
Abstract
A sensitive and accurate ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) method was developed and validated for the determination of EVT201 and its two metabolites, Ro46-1927 and Ro18-5528, in human urine. Different sample preparation methods were compared, and solid-phase extraction (SPE) was finally employed. Separation conditions and mass spectrometry parameters were optimized to achieve complete separation and enough sensitivity. Finally, the three analytes were separated on an Acquity BEH C18 column (2.1 mm × 50 mm, 1.7 μm) with a gradient mobile phase. The gradient elution consisted of acetonitrile (containing 0.1% formic acid) and 10 mM ammonium formate (containing 1% acetonitrile and 0.1% formic acid) at a flow rate of 0.50 mL/min. Detection was performed on an electrospray ionization (ESI) source in positive mode with multiple reaction monitoring (MRM). The method was fully validated in accordance with the bioanalysis guidance in Chinese Pharmacopoeia. It showed satisfying linearity, accuracy, and precision in the range of 0.2–200 ng/mL for all the three analytes. The mean extraction recoveries were 85.2%, 65.6%, 87.9%, and 86.4% for EVT201, Ro46-1927, Ro18-5528, and the IS, respectively. The method was successfully applied to the bioanalysis of 833 urine samples to determine the concentration of EVT201 and its two metabolites simultaneously.
Similar content being viewed by others
Introduction
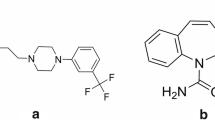
Insomnia is the most prevalent sleep disorder, and it is disturbing many people in the world. Difficulty with sleep onset and sleep maintenance is the most common sleep-related complaint encountered in primary care and many medical practices (Neubauer et al. 2018). For people with insomnia, alterations in both protein structure and function in the central nervous system occur and these effects are aggravated with advancing chronological age (Naidoo et al. 2005; Naidoo et al. 2008). To relieve the symptoms of insomnia, many new anti-insomnia drugs have been developed, such as barbiturates, benzodiazepines, and melatonin receptor agonists. An ideal anti-insomnia drug should help patients in improving the quality of sleep and leaving no sequel in the next day. As a partial positive allosteric modulator of the gamma-aminobutyric acid type A (GABAA) receptor, EVT201 was developed for the treatment of insomnia by Zhejiang Jingxin Pharmaceutical Co. LTD (see the structure in Fig. 1). It could improve the sleep quality and significantly reduce the daytime physiological sleep tendency (Walsh et al. 2010). As a medium-acting benzodiazepine receptor agonist (BzRAs), the elimination half-life of EVT201 ranged from 3 to 4 h. Therefore, it has many advantages over the traditional short-acting or long-acting BzRAs. First, it has a longer elimination half-life than the short-acting BzRAs and thus sustains a longer sleep time than the latter. However, it sustains a weaker residual side effect and less risk of tolerance than other long-acting BzRAs drugs (mean half-lives > 6 h) (Walsh et al. 2009). In addition, it is a partial GABAA receptor agonist, which may result in less undesired effects. Due to its obvious merits in dyskinesia, residual side effect, tolerance, drug-drug interaction, and memory impairment, EVT201 was a promising new drug to treat insomnia. The metabolism of EVT201 was mediated by tyrosine monoxides and cytochrome CYP2D6 to produce several metabolites, including Ro46-1927 and Ro18-5528 (the structures of the three compounds are shown in Fig. 1). As a new drug, many studies are being processed to prove its effectiveness and safety, as well as its mechanism. To support these studies, accurate and reliable determination methods of EVT201 and its metabolites in different biological fluids had to be developed. In a previous study, we developed an ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) method for the analysis of EVT201 and its two metabolites in human plasma (Zhang et al. 2018) and successfully applied it to a phase I clinical trial. To support the elimination study, the analysis method of EVT201 and its metabolites in human urine had to be developed.

