
Abstract
Fibrinolytic enzymes are agents administered for the treatment of myocardial infarctions, strokes, cardiac and respiratory failure. Although several microorganisms are known to produce these fibrinolytic enzymes, only a few of such enzymes, along with the age-old oral anticoagulants, have been employed in the clinical and therapeutic applications in humans. The use of these agents is associated with drawbacks such as allergic reactions and bleeding complications; therefore, it necessitates frequent monitoring of drug levels in the blood. Due to this, there is an impetus on the current effort to identify newer potential candidates from the novel microbial sources which show longer half-life, higher fibrin specificity, higher therapeutic index and lesser allergic reactions. Various methods are available for the preliminary evaluation of a potential drug candidate for the therapeutic use. Choosing the right combination of in vitro and in vivo methods would give crucial insight on the therapeutic potential of the chosen test compound. This article discusses various assay techniques, in vitro trails and in vivo models available, to help researchers in choosing right biological methods and its combinations to evaluate efficacy of potential drug candidate.
Similar content being viewed by others


Introduction
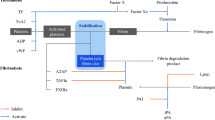
Fibrinolytic enzyme (EC 3.4.) catalyses the breakdown of fibrin clot, by either converting the inactive circulating plasminogen to active plasmin or directly lysing the fibrin into fibrin degradation products, thereby restoring the normal vascular architecture (Kotb 2013). This indicates the therapeutic potential of the fibrinolytic agent. Under normal physiological conditions, constant dynamic equilibrium is maintained in the tissues between blood coagulation and fibrinolysis (Nieuwenhuizen et al. 2012). However, when this equilibrium is disturbed, due to several reasons such as tissue injury or genetic variation or in disease-like diabetes mellitus (Fattah et al. 2004), the need for administering a fibrinolytic agent arises so as to refurbish uniform blood flow and thus aid in the healing process. Deficiencies in coagulation inhibitors such as heparin cofactor II and antithrombin III, inadequate release of plasminogen activators and uncontrolled production of plasminogen activation inhibitors are few pathophysiological conditions that could lead to the formation of blood clots (Gallimore and Friberger 1991) inside the blood vessels, clinically known as thrombosis.
Haemostasis is the process by which bleeding is arrested, when blood vessels are damaged. It occurs in three steps rapidly: vasoconstriction, platelet plug formation and coagulation (fibrin clot formation) (Marieb and Hoehn 2010). Vasoconstriction or vascular spasm involves the constriction of vessels in order to prevent the blood flow. This is followed by the aggregation of platelets, which form a temporary plug at the site of injury, releasing certain chemical messengers such as thromboxane A2 (TXA2), adenosine diphosphate (ADP) and serotonin. This causes more platelets to arrive at the site of injury, clump together leading to platelet plug formation and is termed as primary haemostasis. The final step of haemostasis (secondary haemostasis) is the conversion of fibrinogen to fibrin threads. These fibrin threads along with platelet plug forms a solid clot to seal the injured vessel wall to prevent blood loss (Porrett et al. 2005; Zdanowicz 2002).
In general, fibrinolytic agents can be broadly categorised into two types based on their route of administration. The first type comprises of the oral anticoagulants (OACs) which as the name suggests are administered to the patients orally. Examples of drugs that fall under this category include warfarin, heparin, dabigatran, rivaroxaben and nattokinase. The second type of fibrinolytic agents consists of drugs that are intravenously administered. Some of the drugs that belong to this type are streptokinase, urokinase, alteplase and tenecteplase. Streptokinase (Streptase), nattokinase (microbial origin) and urokinase (Abbokinase) are called as first-generation thrombolytic drugs. These are systemic activators which convert inactive plasminogen to active plasmin in circulation. The second generation of thrombolytics was produced with the help of genetic engineering, consists of fibrin binding domains that target the activation of plasminogen and thereby fibrin clot degradation. A well-known example being tissue plasminogen activator (tPA) produced by recombinant deoxyribonucleic acid technology (rDNA), such as alteplase (Activase) and tenecteplase (TNKase) (Longstaff and Thelwell 2005). Based on these, the third generation of thrombolytics was engineered, wherein the fibrin-binding domains were modified with the aim of improved fibrin binding, zymogenecity, resistance to certain natural inhibitors and longer plasma half-life (Verstraete 2000). Despite the availability of these drug molecules, the scientific data available on second and third generation of thrombolytic drugs, including its in vitro, in vivo studies have not been translated successfully into clinical trials, hence could be rendered as a failure (Longstaff and Thelwell 2005). The only milestone that could be achieved by these drugs is to do with its pharmacokinetics, with extended plasma half-life, but not fibrin specificity or inhibitor susceptibility (Longstaff and Thelwell 2005). This has paved way for the discovery of newer microbial sources for the production of fibrinolytic and/or thrombolytic agents with increased fibrin specificity and longer half-lives (Peng et al. 2005).
