbonyl carbon atoms. The chemical shift value of olefinic carbon atoms were δC 142.4 (C-7) and 119.6 (C-8) while two carbonyl carbon resonated at δC 172.1 (C-9) and 174.1 (C-9′). The carbon atoms of five membered ring of dihydrobenzofuran resonated at δC 84.1 (C-7′) and 59.2 (C-8′) while chemical shift values of aromatic carbon atoms has been shown in Additional file 1: Table S2.
The placements of various substituents at different positions were assign with the help of HMBC spectrum and was quite identical to compound 1. The methyl group at position 10′ showed HMBC correlation with C-9′ while aliphatic proton at C-8′ showed correlation with C-2, C-7′ and C-9′. The aromatic protons at position 2′ and 6′ showed strong correlation with 1′ and 7′ confirming the attachment of dihydrobenzofuran ring. In addition both olefinic protons (H-7 and H-8) showed strong HMBC correlation with C-9, and C-1. Finally, the structure of compound 2 was deduced as (E)-3-((2S,3S)-2-(3,4-dimethoxyphenyl)-7-methoxy-3-(methoxycarbonyl)-2,3-dihydrobenzofuran-4-yl)acrylic acid on the basis of all spectral data and literature comparison [34].
Compound 3
Compound 3 was isolated as yellow gummy solid, that showed molecular ion peak of 386.997 in HR-EI-MS suggesting a molecular formula as C20H18O8. Other EI-MS peaks obtained were at m/z 386, 368, 342, 338, 306, 184, 140, 126 and 96. The absorption bands of UV spectrum were obtained at λmax 320 (2.1), 270 (3.1) and 210 (1.9) nm. The IR spectrum showed absorption band at 3370, 2950, 2820, 1710, 1640, 1480 and 1240 cm−1 suggesting presence of different functional groups like hydroxyl group, alkene, carboxylic acid and ether moiety.
The 1H-NMR spectrum showed signals for 1, 2, 3, 4-tetra substituted benzene ring fused with furan ring at δH 6.89 (d, J = 8.8 Hz, H-5), 7.27 (d, J = 8.8 Hz, H-6) and two trans protons appeared at δH 5.91 (d, J = 5.5 Hz, H-7′), δH 4.74 (d, J = 5.5 Hz, H-8′) respectively. Similarly, trans-disubstituted double bond protons appeared at δH 7.78 (d, J = 17.1 Hz, H-7) and δH 6.48 (d, J = 17.1 Hz, H-8) with extended conjugation with carbonyl carbon of acid moiety.
Similarly,13C-NMR data was also in close agreement with those of compound (1–2). The 13C-NMR spectra suggested the presence of two carboxylic acid groups in compound 3, which appeared at δC 173.1 (C-9) and δC 175.6 (C-9′). It was also evident that the fused benzene ring with furan ring was substituted with hydroxyl group at position 4 with chemical shift value of δC 146.6.
The 1H-NMR and 13C-NMR spectra of compound 3 were quite similar to compounds 1 and 2. The differences observed were presence of carboxylic acid instead of ester substituent at position-8 while at position-4 a hydroxyl (–OH) group was observed as substituent rather than methoxy group. The difference was retrieved from UV, IR and NMR data which was further supported by mass spectrum. The chemical shift values of both 1H-NMR and 13C-NMR spectra have been given in Additional file 1: Tables S1 and S2. Similarly, HMBC correlation was also found similar to compound 1 and 2 as shown in Fig. 2. All spectra data and comparison of literature suggested the structure of compound 3 as (2S,3S)-4-((E)-2-carboxyvinyl)-2-(3,4-dimethoxyphenyl)-7-hydroxy-2,3-dihydrobenzofuran-3-carboxylic acid.