Chemical structures of EVT201 (a), Ro46-1927 (b), Ro18-5528 (c), and the IS (d)
At the beginning, we tried to transfer the determination method from human plasma samples to urines samples. The method exhibited well for the spiked samples. However, for the real samples from the volunteers, there were some small interferences for the two metabolites. Even worse, the incurred sample reproducibility (ISR) test failed in urine for all the three analytes. To solve this problem, an investigation was conducted. Finally, it was found that adsorption was the main cause. Therefore, urine samples were subject to ultrasound to desorption before aliquot. Sample extraction procedures and analytical method were also re-developed. There are some papers on the determination of benzodiazepines and analogs in urine with liquid chromatography-tandem mass spectrometry (LC-MS/MS) or gas chromatography-tandem mass spectrometry (GC/MS) method (Salomone et al. 2011; De Boeck et al. 2017; Ming and Heathcote 2011; Perez et al. 2016; de Bairros et al. 2015; Pebdani et al. 2016; Saito et al. 2014; Bugey and Staub 2007). However, there were no published papers on the determination of EVT201 or its metabolites in human urine up to now. Further analysis of the published methods for other benzodiazepines showed that they could not be employed in the determination of EVT201 and its metabolites. For example, thirteen benzodiazepines in human urine were determined with UHPLC/MS/MS method after precipitation with methanol (Ming and Heathcote 2011), with a lower limit of quantifications (LLOQ) of 20 ng/mL for all the analytes. The sensitivity was not sufficient for our study. Besides, the matrix effect of protein precipitation would probably affect the robustness of the method when the sample size was large. Perez et al. compared LC-MS/MS and GC/MS method (Perez et al. 2016) to determine benzodiazepines in urine with solid-phase extraction (SPE). However, the sensitivity was still insufficient even if 1 mL of urine was used to concentrate. Some novel extraction procedures coupled to LC/MS/MS or GC/MS method were reported as well, such as hollow-fiber liquid-phase microextraction (De Bairros et al. 2015) or dispersive nanomaterial-ultrasound-assisted microextraction method (Pebdani et al. 2016). Though high sensitivity could be obtained with these methods, the procedures were too comprehensive and the throughputs were rather low, which restricted their application. Considering the high sensitivity, robustness demand, and throughput, we decided to use “cleaner” SPE method to purify the urine samples. The UHPLC-MS/MS conditions were re-optimized to separate possible interferences in urine as well.
In this paper, we described a sensitive and robust UHPLC-MS/MS method coupled to SPE for the simultaneous quantification of EVT201 and its two metabolites in human urine. EVT201-d6 was used as the internal standard (IS). Factors which affected the extraction, chromatographic separation, and MS/MS detection were optimized. The new developed method was fully validated with reference to the Chinese Pharmacopoeia guidance. To increase the throughput, Oasis® HLB 96-Well Plate was used for SPE. The method was applied to the analysis of 833 urine samples from an elimination study. ISR was also conducted to evaluate the method reproducibility.
Experimental
Reagents and solutions
Reference standards of EVT201 (lot number: 75184 01/02, purity 99.5%), Ro46-1927 (lot number: CM135-34, purity 95.0%), Ro18-5528 (purity 95.0%), and EVT201-d6 (lot number: 01, purity 95.0%) were obtained from Zhejiang Jingxin Pharmaceutical Co. LTD (Zhejiang province, China). HPLC grade methanol and acetonitrile were purchased from Fisher Chemical (USA). HPLC grade formic acid was purchased from Dikma, and HPLC grade ammonium formate was from Fluka. GR grade ammonium hydroxide was from Sino Pharm Chemical Reagent Co. Ltd. Ultra-pure water (> 18 MΩ/cm) was prepared in-house using a Pall purification system (New York, USA). Different lots of blank human urine were collected from healthy volunteers.
Instrumentation and UHPLC/MS/MS conditions
Analysis was performed on an ABSciex 4000+ triple quadrupole system (ABSciex Corp., USA) equipped with an electrospray ionization source. The UHPLC system consisted of two Shimadzu 30A delivery pumps, an auto-sampler, and a column oven. Data acquisition and quantitative processing were accomplished with Analyst (version 1.6.2) software. Oasis® HLB 96-well plates (30 mg, 30 μm) were used to increase the throughput.