Some of the well-established oral anticoagulants used are warfarin (Coumadin), heparin (Freedman 1992) and newer anticoagulants such as dabigatran (Pradaxa) and rivaroxaban (Xarelto) which are direct thrombin and factor Xa inhibitors (Gonsalves et al. 2013). One of the primary reasons for the restricted use of these oral anticoagulants, especially in the aged populations, is due to the intra-cranial bleeding associated with their administration (Miesbach and Seifried 2012). The OACs such as warfarin and heparin which are currently in use have delayed onset of action and also exhibit certain food and drug interactions along with varied pharmacokinetics and pharmacodynamics property. Therefore, their usage requires routine laboratory monitoring and dosage adjustment in order to maintain the international normalised ratio (INR) within the therapeutic range (Bauer 2011). Particularly, warfarin, which is a vitamin K antagonist, shows significant inter- and intra-patient variability of effective dose (Lip et al. 2011). Albeit the newer OACs have an edge over the well-established OACs in terms of immediate onset of action and absence of interactions with food, their renal elimination indicates the need for dosage reduction. In addition, the lack of availability of assays to measure the drug levels, high acquiring cost of the drug, short half-life which leads to a swift decrease in its anticoagulant effect and unavailability of specific antidote in case of excessive bleeding are some of the other drawbacks (Bauer 2011).
The in vivo lysis of the blood clot has led to the enzyme-based therapy, involving fibrinolytic enzymes which differ in their working mechanisms (Kotb 2013). One being the plasmin-like protein, such as nattokinase, which converts prourokinase to urokinase and increases tissue plasminogen activator in vivo (Sumi et al. 1987, 1990); the other set of enzymes being the ones that convert inactive plasminogen to active plasmin, resulting in lysis of fibrin clot, examples are urokinase and streptokinase (Kotb 2013). In the recent decade, these fibrinolytic enzymes possibly have attracted more attention despite the presence of their oral thrombolytic counterparts (Peng et al. 2005). However, these suffer from several shortcomings such as short half-life span, lesser fibrin specificity, higher therapeutic dose, allergic responses, substantial risk of bleeding complications and increased costs (Blann et al. 2002; Turpie et al. 2002). This, nonetheless, has not ended the lookout for newer, improved fibrinolytic drug candidates, which would address the issues stated above. The current review would shed light in a very precise and concise manner on the various biological methods (in vitro and in vivo assay techniques) which become quintessential for the evaluation of a potential drug candidate for therapeutic use.
Review
In vitro assays
In vitro studies become the indispensable preliminary step in the evaluation of a microbial enzyme that has the potential to be developed into a drug. The in vitro assays discussed in this review may provide critical inputs before proceeding to in vivo testing using animal models and clinical studies.
Fibrinolytic assays
Several fibrinolytic assays are available (Table 1) to assess the clot lysing potential of an enzyme of microbial origin. These estimation methods have been reviewed by Kotb (2013). The assays comprise of either fibrin being used as the substrate directly or fibrin clot being produced by the addition of thrombin to fibrinogen. Although all of these assays share the same aim of measuring the clot lysing ability of the potential fibrinolytic agent, these differ in their ways of measuring the products of clot lysis, in other words the fibrin degradation products. Various methods adopted to measure the end point of lysis resulting in the fibrin degradation products are by measuring the zone of lysis, colorimetry and nephelometry. However, these assays alone do not represent fibrinolysis in vivo and hence will not be sufficient to evaluate the efficacy of the test compound. Therefore, in vivo studies have to be carried out taking into account the physiological conditions.