In vitro anticancer activity
As widely spread natural compounds, benzofuran derivatives have been extensively studied for their biological activities [23]. Previously, a series of synthetic dihydrobenzofuran derivatives showed in vitro anticancer potential [23]. Most of these compounds have been reported to have cytotoxic potential against many human cancer cell lines including leukemia (HL-60-TB and K-562), breast cancer (MDA-MB-435. MDA-N, BT-549 and MCF-7), human cervical cancer (HeLa) and human lung carcinoma (A549) cell lines [23, 28]. Based on the previous reports, we screened the anticancer potential of the three isolated dihydrobenzofuran derivative from P. barbatum against oral (CAL-27) and lung cancer (NCI-H460) cells. Lung and oral cancer was selected by considering their prevalence in Pakistan due to excessive smoking, tobacco and beetle nut chewing. Oral and lung carcinoma ranked 2nd and 3rd respectively out of top 10 cancer related malignancies in Pakistan [31]. Among isolated compounds 1 and 3 showed better anticancer activity against oral cancer cell line (CAL 27) with IC50 48.52 ± 0.95 and 86.95 ± 4.39 respectively which was lower than the standard drug (5-Fluorouracil; Table 1). Whereas, these two compounds also showed anticancer activities against lungs cancer cells (NCI-H460) at higher doses as compared to control drug (Cisplatin; Table 1). Compound 1 was selected for further apoptosis analysis by observing morphological changes and FACS analysis considering its IC50 which was almost half the IC50 of standard drugs. After treatment with compound 1 at IC50 concentration cell morphology started to change after 24 h and with the passage of time cell detach from the surface with loss in cell shape suggesting apoptosis as compared to control. On the other hand, at higher concentration, i.e. IC70 cells were undergone apoptosis after 24 h (Fig. 3). Flow cytometry analysis (FACS) also correspond with the morphological data that after 48 h of treatment significant apoptosis (either early or late) and necrosis was observed (Fig. 4).

Morphological changes in CAL-27 cells after 24 and 48 h of treatment with compound 1 by using phase contrast microscope at ×10 magnification

Compound 1 induced apoptosis in CAL-27 cell line in a dose-dependent manner. a–c Flow cytometric analysis of the apoptosis induced by the compound 1 at IC50 and IC70 concentrations. An increased number of apoptotic cells were observed after treatment as compared to control. d Graphical representation of the cells in different phases of apoptosis after 48 h of treatment. Statistical significance were expressed as **p < 0.01 and ***p < 0.001 as compared to the untreated control
Antiangiogenic studies
Chicken chorioallantoic membrane (CAM) is a low cost in vivo tumor biology model and is used widely for the tumor angiogenesis studies. Tumor growth depends on blood supply which in turn depends on the number of vessels. Hence, the inhibition of neovascularization is one of the targets. We recently reported dihydropyrimidine-based compounds that have potential to inhibit of new blood vessels formation [15]. Moreover, we have also employed CAM assay for the antitumor and anti-angiogenic potentials of crude extracts of some medicinal plants. In current research, we also studied the potential of our isolated dihydrobenzofuran derivatives on normal vascular development in chick embryos using CAM model. Normal saline was used as negative control. Dexamethasone and methotrexate was used as positive control [15].
The antiangiogenic activities of the isolated compounds 1, 2 and 3 were based on the number of blood vessels formed in the chorioallantoic membrane (CAM) of chick embryos. Results of anti-angiogenic assay of isolated compounds are shown in Table 2. Additional file 1: Table S3 showed the average number of blood vessels of each concentration in the CAM of chick embryo using different five concentration of each compound. Chick embryos treated with distilled water yielded the highest mean of 30.4 blood vessels formed. This indicates that water, being the negative control, is the least antiangiogenic among the five treatments. The compound 3 (IC50 = 57.7 ± 0.3 μM, Table 2), yielded moderate number of blood vessels with average mean of about 10.8. The compound 2 (IC50 = 13.4 ± 1.1 μM) yielded an average of 8.6 blood vessels formed. While, the compound 1 (IC50 = 8.2 ± 1.1 μM) yielded a mean of only 5.0 blood vessels which were most active against antiangiogenic activity as given in Fig. 5. The IC50 value was calculated via regression analysis of dose response curve by adding all the concentrations and percent activities (Figure S1 in Additional file 1).

Anti-angiogenic activity of isolated dihydrobenzofuran derivatives along with positive and negative control. Positive control CAM was applied with dexamethasone and methotrexate
Docking studies
In cancer research, cancer cell lines have emerged as an alternative tool for the study of biological mechanisms. However, to hypothesize a biomolecular target that help us to explore the mechanism is a difficult task. The standard drugs used in current research are cisplatin and 5-fluorouracil. Cisplatin is a chemotherapeutic agent that binds with DNA to form a bifunctional 1,2-intrastand cross link. While, 5-fluorouracil is a thymidylate synthase (TS) inhibitor that interrupt its action resulting in inhibition of TS and DNA replication. We extended our investigations and use docking studies to explore the mechanism of anticancer activity.
The crystal structure of DNA duplex receptor was retrieved from Protein Data Bank (PDB code 3LPV) (Fig. 6a). All these compounds targeted major groove in the cisplatin binding site (Fig. 6b). Carbonyl oxygen of ester of compound 1 forms hydrogen bond interactions (HBI) with cytosine and adenine (Fig. 7a). Methoxy group of compound 2 forms π–H interactions with thymine nucleoside (Fig. 7b). While hydroxyl group and carbonyl group of compound 3 forms HBI with guanine A6 and guanine A7 respectively (Fig. 7c). The computed binding affinity of compounds 1–3 are − 6.4355, − 5.9216, − 5.9376 kcal/mol respectively.