Separation was performed on a UHPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm) maintained at 30 °C. Solvent A consisted of 0.1% formic acid in acetonitrile. Solvent B was 10 mM ammonium formate buffer (containing 0.1% formic acid and 1% acetonitrile). The gradient procedures of the mobile phase were re-optimized to separate the interferences in the urine. The total flow rate was 0.5 mL/min, and the injection volume was 5 μL.
Mass spectrometry detection was operated in positive mode. The source conditions were as follows: source temperature, 600 °C; curtain gas, 40 psi; nebulizer gas, 60 psi; and heated gas, 60 psi. The detection was carried out with multiple reaction monitoring (MRM) at unit resolution. The dwell time was set at 80 ms. The MRM transitions were m/z 373.2→58.0 for EVT201, m/z 359.2→316.1 for Ro46-1927, m/z 291.2→274.1 for Ro18-5528, and m/z 379.3→64.0 for the IS. Collision energies were 18, 18, 22, and 31 V, and collision exit potentials were 10, 8, 15, and 11 V for the corresponding compounds. The analysis time was 8 min per sample.
Calibration standards and quality controls
Calibration and quality control (QC) stock solutions (500 μg/mL) were prepared in 50% methanol for all the analytes. Mixed working solutions of the calibration and QCs were prepared by diluting the stock solutions in 50% methanol. The IS stock and working solutions were also prepared in 50% methanol. All the standard solutions were stored at 4 °C. Calibration standards were prepared with working solutions in drug-free human urine at levels of 0.2, 0.5, 1.0, 5.0, 10.0, 50.0, 100.0, and 200.0 ng/mL. QC standards were prepared in the same way at five levels: lower limit of quantification (LLOQ), low QC (LQC), two medium QC (MQC1 and MQC2), and high QC (HQC). The corresponding concentrations were 0.2, 0.6, 8, 80, and 160 ng/mL.
Sample preparation
After being thawed at room temperature, the urine samples were subjected to ultrasound for 20 min and vortex for 5 min. Then, an aliquot of 200 μL urine was mixed with 50 μL of IS working solution, and 200 μL of ammonium hydroxide was added to alkalize. The Oasis® HLB 96-Well plate was pre-conditioned with 1 mL of methanol and 1 mL of water in sequence. Then, the alkalized urine samples were loaded. The plate was washed with 1 mL of 10% methanol twice and then was dried under nitrogen for 25 min. Finally, the analytes were eluted to a receiver plate with 250 μL of methanol (containing 2% of formic acid) and the eluting procedure repeated twice. The eluents were combined and then were dried in a vacuum oven at 45 °C. The residue was reconstituted in 150 μL of 50% methanol and was centrifuged at 15,000 rpm for 5 min. The receiver plate was placed into the auto-sampler for analysis, and the injection volume was 5 μL.
Method validation
The method was fully validated according to the Chinese Pharmacopoeia guidance (2015).
Selectivity and calibration
Blank urine samples from six individuals were used to evaluate the possible endogenous interferences at the peak regions of the analytes and the IS. Eight non-zero calibrators were prepared as described in the “Calibration standards and quality controls” section and were processed with the study samples, in duplicates at the lower and upper limits of quantification. Least square linear regression was used to define the calibration curve over the range of 0.2–200 ng/mL for all the three analytes. The weighting factors were 1/x2.
Precision and accuracy
Intra-/inter-run precisions and accuracies were evaluated at LLOQ and four QC levels in six replicates in four runs. The precision was expressed using the coefficient variance (CV%), and the accuracy was defined as relative bias (RE%). The acceptable criteria for CV% and RE% were within 15% except for LLOQ, where the acceptable deviation is up to 20% for accuracy and precision.