Most of the fibrinolytic enzymes of microbial origin exhibit multispecificity towards various substrates (Yogesh and Halami 2015; Wang et al. 2006), and majority of the assays listed (Table 1) do not help in determining this. Thus, it becomes necessary to address this issue. The type of specificity exhibited by the test enzyme can be determined by performing tests such as protease assay, amidolytic assay and kinetic studies with various blood/plasma proteins as substrates (Kotb 2013). This could further be substantiated by kinetic studies, wherein parameters such as the Michaelis constant (Km), rate of reaction (Vmax) and the turn over number (Kcat) of the test enzyme for various substrates could be evaluated. These parameters would help to determine the affinity and specificity of the test enzyme for a particular substrate. Amidolytic assays involve synthetic peptides (specific peptides for a particular substrate), which are mostly tripeptides or quadrapeptides linked via an amide bond to a fluorophore/chromophore such as para-nitroaniline (pNA). This pNA is later cleaved at the C-terminal amide bond, liberating the fluorophore/chromophore, the fluorescence/absorbance of which can be measured spectrophotometrically. Amidolytic activity is expressed as the amount of p-nitroaniline released per minute by the enzyme. This assay is commonly carried out to evaluate the most sensitive/specific substrate for the test enzyme (Kotb 2013).
Anticoagulant and thrombolytic activity
This assay involves the visualisation of the anticoagulant and thrombolytic ability of a test compound. Anticoagulant action of a particular compound could be defined as the ability to prevent the formation of blood clot. On the other hand, thrombolytic action of a compound would indicate its ability to dissolve an already existing blood clot. It is essential that these aspects be addressed while evaluating the therapeutic potential of a probable drug candidate. According to this assay, fresh blood is collected and incubated with the test compound, along with a negative (saline) and positive control (such as streptokinase) respectively. The mixture is then allowed to stand at physiological temperature (37 °C) for a fixed time interval, and the clot formation and its subsequent dissolution is observed (Lu et al. 2010; Ju et al. 2012).
In addition to the mere visualisation, the percentage of clot lysis can also be determined based on the weight difference between formed clot versus lysed clot (Prasad et al. 2006; Narasimhan et al. 2015). This would indicate the anticoagulant nature of the test compound. On the other hand, to examine the thrombolytic property of a potential drug candidate, fresh blood could be replaced by pre-formed blood clot with the other steps maintained the same as above. The ease of performing this assay can be considered as a major advantage when compared to the other assays described in this report. However, it should be noted that to strengthen the candidature of a potential drug, performing this assay alone will not suffice and hence a combination of assay as described in this report must be adopted.
Clot formation and lysis assay (CloFAL)
It is a global assay of haemostasis widely used in the management of thrombosis and thrombophilia via quantitative measurement of clot lysis periodically. This assay is used clinically to evaluate the net haemostatic balance which is unique to an individual at any given time (Goldenberg et al. 2008). It was reported to be used as a pre-operative screening test in which the clot formation as well as fibrinolysis would be measured simultaneously. In addition, it was used to evaluate both clotting and bleeding disorders in children and adults (Chitlur and Simpson 2015).
In this assay, formation of fibrin clot and the subsequent dissolution of the fibrin clot formed is determined by turbidity measurements (Goldenberg et al. 2008). The assay is performed using platelet poor plasma (PPP), to which either calcium or tissue factor or phospholipid is added to initiate the process of coagulation, while tPA is added to begin fibrinolysis. tPA could be replaced by the test compound for which the fibrinolytic activity has to be measured. The absorbance of the resulting mixture is read at dual wavelength of 405 and 605 nm for a period of 3 h using a spectrophotometer. Here, increase in turbidity would indicate the increase in the aggregation of fibrin fibres, which in turn represents the coagulation process. This phase would be followed eventually by a decrease in the turbidity, thereby representing the fibrinolytic ability of the test compound. A waveform is generated describing the coagulation and fibrinolysis with information regarding the coagulation index, fibrinolytic index and maximum amplitude (Peng and Zhao 2009).
This assay has been used as a rapid adjunctive test for the routine measurement of factor VIII (anti-hemophilic factor) (AHF)/factor IX (Christmas factor) activity in the treatment of haemophilia A/B. Major advantage of this assay is its sensitivity to the components of coagulation and fibrinolytic systems and also to physiological changes in haemostasis. However, the demerits of this assay includes its reduced sensitivity in the presence of platelet poor plasma and its incapability to detect mild deficiency in factor VIII (Chitlur and Simpson 2015).
Euglobulin clot lysis assay
This assay helps in measuring the fibrinolysis within the euglobulin fraction. Addition of acetic acid to PPP leads to precipitation of certain clotting factors, called the euglobulin fraction. Typically, this euglobulin fraction comprises of plasminogen, tissue plasminogen activators, AHF and fibrinogen. The inhibitors of fibrinolysis such as anti-activators and anti-plasmins remain in the supernatant (Nordby et al. 1980).