a Representation of the crystal structure of DNA duplex receptor was retrieved from Protein Data Bank (PDB code 3LPV) containg cisplatin 1,2-d(GpG) intra-strand cross-link. b Overlaid diagram of all the compounds targeting major groove in the cisplatin binding site

a Two-dimensional interaction plot of the synthesized compounds with DNA a compound 1; b compound 2 and; c compound 3
For the study of another target, thymidylate synthase (TS), its crystal structure retrieved from PDB (code 1JU6) with LYA as native ligand. The three-dimensional (3D) overlaid diagram of native ligand LYA and compounds 1–3 is shown in Fig. 8a. While, the 3D interaction diagram of compounds 1–3 is shown in Fig. 8b–d. Carbonyl oxygen of ester of compound 1 form HBI with His256. While oxygen atom of –OC2H5 forms HBI with Tyr258. Met311 for π–sulfur interactions with dimethoxyphenyl ring. Dimethoxyphenyl ring also forms π–π stacking interactions with Phe225. Another HBI was found between –OCH3 and Asn226. The ligand–enzyme complex also stabilize through π–alkyl interactions (Fig. 8b). For compound 2, three HBI were found. The two OCH3 groups forms HBIs with His256 and Tyr258. While, carbonyl oxygen forms HBI with Asn226. Compound 3 also forms three HBIs and two π-alkyl interactions. Carboxylic acid group (HO–C=O) establishes HBIs with Ser216, His256 and Tyr258. Cys195, Ile108 and Leu221 forms π–alkyl interactions. The binding affinity of compounds 1–3 is − 7.9363, − 7.1656 and − 7.7879 kcal/mol respectively. Here, we can conclude that compound 1 has with two ester groups oriented into the binding site to show maximum interactions and lowest energy (most stable ligand enzyme complex).

a The overlaid ribbon diagram of Native ligand (green), compound 1 (pink), compound 2 (yellow) and compound 3 purple) into the binding site of hTS (PDB code 1JU6); b–d Close-up depiction of the lowest-energy three-dimensional (3-D) docking pose of compounds 1–3 respectively
Conclusions
The phytochemical investigation of ethyl acetate fraction of P. barbatum resulted in the isolation of three new derivatives (1–3) of dihydrobenzofuran. Various spectroscopic techniques like mass spectrometry, UV, IR, 1D and 2D-NMR spectroscopy (1H-NMR, 13C-NMR, HMBC, NOESY) were employed for characterization of compounds (1–3). The structures of compound (1–3) were deduced to be methyl (2S,3S)-2-(3,4-dimethoxyphenyl)-4-((E)-3-ethoxy-3-oxoprop-1-en-1-yl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylate (1), (E)-3-((2S,3S)-2-(3,4-dimethoxyphenyl)-7-methoxy-3-(methoxycarbonyl)-2,3-dihydrobenzofuran-4-yl)acrylic acid (2) and (2S,3S)-4-((E)-2-carboxyvinyl)-2-(3,4-dimethoxyphenyl)-7-hydroxy-2,3-dihydrobenzofuran-3-carboxylicacid (3). Among the isolated compounds 1 and 3 were found to be more active against oral cancer cell lines than lung cancer cells as compared to standard drugs. Further investigations via in vivo antiangiogenic activity using Chicken chorioallantoic membrane (CAM) model showed that isolated compound 1 with IC50 of 8.2 ± 1.1 μM showed excellent antiangiogenic activity. Herein, we can conclude that in future, these compounds can be further studied to understand the mechanism of action involved in their anticancer potential.
Abbreviations
- UV:
-
ultravoilet visible spectroscopy
- IR:
-
infrared spectroscopy
- NMR:
-
nuclear magnetic resonance spectroscopy
- MTT:
-
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
- TLC:
-
thin layer chromatography
- MHz:
-
mega hertz
- HR-EI-MS:
-
high resolution electron impact mass spectroscopy
- s:
-
singlet
- d:
-
doublet
- t:
-
triplet
- q:
-
quartet
- dd:
-
doublet of doublet
- J :
-
coupling constant
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- CAM:
-
chorioallantoic membrane
- HMBC:
-
heteronuclear multiple-bond correlation
References
Ahmad S, Ullah F, Ayaz M, Zeb A, Ullah F, Sadiq A. Antitumor and anti-angiogenic potentials of isolated crude saponins and various fractions of Rumexhastatus D.Don. Biol Res. 2016;49(1):18.