Recovery and matrix effect
Extraction recovery was investigated at LQC, MQC1, MQC2, and HQC levels. Recovery was expressed as the peak area ratio of the analyte to the IS in an extracted sample to a non-extracted sample at each level in triplicates. The extracted sample was prepared as in the “Sample preparation” section, except the IS was added after extraction. The non-extracted sample was prepared by spiking equivalent amount of analytes and the IS into the reconstitution solvent. The final volumes of the reconstitution solution for extracted and non-extracted samples were the same. Recovery of the IS was tested against EVT201 at MQC1 level. Its recovery was calculated as the peak area ratio of the IS to EVT201 in an extracted sample to a non-extracted sample in triplicates. Matrix effect was assessed with blank urine samples from six individuals at LQC and HQC levels. The matrix factor (MF) was calculated by the peak area ratio in the presence or absence of matrix with the same amount of analytes and the IS. The IS normalized matrix factor (ISMF) was assessed by comparing MF of the analytes with that of the IS.
Dilution integrity
Dilution integrity was determined by diluting the dilution QC (DQC, 800 ng/mL) sample with blank urine in five replicates for each dilution factor. Carry-over was examined by injecting a double blank sample immediately after the ULOQ. The peak areas at the retention regions of the analytes and the IS were compared with those in the LLOQ.
Stability
Stability of the stock and working solutions of the analytes and the IS was evaluated in our previous paper (Zhang et al. 2018), and the solutions used in this study were all within the validity period. Benchtop stability of urine samples was investigated at LQC/HQC levels. The spiked samples were placed on benchtop at room temperature for 24 h and were analyzed against a fresh calibration curve. For process stability, processed samples at LQC/HQC levels were stored at 4 °C for 192 h upon completion of sample preparation and were analyzed against the fresh calibration curve. Freezing thaw stability was assessed by testing spiked urine samples subjected to different freezing/thaw cycles in triplicates at LQC/HQC levels. For each cycle, spiked samples were frozen over 12 h at − 80 °C and then were thawed completely at room temperature. Long-term stability was assessed by storing the LQC/HQC samples at − 80 °C and testing them periodically against newly prepared calibrations. All the stability results were calculated against the nominal values.
Application
The validated method was applied to the sample analysis from an elimination study in humans, and 833 urine samples were analyzed. The study was approved to be conducted by CFDA (approval number: 2013L00763) and the Ethics Committee for Drug Clinical Trials in the 307th Hospital of PLA (ethics audit code: 2014-12-172-1). Eighty-six volunteers had taken EVT201 capsules at various dosages, that is, 0.5, 1, 1.5, 2.5, 5, 7.5, and 10 mg for the single-dose group, and 1.5, 2.5, and 5.0 mg for the multiple-dose group. Half male and half female volunteers were involved in each dosage. Urine samples were collected at intervals of 0~2, 2~4, 4~8, 8~12, 12~24, 24~36, and 36~48 h after oral administration for single doses. For multiple doses, urine samples were collected at intervals of 0~2, 2~4, 4~8, 8~12, and 12~24 h on the first day and at the same intervals on the fifth day. All the urine samples were frozen at − 80 °C until analysis. Ninety samples were tested for ISR. Differences between the original and the re-assay results were calculated. Large differences between the two results would indicate analytical issues and would be investigated.
Results and discussions
Method development and optimization
An electrospray ionization (ESI) source in positive mode was used for the MS/MS detection, and [M + H]+ were used as the precursor ions for all the analytes. MRM parameters were optimized for each compound to obtain suitable signals for quantification. Detailed parameters were described in the “Instrumentation and UHPLC/MS/MS conditions” section.
For the chromatographic conditions, we tried the conditions described in the previous paper at the beginning (Zhang et al. 2018); however, interferences could not be separated completely for urine samples, especially for the two metabolites, Ro46-1927 and Ro18-5528. Therefore, chromatographic conditions were re-developed to get better resolution. An Acquity BEH C18 (50 × 2.1 mm, 1.7 μm) column was employed with a mobile phase consisting of 10 mM ammonium formate buffer (A) and acetonitrile (B). Acidic mobile phase would improve the sensitivity of the analytes, and different acids were tested and were compared, including formic acid and acetic acid. The noise was higher when using acetic acid; therefore, 0.1% of formic acid was added to both the aqueous and organic solvents. Addition of 1% of acetonitrile in the aqueous phase could reduce bacterial growth. The gradient was investigated carefully to obtain a satisfying peak shape and better separation with the interferences. High initial aqueous phase could increase the sensitivity, and the percentage of B was set at 95%. It maintained for 0.8 min. Then, it descended slowly to 84% in 3.4 min to get complete separation of the analytes and the interferences. It changed to 2% rapidly in 0.2 min and maintained 1.1 min to wash the column. It went back to the initial percentage at 5.6 min and stopped at 8 min. The total run time was 8 min per injection.