Venous blood is collected into pre-chilled tubes containing an anticoagulant which is centrifuged to obtain the PPP. The PPP is then acidified by adding acetic acid to precipitate euglobulin fraction, and the supernatant is discarded. The euglobulin fraction is resuspended in borate solution, and clotting is initiated by the addition of thrombin or calcium chloride at 37 °C and observed for complete clot lysis upon addition of the test compound; this is referred to as the euglobulin lysis time (Nordby et al. 1980). In the recent times, this assay has been modified using a computerised kinetic spectrophotometric microtitre plate reader, wherein the absorbance of the recalcified euglobulin fraction over time is determined (Smith et al. 2003).
The primary advantage of this assay is that it is a rapid and sensitive method to detect the fibrinolysis after venous stasis (Nordby et al. 1980). Although this assay measures fibrinolysis alone, the major drawback is that it is affected by the activator content, levels of inhibitors, the fibrinogen and plasminogen content present in the sample (Lisman et al. 2005). This assay result is affected during pregnancy and in medical condition such as liver diseases where increase in plasminogen, fibrinogen and plasminogen inhibitor content is observed. During pregnancy, the clot lysis time is found to be reduced. The euglobulin clot lysis time also depends on the factor XII deficiency as the fibrin clot formed would be poorly cross-linked and its dissolution would be rapid. Additionally, this test also depends on the fibrinogen concentration, as fibrinogen levels below normal will lead to formation of a smaller clot which in turn leads to lesser time of lysis. Whereas if the fibrinogen concentration is above 600 mg/dL, the fibrin clot formed provides excessive substrate for the plasmin formed, subsequently resulting in increased clot lysis time (Johns and Cohen 2003). Thus, the euglobulin lysis time is dependent on the composition of blood sample taken for assay.
Anti-platelet aggregation activity
Anti-platelet aggregation assay is used to evaluate the ability of the test compound to inhibit or reduce the aggregation of platelets. Platelet aggregation is an important phenomenon witnessed during the formation of a blood clot. On the other hand, anti-platelet aggregation is a process which involves the decrease in platelet aggregation, thereby inhibiting the thrombus (blood clot) formation. It mainly functions by interfering with the platelet activation step in the primary haemostasis. This assay involves the passage of light through a stirred turbid suspension of platelets. Upon addition of an aggregatory agent, the platelets aggregate and clump together reducing the amount of light being scattered, thereby reducing the optical density of the suspension. The amount of transmitted light recorded gives a measure of the platelet suspension (Born 1962). This assay when performed in combination with other thrombolytic and anticoagulant assays could broaden the use of the test compound in the clinical field. Han et al. (2005) had demonstrated the anticoagulation property along with the anti-platelet aggregation activity for the polysaccharide produced by a marine fungus Keissleriella sp. YS4108. It has been quoted by Farrell et al. (1992) that platelet aggregation is mediated by the binding of plasma protein fibrinogen to a platelet cell surface receptor, when the required recognition sequence is present on the fibrinogen ligand.
The assay is performed by incubating the test compound with platelet-rich plasma (PRP) at 37 °C for a certain time interval, followed by the addition of an aggregatory agent. The sample is then analysed through a turbidometer, the change in turbidity is recorded and percentage aggregation is calculated. Usually, heparin is used as the reference standard (Born 1962). The main advantage of this assay is its simplicity and ease of use.
However, this assay suffers from certain shortcomings such as (1) the platelet function in vitro does not reflect the platelet function in vivo at all times; (2) sample ageing occurs due to the time required for the PRP preparation which leads to variation in the result obtained; (3) as it is a turbidometric assay, substances such as lipids present in the PRP or PPP can alter the absorbance; and (4) since the platelets are heterogeneous in size and density, it is likely that some of them could be lost during the preparation of PRP (Gresele et al. 2007). Some of the clinically approved anti-platelet agents are aspirin, clopidogrel, prasugrel and also certain anticoagulants such as heparin (Xiang et al. 2008). Nevertheless, it has been reported that most of these drugs exhibit irreversible inhibition which raises concern with regard to their safety and efficacy. Aspirin, clopidogrel and prasugrel solely inhibit the activation of platelet generated by TXA2 and ADP respectively. This single target and/or irreversible nature results in excessive bleeding complications (Hsieh et al. 2007; Fabre and Gurney 2010). Other limitations associated with these drugs are drug-drug interaction, anti-platelet resistance and affordability (Xiang et al. 2008).