Ahmad S, Ullah F, Zeb A, Ayaz M, Ullah F, Sadiq A. Evaluation of Rumexhastatus D. Don for cytotoxic potential against HeLa and NIH/3T3 cell lines: chemical characterization of chloroform fraction and identification of bioactive compounds. BMC Complement Altern Med. 2016;16(1):308.
Ali M, Ali S, Khan M, Rashid U, Ahmad M, Khan A, Al-Harrasi A, Ullah F, Latif A. Synthesis, biological activities, and molecular docking studies of 2-mercaptobenzimidazole based derivatives. Bioorg Chem. 2018;80:472–9.
Ayaz M, Muhammad J, Farhat U, Abdul S, Fazal S, Mir AK, Waqar A, Gowhar A, Muhammad I, Sajjad A. Molecularly characterized solvent extracts and saponins from Polygonum hydropiper L. show high anti-angiogenic, anti-tumor, brine shrimp, and fibroblast NIH/3T3 cell line cytotoxicity. Front Pharmacol. 2016;7:74.
Ayaz M, Junaid M, Ullah F, Sadiq A, AzamKhan M, Ahmad W, RazaShah M, Imran M, Ahmad S. Comparative chemical profiling, cholinesterase inhibitions and anti-radicals properties of essential oils from Polygonum hydropiper L.: a preliminary anti-Alzheimer’s study. Lipids Health Dis. 2015;14(1):141.
Ayaz M, Junaid M, Ullah F, Sadiq A, Ovais M, Ahmad W, Zeb A. Chemical profiling, antimicrobial and insecticidal evaluations of Polygonum hydropiper L. BMC Complement Altern Med. 2016;16(1):502.
Ayaz M, Junaid M, Ullah F, Subhan F, Sadiq A, Ali G, Ovais M, et al. Anti-Alzheimer’s studies on β-sitosterol isolated from Polygonum hydropiper L. Front Pharmacol. 2017;8:697.
Ayaz M, Junaid M, Ullah F, Sadiq A, Shahid M, Ahmad W, Ullah I, Ahmad A, Syed N-i. GC–MS analysis and gastroprotective evaluations of crude extracts, isolated saponins, and essential oil from Polygonum hydropiper L. Front Chem. 2017;5:58.
Brown LL, Larson SR, Sneden AT. Vanicosides C–F, new phenylpropanoid glycosides from Polygonum pensylvanicum. J Nat Prod. 1998;61(6):762–6.
Chaudhry BA, Syad MY, Janbaz KH, Dasti AA, Loothar BA. Biological activities of Polygonum barbatum. J Res Sci. 2003;14(2):169–71.
Chopra R, Nayar S, Chopra I. “Glossary of Indian medicinal plants (including the supplement). New Delhi: Council of Scientific and Industrial Research; 1986. p. 2–79.
Dassault Systèmes BIOVIA. Discovery studio modelling environment, release 4.5. San Diego: Dassault Systems; 2015.
Farooq U, Naz S, Zehra B, Khan A, Ali SA, Ahmed A, Sarwar R, Bukhari SM, Rauf A, Ahmad I. Isolation and characterization of three new anti-proliferative sesquiterpenes from Polygonum barbatum and their mechanism via apoptotic pathway. BMC cancer. 2017;17(1):694.
Hartwell JL. Plants used against cancer. survey Lloydia. 1967;30:379–436.
Iftikhar F, Yaqoob F, Tabassum N, Jan MS, Sadiq A, Tahir S, Batool T, Niaz B, Ansari FL, Choudhary MI, Rashid U. Design, synthesis, in-vitro thymidine phosphorylase inhibition, in-vivo antiangiogenic and in-silico studies of C-6 substituted dihydropyrimidines. Bioorg Chem. 2018;80:99–111.
Isobe T, Kanazawa K, Fujimura M, Noda Y. Flavonoids of Polygonum sieboldi and P. filiforme. Bull Chem Soc Jpn. 1981;54(10):3239.
Kamal Z, Ullah M, Ahmad S, Ullah F, Sadiq A, Ayaz M, Zeb A, Imran M. Exvivo antibacterial, phytotoxic and cytotoxic, potential in the crude natural phytoconstituents of Rumexhastatus D. Don. Pak J Bot. 2015;47:293–9.
Khan S, Farooq U, Khan A, Naz S, Sarwar R, Mirza N. Antimicrobial potential and physio-chemical analysis of Polygonum barbatum L. J Chem Soc Pak. 2014;36(4):687.
