
Abstract
Background
The Hainan Partridge (Arborophila ardens, Phasianidae, Galliformes) is an endemic species of Hainan Island, China, and it is classified as globally vulnerable species. There are at least 16 species in genus Arborophila and no genome sequence is available.
Methods
The whole genome of Hainan Partridge was de novo sequenced (with shotgun approach on the Illumina 2000 platform) and assembled.
Results
The genome size of Arborophila ardens is about 1.05 Gb with a high N50 scaffold length of 8.28 Mb and it is the first high quality genome announced in Arborophila genus. About 9.19% of the genome was identified as repeat sequences and about 5.88 million heterozygous SNPs were detected. A total of 17,376 protein-coding genes were predicted and their functions were annotated. The genome comparison between Hainan Partridge and Red Junglefowl (Gallus gallus) demonstrated a conserved genome structure. The phylogenetic analysis indicated that the Hainan Partridge possessed a basal phylogenetic position in Phasianidae and it was most likely derived from a common ancestor approximately 36.8 million years ago (Mya). We found that the Hainan Partridge population had experienced bottleneck and its effective population decreased from about 1,040,000 individuals 1.5 Mya to about 200,000 individuals 0.2 Mya, and then recovered to about 460,000 individuals. The number of 1:1 orthologous genes that were predicted to have undergone positive selection in the Hainan Partridge was 504 and some environmental adaptation related categories, such as response to ultraviolet radiation were represented in GO distribution analysis.
Conclusions
We announced the first high quality genome in Arborophila genus and it will be a valuable genomic resource for the further studies such as evolution, adaption, conservation, not only on Hainan Partridge but also on Arborophila or Phasianidae species.
Similar content being viewed by others


Background
The Hainan Partridge (Arborophila ardens, Phasianidae, and Galliformes) is a species endemic to Hainan Island of China, distributed mainly in tropical evergreen forests between 600 and 1600 m above the sea level (Gao 1998; Yang et al. 2011). During the past few decades, the population of this partridge was reported to have rapidly declined owing to their poor flying ability and ground-dwelling nature, making them vulnerable to human activities, non-indigenous predators and rapid loss of habitat (Chang et al. 2012; Liang et al. 2013; Rao et al. 2017). Therefore, it was listed as a first class protected species in China (Zhang et al. 2003) and global vulnerable species (IUCN 2018). Historical and ongoing population declines (Chang et al. 2012; Chen et al. 2015a) and a suite of persistent and novel threats (Liang et al. 2013) have led to governmental protection of this species in much of their range. However, the whole genome of Hainan Partridge is not currently available and few studies have been conducted for the exploration related to the genetic mechanisms of the environmental adaption to Hainan Island of the Hainan Partridge. To provide genome-scale insights into the vulnerable Hainan Partridge, facilitate comparative studies of avian genomics and further the development of genetic tools for Hainan Partridge research and conservation, we sequenced the genome of the Hainan Partridge. The results will provide a better understanding of the factors that shape the evolutionary history of the Hainan Partridge and eventually improve its conservation.
Methods
Sampling and sequencing
Muscle sample was collected from a wild dead male Hainan Partridge which was preserved in the Natural History Museum of Sichuan University (NCBI Taxonomy ID: 1206065). A whole genome shotgun approach on the Illumina 2000 platform was performed to sequence the genome. Two paired-end libraries with insert sizes of 230 bp and 500 bp, as well as three mate-paired libraries with insert sizes of 2 kb, 5 kb and 10 kb were constructed.
Genome size estimation, genome assembly and completeness evaluation
Before assembly, a 17-Kmer analysis was performed to estimate the genome size and the assembly was firstly performed by SOAPdenovo2 (Luo et al. 2012) with the parameters set as “all -d 5 –M 3 –k 25”. After using SSPACE (Boetzer et al. 2011) to build super-scaffolds, intra-scaffold gaps were then filled using Gapcloser (Tigano et al. 2018), which is distributed with SOAP, with reads from short-insert libraries. In order to verify the correctness of the assembly, we aligned it to the Red Junglefowl (Gallus gallus) reference genome. Visualization of the Hainan Partridge/Red Junglefowl genome alignment was performed using LAST (Kiełbasa et al. 2011) with the settings suggested by the software developers for similarly distantly related taxa. We used CEGMA (Parra et al. 2007) and BUSCO (Simão et al. 2015) to evaluate the genome completeness.