Thromboelastography
Thromboelastography (TEG) is performed to monitor clot development, clot stabilisation and its dissolution, representing in vivo haemostasis. It was first described by Hartert (1948). Thromboelastography assay involves the measurement of the viscoelastic properties of the whole blood clot under shear stress (Artang et al. 2014). The TEG equipment comprises of a small plastic cup and a pin suspended by a torsion wire connected to a transducer. The clot formation occurs between the cup and the pin; it generates torque on the wire which is measured and translated into the time taken for the clot formation to begin, total time taken for the clot to reach up to a specified level of strength, time taken for the fibrin to build up and the integrity of the clot. Based upon the clot formed, if any, viscoelastic properties of the clot and clot lysing potential of the test compound are evaluated.
The blood sample, with and without the test compound, is collected and placed in the cup, which is kept at 37 °C, and the pin is lowered in to the sample (Fig. 1). The cup then oscillates through a 4°45 arc with each oscillation angle lasting for a certain time. Depending upon the clot formed, torque generated on the wire is measured and translated into several parameters as mentioned above (Donahue and Otto 2005).

Thromboelastography mechanism (modified from Donahue and Otto 2005)
This assay has been reported to be advantageous due to its sensitivity to the changes in the fibrin polymerization and platelet count, the two most vital parameters responsible for clot fprmation (Bolliger et al. 2012). Compared to the currently available diagnostic tests such as D-dimer, TEG could provide information on identifying the unpredictable changes in coagulability which occurs in conditions such as sepsis and disseminated intravascular coagulation (Donahue and Otto 2005). In addition, TEG determines the level of functioning of platelets effectively which is more important than knowing the total number of available platelets. It is shown to be more reliable diagnostic tool for monitoring postoperative hypercoagulability and also monitor the patients receiving anticoagulant therapy (Mahla et al. 2001). The demerits of this assay include the high cost of the equipment, in case of using the whole blood. It is recommended that to get reliable results, the delay between the drawing of blood and testing of sample must be minimised (Donahue and Otto 2005).
In vivo assays
In order to understand the pathophysiology of thrombosis, use of animal models becomes pivotal. In vivo studies help in comprehending the entire process of initiation, propagation and resolution of thrombosis. It also serves as a platform to evaluate novel therapeutic agents for prophylaxis and treatment of thrombus-related conditions (Diaz et al. 2012). The in vivo models discussed in this review are based upon initiation of thrombus using different approaches except the d-dimer test, where fibrinolysis is measured indirectly.
d-Dimer test
d-Dimer assay serves as a tool in monitoring fibrinolysis with the help of cross-linked d-dimer (XDP) domains that are present in the fibrin derivatives. The final step in the coagulation cascade involves the action of thrombin in converting fibrinogen to fibrin monomers. These fibrin monomers self-assemble into fibres and get covalently cross-linked at the outer 2D domains of the adjacent fibrin monomers and the central E domain of the third fibrin monomer. Later, the enzyme plasmin cleaves the cross-linked fibrin polymer at various sites resulting in the release of d-dimer fragments (XDPs) (Olson et al. 2013). Some of the commercially available assays used to detect these XDPs are latex agglutination, immunoturbidimetry and enzyme-linked immunosorbent assay (ELISA).
This assay is commonly performed before and after treatment with the test compound. Blood samples are withdrawn, and XDPs released are detected by immunoassays consisting of monoclonal antibodies specific for d-dimer domain in the fibrin molecule. The presence of d-dimers indicates the occurrence of fibrinolysis due to the plasmin-like action of the test compound. The d-dimer is usually reported as mass units, and there are two different types of units: the fibrinogen equivalent unit (FEU) at 340 kDa and the d-dimer unit (DDU) at 195 kDa (Olson et al. 2013).
The applicability of the d-dimer test helps in determining the duration of the anticoagulation therapy serves as an advantage (Olson et al. 2013). During medical conditions such as arterial, venous thrombosis and pulmonary embolism, the d-dimer levels are elevated (Mavromatis and Kessler 2001; Wakai et al. 2003; Adam et al. 2009). However, few of the drawbacks associated with this assay are (i) its variation with respect to the types of d-dimer unit that differ with onefold difference in mass and the magnitude of units reported clinically leads to confusion, particularly when this assay is used to exclude conditions such as venous thromboembolism; (ii) this assay is not applicable for fibrinogenolysis, as the derivatives of fibrinogen degradation lack d-domain despite being present in high concentration; and (iii) it has also been reported that the various quantitative d-dimer assays currently in use vary in their reproducibility (Olson et al. 2013).