Sample preparation procedures were also investigated. Liquid-liquid extraction (LLE) with ethyl acetate was tried at the beginning to get rid of salts and possible endogenous interferences, with reference to the plasma extraction procedures (Zhang et al. 2018). Satisfying results were obtained at the stage of validation and sample analysis. However, incurred sample reproducibility (ISR) test failed for all the three analytes, especially for the two metabolites. It indicated that the method was not applicable for urine samples, and investigation had to be conducted to find the main reason. Matrix effect was the first suspected cause. However, re-evaluation of the matrix effect excluded this reason. Besides, the same batch of real sample was injected for three times, and the CV% was less than 4.8%, 7.1%, and 11.9% for EVT201, Ro46-1927, and Ro18-5528. The results proved that the matrix effect was negligible, and it was not the main reason. The influence of adsorption was assessed in the following. A urine sample was transferred from one tube to another new one for 5 times and stood still for at least 10 min each time. Then, the urine in the first tube and the last tube were analyzed, and the peak areas were compared. Large differences were found between the two samples for EVT201 (− 31.5%) and Ro46-1927 (− 26.3%). While for Ro18-5528, it was only 2.0%. Therefore, adsorption might be the main cause of ISR failure. This problem had been ignored at the validation stage and the sample assay stage as well; for the calibrators and QC, samples were spiked and prepared individually and freshly. To solve this problem, 0.2% of Triton-X was tried to reduce the adsorption. However, trace amount of Triton-X would suppress the signal greatly, and the suppression effect would last for a rather long time. Methanol was also tested to reduce the adsorption. However, high ratio of methanol in the samples restricted the application of LLE in extraction step because the extractant could not be separated clearly from the aqueous phase. Finally, satisfying results were obtained with ultrasound-assisted desorption for 20 min. It might be explained by the unstable adsorption of the analytes to the insoluble impurities in urine. Therefore, ultrasound-assisted desorption step was performed before the extraction. To get cleaner extracts, solid-phase extraction (SPE) was used for sample preparation. In this study, Oasis HLB 96-Well plate (30 mg, 30 μm) was used to get high extraction recovery and to improve the throughput as well. Alkalized urine samples were loaded after the SPE plate was conditioned with methanol and water. The plate was washed twice with 1 mL of 10% methanol. Then, it was dried under nitrogen gas flow for 25 min, and 250 μL of acidic methanol (containing 0.2% formic acid) was used to elute the analytes twice. The eluent was combined and was dried in a vacuum oven. The residue was reconstituted in 50% methanol. In total, the urine samples were preconcentrated 1.33-folds during sample preparation.
Method validation
Selectivity and linearity
Urine samples from six different individuals were analyzed for specificity. No obvious interferences were observed at the peak regions of the analytes and the IS for both spiked samples and the real samples (Fig. 2). It indicated that the method was specific enough for the determination of urine samples.

Representative chromatograms of A double blank urine, B single blank urine (IS conc. 100 ng/mL), C a spiked sample at LLOQ (analytes conc. 0.2 ng/mL; IS conc. 100 ng/mL), D a real sample predose, and E a real sample after oral administration of 5 mg EVT201 capsule (8–12 h interval)
Eight calibrators were used to construct the calibration curve with a weighting factor of 1/x2. The assay was found to be linear over the tested calibration range (0.2–200 ng/mL) for all the three analytes with an LLOQ of 0.2 ng/mL. The coefficient values were all over 0.9900. For all the accepted runs, the accuracy of the calibrators ranged from 92.6 to 105.2% for EVT201, 96.4 to 102.4% for Ro46-1927, and 97.3 to 104.6% for Ro18-5528. Precision ranged from 1.2 to 5.6% for EVT201, 2.6 to 6.0% for Ro46-1927, and 1.4 to 7.5% for Ro18-5528.