Gene prediction and annotation
We combined the de novo and homology-based prediction to identify protein-coding genes (PCGs) in the genome. The de novo prediction was performed on the assembled genomes with repetitive sequences masked as “N” based on the HMM (hidden Markov model) algorithm. AUGUSTUS (Stanke et al. 2006) and GENSCAN (Burge and Karlin 1997) programs were executed to the PCGs using appropriate parameters. For the homology prediction, proteins of the Red Junglefowl, Turkey (Meleagris gallopavo), Zebra Finch (Taeniopygia guttata), and human (Homo sapiens) were mapped onto the genome using TblastN (Altschul et al. 1997) with an E-value cutoff of 1E−5. To obtain the best matches of each alignment, the results yielded from TblastN were processed by SOLAR (Yu et al. 2006). Homologous sequences were successively aligned against the matching gene models using GeneWise (Birney et al. 2004). We used EVidenceModeler (EVM) (Haas et al. 2008) to integrate the above evidence and obtained a consensus gene set.
Functional annotation
Functional annotation of all genes was undertaken based on the best match derived from the alignments to proteins annotated in Swissprot and TrEMBL databases (Boeckmann et al. 2003) and BlastP tools with the same E-value cut-off of 1E−5 was applied. Descriptions of gene products from Gene Ontology ID were retrieved from the results of Swissprot. We also annotated proteins against the NCBI non-redundant (Nr) protein database. The motifs and domains of genes were annotated using InterProScan (Hunter et al. 2008) against publicly available databases, including ProDom (Bru et al. 2005), PRINTS (Attwood et al. 2000), PIRSF (Wu et al. 2004), Pfam (Finn et al. 2013), ProSiteProfiles (Sigrist et al. 2002), PANTHER (Thomas et al. 2003), SUPERFAMILY (Gough and Chothia 2002), and SMART (Letunic et al. 2004). To find the best match and involved pathway for each gene, all genes were uploaded to KAAS (Moriya et al. 2007), a web server for functional annotation of genes against the manually corrected KEGG genes database by BLAST, using the bi-directional best hit (BBH) method.
Analyses of gene family, phylogeny, and divergence
We used orthoMCL (Li et al. 2003) to define orthologous genes from 10 avian genomes (Hainan Partridge, Red Junglefowl, Turkey, Chinese Monal Lophophorus lhuysii, Japanese Quail Coturnix japonica, Rock Dove Columba livia, Mallard Anas platyrhynchos, Peregrine Falcon Falco peregrinus, Zebra Finch, Ostrich Struthio camelus). Phylogenetic tree of these 10 birds was constructed using nucleotide sequences of 1:1 orthologous genes. Coding sequences from each 1:1 orthologous family were aligned by PRANK (Nick and Ari 2010) and concatenated to one sequence for each species for building the tree. Modeltest (Posada and Crandall 1998) was used to select the best substitution model for the whole concatenated sequence. RAxML (Stamatakis 2014) was then applied to reconstruct the maximum likelihood (ML) phylogenetic tree with 1000 bootstrap replicates. Divergence time estimation was performed by PAML MCMCTREE (Yang 2007).
Positive selection analysis
The above alignments of 1:1 orthologous genes and phylogenetic tree were used to estimate the ratio of the rates of non-synonymous to synonymous substitutions (ω) using the codeml program within PAML under the branch-site model. We then performed a likelihood ratio test and identified the positively selected genes (PSGs) by means of FDR adjustment with Q-values < 0.05.