Ferric chloride-induced thrombosis model
It is one of the most widely used model to evaluate the efficacy of anticoagulant and anti-platelet drugs in vivo (Eckly et al. 2011; Yuan et al. 2012; Surin et al. 2009). This involves the application of ferric chloride topically to an intact vessel, leading to vascular wall injury (Eckly et al. 2011), endothelial dysfunction, platelet activation and eventually an intravascular thrombus formation (Yuan et al. 2012). The ability of the test compound to prevent the clot/thrombus formation is evaluated in this assay.
The test compound is injected intraperitoneally prior to the application of ferric chloride. Upon application of ferric chloride solution to the intact vessel such as the carotid artery, the blood flow is continuously monitored with the help of a Doppler flowmeter, which reflects the occlusion time in the carotid artery. Alternatively, it can also be observed under intravital microscopy. The measurable parameter in this model is the time elapsed from initial injury to entire vessel destruction (Li et al. 2013).
The merits of this assay are as follows: (i) the thrombus formation observed in this case has been reported to be closest to the human pathological condition (Yuan et al. 2012) and (ii) a distinctive feature of this model is its sensitivity to various thrombin inhibitors (Eckly et al. 2011). Several in vitro experiments performed indicate that ferric chloride alters the ability of adhesive proteins such as fibrinogen, collagen and von Willebrand factor. Additionally, studies suggest that ferric chloride results in generation of reactive oxygen species which lead to lipid peroxidation, followed by endothelial cell destruction forming a thrombus (Eckly et al. 2011). The only drawback of this method is that the transmural vein wall injury mimics only a smaller number of deep vein thrombosis (DVT) cases clinically and hence must be combined with other assays to diagnose/treat DVT (Diaz et al. 2012).
Carrageenan-induced thrombosis model
This model has been extensively used to test the effect of several anti-thrombus and thrombolytic agents clinically (Yuan et al. 2012; Yan et al. 2009; Kamiya et al. 2010; Simkhada et al. 2012). Carrageenan is a linear sulphur containing polysaccharide, extracted from edible red seaweeds. They comprise of repeating units of galactose and anhydrogalactose (both sulphated and nonsulpahted) adjoined by glycosidic linkages. Commercially, there are three major classes of carrageenan available, kappa, iota and lambda carrageenan, differing in the number and position of the sulphur groups on the galactose units (Barbeyron et al. 2000). However, kappa-carrageenans were reported to be the most potent thrombogen (Bekemeier et al. 1985). As shown in Fig. 2, the disappearance of the wine-coloured thrombus due to thrombolytic action of the test drug is monitored. The underlying mechanism of action of the carrageenan-induced thrombosis is believed to be primarily via the activation of Hageman factor (An and Freidman 1998) followed by intravasal coagulation (Eckly et al. 2011). Furthermore, histological studies demonstrate that the occurrence of thrombocyte aggregates and leukostasis trigger disseminated intravasal coagulation (Yan et al. 2009).

Carrageenan-induced thrombosis model
In this assay, the mice are intravenously injected with sterile carrageenan dissolved in physiological saline. Suitable dosage required to cause the thrombus formation is determined. This is followed by immersing the tails in ice-water bath and observing the appearance of wine-coloured thrombus in tail (Fig. 2). The test compound is injected intraperitoneally prior to carrageenan injection while using a standard commercial fibrinolytic agent as positive control and saline as negative control (Yan et al. 2009).
The advantages of using this model lies in its simplicity and reproducibility with continuous measurement of thrombus range and extent without sacrificing the animal (Yan et al. 2009).
Rat groin flap model
This model serves to be a significant tool to assess and compare the effects of various anticoagulants in addition to vasomotor drugs (Wallmichrath et al. 2014). The single, microvascular pedicle present in the flap facilitates several experimental procedures to be performed (Tan and Green 1987). A laser Doppler perfusion monitoring system (LDPM) is employed to monitor the blood flow in the feeding artery and flap microcirculation.
A laser Doppler probe is used commonly for skin measurements. It is affixed to the flap along with an invasive needle probe to record artery blood perfusion, corpuscular movements and thereby monitor the entire vessel lumen. The vessel wall is stimulated using a bipolar stimulating electrode, with a systematic measuring of the blood pressure with a pressure transducer. Care is taken to avoid stimulation-induced artery contraction. Once blood perfusion is stabilised, the epigastric artery proximal to the laser Doppler flowmeter (LDF) reading area is stimulated until thrombus formation is observed. The test compound should be infused through the jugular artery 5 min prior to the stimulation. Following which, using LDPM measurements and a video camera system, the course of thrombus formation is observed. If the test compound has an anticoagulant action, thrombus formation as a result of stimulation induced by the electrode would be prevented. Additionally, stimulated sections of the flap artery could be processed and microscopically examined at various time intervals. Statistical approaches can be followed to determine or compare the percentage changes of blood flow (Peng and Zhao 2009).