Precision and accuracy
Intra-/inter-run precision and accuracy results of LLOQ and QCs are shown in Table 1. As can be seen, the intra-/inter-run precision of LLOQ was no more than 9.4%, 9.4%, and 15.2% for EVT201, Ro46-1927, and Ro18-5528, respectively. The corresponding accuracy (RE%) was within − 12.0%, 2.0%, and 5.5%. Signal to noise ratios (S/N) for LLOQ were 18, 13, and 13 for the corresponding analytes. As for the QCs, the intra-/inter-run precisions were no more than 5.0% for EVT201, 10.6% for Ro46-1927, and 5.8% for Ro18-5528. The accuracy was within 11.9%, 11.6%, and − 8.1% for the corresponding analytes.
Matrix effect and recovery
Matrix effect at LQC/HQC level was assessed with six different lots of urine. The results of the matrix effect are shown in Table 2. As can be seen, the CV% of the ISMF was less than 6.0%, 9.5%, and 7.9% for EVT201, Ro46-1927, and Ro18-5528. We also compared the results of the SPE method with those of LLE. The results could be accepted for both methods. It proved that the matrix effect was not the main cause of ISR failure with the LLE method as well.
The detailed results of extraction recovery with SPE and LLE are compared in Fig. 3. Increased recovery could be obtained with the SPE method for all the analytes. With the SPE method, the mean recoveries were 85.2%, 65.5%, and 87.9% for EVT201, Ro46-1927, and Ro18-5528, respectively. Higher extraction recovery would lead to relatively lower variation, which would benefit the robustness.

Comparison of extraction recovery with SPE and LLE
Dilution integrity and carryover
The results of dilution integrity experiments showed that after being diluted with blank urine for ten folds, the RE% of DQC was − 7.0%, − 1.4%, and − 17.7% for EVT201, Ro18-5528, and Ro46-1927, respectively. Therefore, the former two compounds in urine samples could endure 10-folds dilution while Ro46-1927 could not. The dilution integrity of Ro46-1927 was further tested. It was found to endure five folds of dilution, and the RE% was only − 0.4%. In the carryover tests, no residue peak was found at the peak regions of Ro46-1927 and Ro18-5528. The highest carryover of EVT201 was 16.4% (compared to the response of LLOQ), and it was only 0.2% for the IS. Hence, carryover was negligible for all the compounds.
Stability
Stability results of LQC/HQC samples are summarized in Table 3. As can be seen, all the analytes were stable in urine at room temperature for at least 24 h on benchtop. No degradation was observed after being placed in auto-sampler at 4 °C for at least 192 h. EVT201 and Ro18-5528 remained stable after ten freezing/thaw cycles while Ro46-1927 could only endure nine cycles. As to the long-term stability, EVT201 and Ro46-1927 were stable in urine for at least 944 and 939 days respectively when being stored at − 80 °C, while Ro18-5528 was stable for only 694 days.
Application
The validated method was applied to the analysis of urine samples from an elimination study. The total number of urine samples was 833, and the samples were analyzed in seventeen runs. To demonstrate the usability of the method, representative drug excretion profiles after single-dose oral administration of EVT201 are shown in Fig. 4. As can be seen, most of EVT201 was excreted within 12 h, and Ro46-1927 within 24 h. However, Ro18-5528 was continually excreted until the end of sample collection. It indicated that the sample collection time was not long enough for Ro18-5528, which was consistent with the plasma results in our previous findings (Zhang et al. 2018). Furthermore, the concentrations of Ro18-5528 were rather low in all the urine samples. It indicated that Ro18-5528 was not a major metabolite in urine. Therefore, the results of Ro18-5528 for volunteers were only used for reference. Total excretion ratios from urine were calculated, and the results are listed in Table 4 for single dosages of EVT201. The total excretion rates were low, and the volunteers’ renal status were all normal according to renal function assay results after taking medicine, which implied that most of EVT201 and the metabolites were not excreted from the kidney. Other excretion pathways and other types of metabolites in urine should be further studied.