SNP distribution and demography from genome data
We used SAMtools and Pairwise Sequentially Markovian Coalescent (PSMC) to detect SNPs between diploid chromosomes (Li et al. 2009) and the demographic history of the Hainan Partridge (Li and Durbin 2011), respectively.
Results
Genome sequencing, assembly and quality assessment
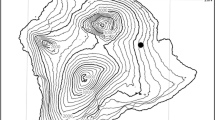
After filtering out low quality and duplicated reads, a total of 221.54 Gb (~ 205-fold coverage) high quality sequences were obtained (Table 1). The genome size of Hainan Partridge is 1.08 Gb on the basis of K-mer analysis and it is similar to the reported avian genomes (Cai et al. 2013). The total length of all scaffolds was 1.05 Gb with the scaffold N50 8.28 Mb (Table 2). The genome completeness was evaluated using CEGMA methods with the results of 83.47% for completeness and 90.32% for partial gene set (Additional file 1: Table S1). A total of 91.4% of the eukaryotic 1:1 genes were captured according to the BUSCO evaluations (Additional file 1: Table S2). At the same time, visual inspection of the alignment of the Hainan Partridge scaffolds against the Red Junglefowl reference genome also indicated high synteny and assembly correctness. Hainan Partridge scaffolds generally aligned entirely to a single Red Junglefowl chromosome, even though detectable inversions were common and some chromosomal rearrangements were evident, especially on chromosome 2 and chromosome 4 of the Red Junglefowl (Fig. 1a, b). Some of the Red Junglefowl microchromosomes were covered almost entirely by a single Hainan Partridge scaffold (Fig. 1c, d).

Alignments of the Hainan Partridge scaffolds to Red Junglefowl chromosomes (a, b) several chromosomal rearrangements between the red jungle fowl and Hainan Partridge are evident in the red jungle fowl chromosome 2 and chromosome 4, while c, d the alignment of the Hainan Partridge scaffolds to Red Junglefowl chromosome 15 and chromosome 26 is an example of scaffolds approaching chromosome size, high synteny and high assembly correctness. Forward alignments are in blue, reverse alignments in red
Repeat sequences and gene prediction
The GC content of the Hainan Partridge genome was approximately 42.17%, similar to other bird species such as the Ground Tit, Red Junglefowl and Zebra Finch (Cai et al. 2013). The repeat sequences are about 9.19% (96.04 Mb) including long interspersed nuclear elements (LINEs, 6.70%), long terminal repeats (LTRs, 1.27%), short interspersed nuclear elements (SINEs, 0.06%), and DNA transposons 1.14% (Additional file 1: Table S3). A total of 17,376 PCGs in Hainan Partridge genome were predicted and most (92.03%) of them were well supported by public protein databases (TrEMBL, Swissprot, Nr, InterPro, GO and KEGG) (Fig. 2b, Additional file 1: Table S4). The average length of genes and coding sequences were 24,359 bp and 1689 bp with an average of 10 exons per gene.

Comparative genomics in avian species studied. a Phylogenetic tree constructed using 1:1 orthologous genes. The time lines indicate divergence times among the species. b Functional annotation of Hainan Partridge genes. c Orthologous gene clusters in five Phasianidae species
Bird phylogeny, divergence and evolution of gene families
We identified 14,668 gene families from 10 available bird genomes (Hainan Partridge, Red Junglefowl, Turkey, Chinese Monal, Japanese Quail, Mallard, Peregrine Falcon, Zebra Finch, Ostrich), of which 5491 represented 1:1 orthologous gene families. Comparison of orthologous gene clusters between the former five Phasianidae species is shown in Fig. 2c. The maximum likelihood phylogeny constructed based on the 1:1 orthologous genes indicated that Hainan Partridge possessed a basal phylogenetic position within Phasianidae and was most likely derived from a common ancestor approximately 36.8 Mya (Fig. 2a).