The only disadvantage is the short length of the pedicle (Acton 2011). It has been reported that the use of antithrombotic agents as a prophylactic measure is the most common strategy adopted to avoid vascular thrombosis resulting from a free flap surgery (Peng and Zhao 2009).
Other models based on thrombus formation
Thrombus formation can also be induced by certain alternative approaches such as phytochemical injury or laser-induced injury. The thrombolytic ability of the test compound is evaluated. Phytochemical injury is generated using a photosensitizing dye such as rose bengal, which produces reactive oxygen intermediates, causing photo excitation, leading to oxidative damage of the vessel wall and subsequent thrombus formation. The test compound is injected prior to the thrombus formation. The dye is then either injected or applied topically and remains inert until it is exposed to 543-nm wavelength light, as there is production of singlet oxygen radicals at the target site (Westrick et al. 2007). This dye is particularly effective for in vivo studies due to its increased phytochemical efficacy and lower systemic toxicity which serves as ab advantage.
In laser-induced injury, thrombosis is induced through heat damage to a defined region of the endothelium, causing morphologic change to the vessel wall. Usually, the test compound is administered prior to the thrombus formation. It has some advantage over the ferric chloride-induced thrombosis model. The endothelial damage caused by former is limited in contrast to the complete denudation of endothelium seen in latter (Furie and Furie 2005).
By and large these agents should decrease the platelet function, reduce blood viscosity and counterwork the thrombin activity on fibrinogen and platelets, thereby increasing the blood flow. Although thrombolytic agents such as streptokinase, urokinase and tPA have been proven to reverse the microvascular thrombosis in the animal models, reports suggesting their efficacy with human subjects are scanty (Askari et al. 2006). Rooks et al. (1994) had observed that the use of urokinase on an established thrombus showed a significant improvement in comparison with heparin or dextran.
A brief summary of the various assays described in the manuscript has been provided in Table 2.
Conclusions
Despite the availability of several assays to evaluate the activity of the microbial enzymes in vitro and in vivo, their use has been limited to just a handful of reports. Most pertinent question is why these methods are not adopted widely? In vitro and in vivo assays although available in plenty, not a single assay completely resembles the pathophysiology of thrombosis in humans. Therefore, it is best that a combination of several assays are to be used to evaluate any new compound of microbial origin, for a clear clinical relevance and pharmacological effectiveness. Albeit the bottlenecks, these assays could be yet another step closer to studying thrombosis and thereby providing a holistic approach in tackling this disease in humans. The specificity of the test compound towards a target molecule (in the blood) is an important factor that needs to be considered. This could be addressed by carrying out kinetic assays in combination with the other assays as discussed in the review. Global in vitro assays such as CloFAL and TEG when performed together, in the presence of the test compound, could give a complete picture on the net haemostatic balance, time taken for the clot formation to occur, integrity of the clot formed and also the level of functioning of platelets. Furthermore, if the test compound shows satisfactory results, it could be applied to any one of the in vivo animal models described in the review. Doing so would help in a better understanding of the role of test compound in alleviating pathophysiology of the disease and would help in determining whether the candidate drug has scientific merit to justify further development. It would also provide information on the mechanism of action of the test compound, as to whether it is a fibrinolytic- or thrombolytic-based compound. Based on the results obtained from these assays, test compound exhibiting a desired pharmacological effect should be subjected to clinical trials involving human volunteers.
Abbreviations
- ADP:
-
Adenosine diphosphate
- AHF:
-
Factor VIII, anti-hemophilic factor
- CloFAL:
-
Clot formation and lysis assay
- DDU:
-
d-Dimer unit
- DVT:
-
Deep vein thrombosis
- ELISA:
-
Enzyme-linked immunosorbant assay
- Factor IX:
-
Christmas factor
- FEU:
-
Fibrinogen equivalent unit
- INR:
-
International normalised ratio
- K cat :
-
Turn over number
- K m :
-
Michaelis constant
- LDF:
-
Laser Doppler flowmeter
- LDPM:
-
Laser Doppler perfusion monitoring system
- OAC:
-
Oral anticoagulant
- pNA:
-
Para-nitroaniline
- PPP:
-
Platelet poor plasma
- PRP:
-
Platelet-rich plasma
- rDNA:
-
Recombinant deoxyribonucleic acid technology
- TEG:
-
Thromboelastography
- tPA:
-
Tissue plasminogen activator
- TXA2:
-
Thromboxane A2
- V max :
-
Maximum rate of reaction
- XDPs:
-
d-Dimer fragments
References
Acton QA. Issues in surgical research, techniques and innovation, reconstructive microsurgery. Georgia: Scholarly Editions; 2011.