Kim H-M. Antiallergy drugs from oriental medicines. Oriental Pharm Exp Med. 2000;1(1):1–7.
Kirtikar KR, Basu BD. Indian medicinal plants. India: International Book Distributor; 1993.
Kott V, Barbini L, Cruanes M, de Munoz J, Vivot E, Cruañes J, Martino V, Ferraro G, Cavallaro L, Campos R. Antiviral activity in Argentine medicinal plants. J Ethnopharmacol. 1998;64(1):79–84.
Lian L. Studies on constituents of antihypertensive plant Polygonum perfoliatum. Kuo Li Chung-Kuo I Yao Yen Chiu So Yen Chiu Pao Kao. 1983;7:103–29.
Li W, Yang ZH, Hu AX, Yan XW, Ding N, Ye J. Design, synthesis, and antitumor activity of (E, Z)-1-(dihydrobenzofuran-5-yl)-3-phenyl-2-(1, 2, 4-triazol-1-yl)-2-propen-1-ones. Chem Biol Drug Des. 2015;86(6):1339–50.
Manandhar NP. Plants and people of Nepal. Timber: Timber Press; 2002.
Mazid MA, Datta BK, Nahar L, Bashar S, Bachar SC, Sarker SD. Antinociceptive, anti-inflammatory and diuretic properties of Polygonum barbatum (L.) Hara var. barbata. Revista Brasileira de Farmacognosia. 2009;19(3):749–54.
Mazid MA, Datta BK, Nahar L, Bashar SK, Bachar SC, Sarker SD. Phytochemical studies on Polygonum barbatum (L) Hara var barbata (Polygonaceae). Records of Natural products. 2011;5(2):143.
Molecular Operating Environment (MOE), 2016.08. Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. Molecular Operating Environment: Montreal; 2018.
Pieters L, Van Dyck S, Gao M, Bai R, Hamel E, Vlietinck A, Lemiere G. Synthesis and biological evaluation of dihydrobenzofuranlignans and related compounds as potential antitumor agents that inhibit tubulin polymerization. J Med Chem. 1999;42(26):5475–81.
Qureshi RA, Ghufran MA, Sultana KN, Ashraf M, Khan AG. Ethnomedicinal studies of medicinal plants of Gilgit District and surrounding areas. Ethnobot Res Appl. 2007;5:115–22.
Ramani A. In-vitro antioxidant activity of Polygonum barbatum leaf extract. Asian J Pharm Clin Res. 2011;4(1):113–5.
World Health Organization Rankings. Health Profile: Pakistan. World Life Expectancy. 2016. UK: World Health Organization. http://www.worldlifeexpectancycom/country-health-profile/pakistan.
World Health Organization. Cancer. World Health Organization; 2018. http://www.who.int/en/news-room/fact-sheets/detail/cancer.
World Health Rankings. Health profile: Pakistan. World Life Expectancy. 2014. http://www.worldlifeexpectancy.com/country-health-profile/pakistan.
Yu Y, Wang Y-R, Dong Z-H, Li W, Li S-M, Huang X-F. New phenolic acids from Salvia yunnanensis CH Wright. Nat Prod Res. 2017;31(21):2505–12.
Authors’ contributions
SN, AS, YR and AA carried out experimental work, data collection and literature search under the supervision of UF and drafted the manuscript. UR performed docking studies. URand AS performed the angiogenesis assay and refined the manuscript for publication. All authors read and approved the final manuscript.
Acknowledgements
We are thankful to Higher Education Commission (HEC) for providing financial assistance under NRPU Grant # 20-2798. AS is thankful to the Higher Education Commission of Pakistan for financial support under Project No 22-1/HEC/R&D/PPCR/2018. UR is thankful to Higher Education Commission for financial support for the purchase of MOE license under HEC-NRPU Project 5291/Federal/NRPU/R&D/HEC/2016.
Competing interests
The authors declare that they have no competing interests.
Availability of materials and data
The whole data is available in the manuscript.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The experiments were carried out as per the approval of the ethical committee, Department of Pharmacy, University of Malakand, Pakistan according to the animals Bye-Laws 2008 (Scientific Procedure Issue-1).
Funding
This project got funding for carrying experimental procedure from Higher Education Commission of Pakistan.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1.
Supporting information.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Farooq, U., Naz, S., Shams, A. et al. Isolation of dihydrobenzofuran derivatives from ethnomedicinal species Polygonum barbatum as anticancer compounds. Biol Res 52, 1 (2019). https://doi.org/10.1186/s40659-018-0209-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-018-0209-0