Positive selection in Hainan Partridge
There are 504 genes in 5491 1:1 orthologous genes which were under positive selection in the Hainan Partridge using the branch-site likelihood ratio test. The KEGG annotation of these PSGs suggested that they were distributed in 46 pathways such as signal transduction (28 genes), folding, sorting and degradation (21 genes), the immune system (20 genes), and transport and catabolism (17 genes) (Additional file 2: Fig. S1a). The Gene Ontology (GO) annotation classified the PSGs into three categories: molecular functions, cellular components and biological processes. Molecular functions included genes mainly involved in binding (291 genes; GO:0005488) and catalytic activity (141 genes; GO:0003824). Genes related to cellular components were primarily cell (420 genes; GO:0005623), cell part (418 genes; GO:0044464), and organelle (357 genes; GO:0043226). Biological process genes were mainly involved in cellular process (366 genes; GO:0009987), metabolic process (250 genes; GO:0008152), and biological regulation (248 genes; GO:0065007) (Additional file 2: Fig. S1b).
We found several PSGs related to environmental adaptation in Hainan Partridge. For example, there are three genes (CASP3, BRCA2, DTL) related to response to ultraviolet (UV) (GO:0009411) and it is possible that they directly respond to the high UV radiation in Hainan Island (Liao et al. 2007). Furthermore, the endoplasmic reticulum (GO: 0005783) plays key roles in crucial processes like protein transport and energy metabolism and the mRNA expression of genes in GO: 0005783 in mice is related to temperature (Yu et al. 2011).
Demography reconstruction
We identified 7,015,181 heterozygous SNPs in the Hainan Partridge genome and their density distribution is shown in Fig. 3a. On the basis of local SNP densities, we performed PSMC analysis and found that Hainan Partridge population had experienced one bottleneck in demographic history during 20 Mya to 10,000 years ago (Fig. 3b). The effective population size decreased from approximately 1,040,000 individuals about 2.5 Mya to a minimum of 200,000 individuals approximately 0.25 Mya and then expand to about 460,000 individuals.

SNP density distribution and demography reconstruction of the Hainan Partridge. a distribution of SNP density across the Hainan Partridge genome. Heterozygous SNPs between the chromosomes were annotated, and heterozygosity density was observed in non-overlapping 50-kb windows. b PSMC inference of Hainan Partridge population history based on autosomal data. The central bold lines represent inferred population sizes. The 100 thin curves surrounding each line are the PSMC estimates that were generated using 100 sequences randomly resampled from the original sequence. The mutation rate on autosomes used in time scaling, was estimated using red jungle fowl autosome data
Discussion
There are more than 250 species in the Galliformes in the world including 63 Galliformes species distributed in China (Shen et al. 2010; Li et al. 2010; Zheng 2017). However, there is a limited number of pheasant genomes sequenced so far. The genus Arborophila is very abundant with at least 16 species (Chen et al. 2015a, b; Clements et al. 2018; del Hoyo et al. 2018) and they all are in IUCN red list (IUCN 2018). However, no genome is available for this genus. In this research, we provided the high-quality genome sequences of Hainan Partridge and it will be a very important resource for the investigation associated with the molecular genetic mechanisms of the environmental adaption to Hainan Island of the Hainan Partridge, facilitating comparative studies of avian genomics and developing the genetic tools for Hainan Partridge protection.
The genome synteny analysis between the Hainan Partridge and Red Junglefowl demonstrated that their genome structures were relatively conserved. Our observation was in line with previous reports of conserved overall synteny between the Ground Tit (Pseudopodoces humilis) and Zebra Finch (Cai et al. 2013), the Zebra Finch and Red Junglefowl (Warren et al. 2010), and also between the Turkey and Red Junglefowl (Yang 2002). However, this inference needs further confirmation with more sequenced avian genomes. The phylogenetic analysis showed that Phasianidae was a monophyly and the Hainan Partridge was given a basal phylogenetic position, branching apparently earlier than other genera within Phasianidae. This is in accordance with the opinion that Arborophila was basal to the phasianines (Crowe et al. 2006). The Hainan Partridge diverged from the other lineages in the Phasianidae around 36.8 Mya, which was much earlier than other genera (Crowe et al. 2006; Chen et al. 2015b).