Representative drug excretion profiles after a single dose of oral administration (dosage of EVT201: 0.5, 1.0, 2.5, and 5 mg)
For urine samples, the non-homogeneous of sample and adsorptive characters of drugs usually reduce the robustness of the quantification method, though it seems that urine is “cleaner” than plasma or other biofluids. To demonstrate the robustness of the validated method, ninety urine samples were selected from the assayed samples to perform ISR test. The ISR results of all of the three analytes met the acceptance criteria of the guidance. It illustrated the robustness and reproducibility of the method.
Conclusion
In this study, a sensitive, selective, and robust UHPLC-MS/MS method coupled to SPE sample preparation procedures were developed and validated for the determination of EVT 201 and its two metabolites in human urine. Two sample preparation methods were compared, and the SPE procedures were more robust with higher extraction recoveries. Besides, considering the low concentration of the three compounds in human urine and the large sample size, highly sensitive UHPLC-MS/MS method with high throughput was used in this study, though it is more expensive. The method exhibited well across the whole calibration range. The applicability of the method was confirmed by analyzing 833 urine samples successfully with satisfying ISR results. In addition, the excretion data were helpful and meaningful for the later investigation of EVT201.
Availability of data and materials
All raw data used in this manuscript are archived in Peking University Third Hospital. It is available and could be supplied upon request.
Abbreviations
- UHPLC-MS/MS:
-
Ultra-high performance liquid chromatography-tandem mass spectrometry
- ESI:
-
Electrospray ionization
- MRM:
-
Multiple reaction monitoring
- ISR:
-
Incurred sample reproducibility
- BzRAs:
-
Benzodiazepine receptor agonist
- LLOQ:
-
Lower limit of quantifications
- ULOQ:
-
Upper limit of quantification
- SPE:
-
Solid-phase extraction
- LLE:
-
Liquid-liquid extraction
- IS:
-
Internal standard
- CV:
-
Coefficient variance
- RE:
-
Relative bias
- MF:
-
Matrix factor
- ISMF:
-
Internal standard normalized matrix factor
- LQC:
-
Quality control at low level
- MQC:
-
Quality control at medium level
- HQC:
-
Quality control at high level
- DQC:
-
Quality control for dilution test
- GABAA :
-
Gamma-aminobutyric acid type A
- LC-MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- GC/MS:
-
Gas chromatography-tandem mass spectrometry
References
Bugey A, Staub C. Application of monolithic supports to online extraction and LC-MS analysis of benzodiazepines in whole blood samples. J Sep Sci. 2007;30(17):2937–78.
De Bairros AV, De Almeida RM, Pantaleão L, Barcellos T, e Silva SM, Yonamine M. Determination of low levels of benzodiazepines and their metabolites in urine by hollow-fiber liquid-phase microextraction (LPME) and gas chromatography-mass spectrometry (GC-MS). J Chromatogr B Anal Technol Biomed Life Sci. 2015;975:24–33.
De Boeck M, Missotten S, Dehaen W, Tytgat J, Cuypers E. Development and validation of a fast ionic liquid-based dispersive liquid-liquid microextraction procedure combined with LC-MS/MS analysis for the quantification of benzodiazepines and benzodiazepine-like hypnotics in whole blood. Forensic Sci Int. 2017;274:44–54.
Ming DS, Heathcote J. A rapid and accurate UHPLC/MS/MS method for the determination of benzodiazepines in human urine. J Chromatogr B Anal Technol Biomed Life Sci. 2011;879(5-6):421–8.
Naidoo N, Ferber M, Master M, Zhu Y, Pack AI. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci. 2008;28(26):6539–48.
Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J Neurochem. 2005;92(5):1150–7.