The Hainan Partridge is only distributed in Hainan Island and there are at least three positively selected genes (CASP3, BRCA2, DTL) related to UV radiation. This evolution of genes may be important for the Hainan Partridge to survive in the high UV radiation environment at low latitude Hainan Island (Liao et al. 2007). This kind of adaptation has also been developed by other birds living at high altitude (Cai et al. 2013). Previous studies reported that CASP3, a central effector of apoptosis, facilitated rather than inhibited radiation-induced genetic instability and carcinogenesis (Liu et al. 2015). Liu et al. (2015) showed that a significant fraction of mammalian cells that were treated with ionizing radiation could survive despite caspase-3 activation, and this sublethal activation of CASP3 promoted persistent DNA damage and oncogenic transformation. Homologous recombination (HR) repair following DNA double-strand breaks (DSB) was a primary, high-fidelity mechanism of radiation repair in cells. An important step in HR was recruitment of the repair protein RAD51 by BRCA2 to the damaged DNA sites; the alteration of these proteins rendered cells resistant to cytotoxic damage (Abaji et al. 2005; Luo et al. 2016). Previous studies found genes that were known to upregulate DNA repair proteins such as BRCA2 to protect cells from radiation-induced DNA damage (Im et al. 2018). Several studies have demonstrated that DTL had an oncogenic function in some cancer types, such as hepatocellular carcinoma, breast cancer, and Ewing sarcoma and it plays an important part in regulating the protein stability of p53 (Kobayashi et al. 2015; Banks et al. 2006; Li et al. 2009).
In the present study, we performed PSMC analysis and the results contradicted the previous studies proposing that the Hainan Partridge populations contracted during the last ice age followed by a warming period expansion (Hewitt 2004). However, our results supported the expectation that the current demography was representative of their past during the Last Glacial Maximum (LGM) (Chang et al. 2012). Previous studies revealed the postglacial expansion events from potential refugia of the Hainan Partridge by climate warming, but this phenomenon was not shown in the present study. The pattern in this study has revealed much similarity to forest community results (species in Southeast Asia once have survived ice ages with comparatively steady demographic history during the LGM) (Chang et al. 2012). These similar results of steady demographic history after postglacial periods were supported by several previous researches (Xu et al. 2010). Overall, it is possible that the Hainan Partridge has experienced local adaptation and dealt with the glacial climate changes, owing to the lack of evidence of effective population size contraction during the LGM.
Conclusions
We sequenced the Hainan Partridge genome and compared it with other avian genomes. Phylogenetic analysis confirmed that the Hainan Partridge possessed a basal phylogenetic position in Phasianidae. Positive selection analysis revealed the environmental adaption of Hainan Partridge to UV radiation in the Hainan Island.
References
Abaji C, Cousineau I, Belmaaza A. BRCA2 regulates homologous recombination in response to DNA damage: implications for genome stability and carcinogenesis. Cancer Res. 2005;65:4117–25.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Attwood TK, Croning MD, Flower DR, Lewis AP, Mabey JE, Scordis P, Selley JN, Wright W. PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res. 2000;28:225–7.
Banks D, Wu M, Higa LA, Gavrilova N, Quan J, Ye T, Kobayashi R, Sun H, Zhang H. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle. 2006;5:1719–29.
Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–95.
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’donovan C, Phan I, Pilbout S. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–70.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–9.
Bru C, Courcelle E, Carrère S, Beausse Y, Dalmar S, Kahn D. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 2005;33:D212–5.
Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA1. J Mol Biol. 1997;268:78–94.
Cai Q, Qian X, Lang Y, Luo Y, Xu J, Pan S, Hui Y, Gou C, Cai Y, Hao M, Zhao J. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 2013;14:R29.
Chang J, Chen D, Ye X, Li S-H, Liang W, Zhang Z, Li M. Coupling genetic and species distribution models to examine the response of the Hainan partridge (Arborophila ardens) to Late Quaternary climate. PLoS ONE. 2012;7:e50286.
Chen D, Chang J, Li S-H, Liu Y, Liang W, Zhou F, Yao CT, Zhang Z. Was the exposed continental shelf a long-distance colonization route in the ice age? The Southeast Asia origin of Hainan and Taiwan partridges. Mol Phylogenet Evol. 2015a;83:167–73.
Chen D, Liu Y, Davision WHG, Dong L, Chang J, Gao SH, Li SH, Zhang ZW. Revival of the genus Tropicoperdix Blyth 1859 (Phasianidae, Aves) using multilocus sequence. Zool J Linn Soc. 2015b;175:429–38.
Clements J, Schulenberg T, Iliff M, Roberson D, Fredericks B, Sullivan B, Wood C. The eBird/Clements checklist of birds of the world: v2018. 2018. http://www.birds.cornell.edu/clementschecklist/download/. Accessed 20 Sept 2018.
Crowe TM, Bowie RC, Bloomer P, Mandiwana TG, Hedderson TA, Randi E, Pereira SL, Wakeling J. Phylogenetics, biogeography and classification of, and character evolution in, gamebirds (Aves: Galliformes): effects of character exclusion, data partitioning and missing data. Cladistics. 2006;22:495–532.
del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E. Handbook of the birds of the world alive. Barcelona: Lynx Edicions. 2018. https://www.hbw.com/node/53455. Accessed 4 Sept 2018.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL. Pfam: the protein families database. Nucleic Acids Res. 2013;42:D222–30.
Gao Y. Conservation status of endemic Galliformes on Hainan Island, China. Bird Conserv Int. 1998;9:411–6.
Gough J, Chothia C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002;30:268–72.
Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:1.
Hewitt GM. Quaternary Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B. 2004;359:183–95.
Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD. InterPro: the integrative protein signature database. Nucleic Acids Res. 2008;37:D211–5.
Im J, Lawrence J, Seelig D, Nho RS. FoxM1-dependent RAD51 and BRCA2 signaling protects idiopathic pulmonary fibrosis fibroblasts from radiation-induced cell death. Cell Death Dis. 2018;9:584.
IUCN. Arborophila ardens. The IUCN red list of threatened species. Version 2018-1. http://www.iucnredlist.org. 2018. Accessed 20 Jul 2018.
Kiełbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–93.
Kobayashi H, Komatsu S, Ichikawa D, Kawaguchi T, Hirajima S, Miyamae M, Okajima W, Ohashi T, Kosuga T, Konishi H, Shiozaki A. Overexpression of denticleless E3 ubiquitin protein ligase homolog (DTL) is related to poor outcome in gastric carcinoma. Oncotarget. 2015;6:36615.
Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32:D142–4.
Liao Y, Wang W, Zhang L, Yang L. Bio-effective UV radiation intensity distribution reaching the land surface in China. Geogr Res. 2007;26:821–7.
Liang W, Cai Y, Yang C. Extreme levels of hunting of birds in a remote village of Hainan Island, China. Bird Conserv Int. 2013;23:45–52.
Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493.
Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Li J, Ng EK, Ng YP, Wong CY, Yu J, Jin H, Cheng VY, Go MY, Cheung PK, Ebert MP, Tong J. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br J Cancer. 2009;101:691.
Li R, Tian H, Li X. Climate change induced range shifts of Galliformes in China. Integr Zool. 2010;5:154–63.
Liu X, He Y, Li F, Huang Q, Kato TA, Hall RP, Li C. Caspase-3 promotes genetic instability and carcinogenesis. Mol Cell. 2015;58:284–96.
Luo K, Li L, Li Y, Wu C, Yin Y, Chen Y, Deng M, Nowsheen S, Yuan J, Lou Z. A phosphorylation–deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Genes Dev. 2016;30:1–15.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5.
Nick G, Ari L. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 2010;11:1–7.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics (Oxford, England). 1998;14:817–8.