Neubauer DN, Pandi-Perumal SR, Spence DW, Buttoo K, Monti JM. Pharmacotherapy of insomnia. J Cent Nerv Syst Dis. 2018;10:1–7.
Pebdani AA, Khodadoust S, Talebianpoor MS, Zargar HR, Zarezade V. Preconcentration and determination of chlordiazepoxide and diazepam drugs using dispersive nanomaterial-ultrasound assisted microextraction method followed by high performance liquid chromatography. J Chromatogr B Anal Technol Biomed Life Sci. 2016;1008:146–55.
Perez ER, Knapp JA, Horn CK, Stillman SL, Evans JE, Arfsten DP. Comparison of LC-MS-MS and GC-MS analysis of benzodiazepine compounds included in the drug demand reduction urinalysis program. J Anal Toxicol. 2016;40(3):201–7.
Saito K, Kikuchi Y, Saito R. Solid-phase dispersive extraction method for analysis of benzodiazepine drugs in serum and urine samples. J Pharm Biomed Anal. 2014;100:28–32.
Salomone A, Gerace E, Brizio P, Gennaro MC. Vincenti M.A fast liquid chromatography-tandem mass spectrometry method for determining benzodiazepines and analogues in urine. Validation and application to real cases of forensic interest. J Pharm Biomed Anal. 2011;56(3):582–91.
Walsh JK, Salkeld L, Knowles LJ, Tasker T, Hunneyball IM. Treatment of elderly primary insomnia patients with EVT 201 improves sleep initiation, sleep maintenance, and daytime sleepiness. Sleep Med. 2010;11(1):23–30.
Walsh JK, Thacker S, Knowles LJ, Tasker T, Hunneyball IM. The partial positive allosteric GABAA receptor modulator EVT201 is efficacious and safe in the treatment of adult primary insomnia patients. Sleep Med. 2009;10:859–64.
Zhang X, Zhou C, Zhang Y, Zhu S, Cai X, Pan C, Wang C, Zhai S, Yang L. Simultaneous quantification of EVT201, a novel partial positive allosteric GABAA receptor modulator, and its two metabolites in human plasma by UHPLC/MS/MS. J Pharm Biomed Anal. 2018;159:282–90.
Acknowledgements
All the authors gratefully acknowledge the staff in the Department of Pharmacy, Peking University Third Hospital, for conducting the bioanalysis work of this study.
Funding
This study was funded by Zhejiang Jingxin Pharmaceutical Co. LTD. At the time the study was conducted, Shuangpeng Zhu, Xiaofeng Cai, Chunmiao Pan, and Chuanzhen Wang were employees of this company. But none of the authors received personal financial support compensation for the writing of the manuscript. And we have no other conflicts of interest regarding the content of this article.
No other fanatical fund was received to conduct this study.
Author information
Authors and Affiliations
Contributions
The manuscript was written through the contributions of all authors. Congya Zhou and Yuanyuan Zhang contributed mainly to sample assay. Congya Zhou was the one who wrote this manuscript with the help of Xianhua Zhang, who developed the method and designed the study. Suodi Zhai and Li Yang were the co-principal investigator of the elimination study who contributed to the study design. Mr. Shuangpeng Zhu and Mr. Xiaofeng Cai were the main participants in study design. Mr. Xiaofeng Cai and Ms. Chunmiao Pan were responsible for the urine sample collection and sample pre-treatment. Mr. Chuanzhen Wang reviewed all the data in this study. Mr. Shuangpeng Zhu reviewed and revised this manuscript. The manuscript has been read and approved by all the named authors, and there are no other persons who satisfied the criteria for authorship but are not listed. We confirm that the order of authors listed in the manuscript has been approved by all of us. We have given due consideration to protect intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing, we confirm that we have followed the regulations of our institutions concerning intellectual property.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, C., Cai, X., Zhang, Y. et al. Accurate quantification of EVT201 and its two metabolites in human urine using UHPLC-MS/MS method with solid phase extraction. J Anal Sci Technol 11, 23 (2020). https://doi.org/10.1186/s40543-020-00222-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-020-00222-w