Rao X, Yang C, Liang W. Breeding biology and novel reproductive behaviour in the Hainan partridge (Arborophila ardens). Avian Res. 2017;8:34.
Shen Y, Liang L, Sun Y, Yue B, Yang X, Murphy RW, Zhang Y. A mitogenomic perspective on the ancient, rapid radiation in the Galliformes with an emphasis on the Phasianidae. BMC Evol Biol. 2010;10:132.
Sigrist CJ, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M, Bairoch A, Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–74.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with 1:1 orthologs. Bioinformatics. 2015;31:3210–2.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:435–9.
Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–41.
Tigano A, Sackton TB, Friesen VL. Assembly and RNA-free annotation of highly heterozygous genomes: the case of the thick-billed murre (Uria lomvia). Mol Ecol Resour. 2018;18:79–90.
Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, et al. The genome of a songbird. Nature. 2010;464:757–62.
Wu CH, Nikolskaya A, Huang H, Yeh LS, Natale DA, Vinayaka CR, Hu ZZ, Mazumder R, Kumar S, Kourtesis P, Ledley RS. PIRSF: family classification system at the protein information resource. Nucleic Acids Res. 2004;32:D112–4.
Xu L, He C, Shen C, Jiang T, Shi L, Sun K, Berquist SW, Feng J. Phylogeography and population genetic structure of the great leaf-nosed bat (Hipposideros armiger) in China. J Hered. 2010;101:562–72.
Yang C, Zhang Y, Cai Y, Stokke BG, Liang W. Female crowing and differential responses to simulated conspecific intrusion in male and female Hainan partridge (Arborophila ardens). Zool Sci. 2011;28:249–53.
Yang Z. Likelihood and Bayes estimation of ancestral population sizes in hominoids using data from multiple loci. Genetics. 2002;162:1811–23.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91.
Yu J, Liu F, Yin P, Zhu X, Cheng G, Wang N, Lu A, Luan W, Zhang N, Li J, Guo K. Integrating miRNA and mRNA expression profiles in response to heat stress-induced injury in rat small intestine. Funct Integr Genomics. 2011;11:203–13.
Yu X, Zheng H, Wang J, Wang W, Su B. Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics. 2006;88:745–51.
Zhang Z, Ding C, Ding P, Zheng G. The current status and a conservation strategy for species of Galliformes in China. Biodivers Sci. 2003;11:414–21.
Zheng GM. Pheasants in China. Beijing: Higher Education Press; 2017 (in Chinese).
Authors’ contributions
CZ, BY designed and supervised the project. CZ, SZ performed the bioinformatic analyses. CZ wrote the manuscript. All authors contributed to revising the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Jiazheng Jin, Qinchao Wen, Haoran Yu and Yang Geng for their valuable advice on bioinformatic analyses.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Genome and DNA sequencing data of the Hainan Partridge have been deposited into the NCBI Sequence Read Archive (SRA) under the ID PRJNA317652. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethical approval
The investigations comply with the current laws of China in which they were performed.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 31702017).
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Statistics of the genome completeness of Hainan Partridge based on 248 CEGs. Table S2. Statistics of the genome completeness of Hainan Partridge based on BUSCO benchmark. Table S3. Statistics of repetitive elements in Hainan Partridge genome. Table S4. Functional annotation of Hainan Partridge genes.
Additional file 2: Figure S1.
Functional distribution of positively selected genes (PSGs). (a) functional distribution of PSGs according to the KEGG pathway database. The y-axis illustrates the KEGG functional categories, while the number of genes in each category is plotted on the x-axis. (b) functional distribution of PSGs according to the gene ontology (GO) database. The y-axis reveals the GO functional categories, while the number of genes in each category is plotted on the x-axis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Zhou, C., Zheng, S., Jiang, X. et al. First complete genome sequence in Arborophila and comparative genomics reveals the evolutionary adaptation of Hainan Partridge (Arborophila ardens). Avian Res 9, 45 (2018). https://doi.org/10.1186/s40657-018-0136-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-018-0136-3