Adam SS, Key NS, Greenberg CS. D-dimer antigen: current concepts and future prospects. Blood. 2009;113:2878–88.
An YH, Freidman RJ. Animal models in orthopedic research. Washington DC: CRC Press; 1998.
Artang R, Galloway G, Nielsen JD. Monitoring novel anticoagulants dabigatran, rivoroxaban and apixaban using thromboelastography. J Am Coll Cardiol. 2014;63:A439. https://doi.org/10.1016/S0735-1097(14)60439-3.
Askari M, Fisher C, Weniger FG, Sean B, Lee WPA. Anticoagulation therapy in microsurgery: a review. J Hand Surg. 2006;31:836–46.
Barbeyron T, Michel G, Potin P, Henrissat B, Kloareg B. ɩ-Carrageenases constitute a novel family of glycoside hydrolases, unrelated to that of κ-carrageenases. J Biol Chem. 2000;275:35499–505.
Barta G. Dyed fibrin plate assay of fibrinolysis. Can J Physiol Pharmacol. 1966;44(2):233–40.
Bauer K. Recent progress in anticoagulant therapy: oral direct inhibitors of thrombin and factor Xa. J Thromb Haemost. 2011;9:12–9.
Bekemeier H, Hirschelmann R, Giessler AJ. Carrageenin-induced thrombosis in rats and mice: a model for testing antithrombotic substances? Agents Actions. 1985;16:446–51.
Blann AD, Landray MJ, Lip GYH. ABC of antithrombotic therapy: an overview of antithrombotic therapy. BMJ. 2002;325:762–5.
Bolliger D, Seeberger MD, Tanaka KA. Principles and practice of thromboelastography in clinical coagulation management and transfusion practice. Transfus Med Rev. 2012;26:1–13.
Born GVR. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194:927–9.
Chitlur M, Simpson ML. Pediatric thrombotic disorders. In: Goldenberg NA, Manco-Johnson MJ, editors. Role of global assays in thrombosis and thrombophilia. United Kingdom: Cambridge University Press; 2015. p. 142–57.
Diaz JA, Obi AT, Myers Jr DD, Wrobleski SK, Henke PK, Mackman N, Wakefield TW. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:556–62.
Donahue SM, Otto CM. Thromboelastography—a tool for measuring hypercoagulability, hypocoagulabillity and fibrinolysis. J Vet Emerg Crit Care. 2005;15:9–16.
Eckly A, Hechler B, Freund M, Zerr M, Cazenave P, Lanza F, Mangin PH, Gachet C. Mechanisms underlying FeCl3-induced arterial thrombosis. J Thromb Haemost. 2011;9:779–89.
Fabre J, Gurney ME. Limitations of current therapies to prevent thrombosis: a need for novel strategies. Mol BioSyst. 2010;6:305–15.
Farrell DH, Thiagarajan P, Chung DW, Davie EW. Role of fibrinogen α and ɣ chain sites in platelet aggregation. Proc Natl Acad Sci U S A. 1992;89:10729–32.
Fattah M, Shaheen M, Mahfouz M. Disturbances of haemostasis in diabetes mellitus. Dis Markers. 2004;19:251–8.
Fossum S, Hoem NO. Urokinase and non-urokinase fibrinolytic activity in protease-inhibitor-deprived plasma, assayed by a fibrin micro-plate method. Immuno Pharmacol. 1996;32:119–21.
Freedman. Oral anticoagulants: pharmacodynamics, clinical indications and adverse effects. J Clin Pharmacol. 1992;32:196–209.
Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–62.
Gallimore M, Friberger P. Chromogenic peptide substrate assays and their clinical applications. Blood Rev. 1991;5:117–27.
Gidron E, Margalit R, Shalitin Y. A rapid screening test for reduced fibrinolytic activity of plasma: streptokinase activated lysis time. J Clin Path. 1978;31:54–7.
Goldenberg NA, Manco-Johnson MJ, Jacobson L, Hathaway WE (2008) Methods for a global assay of coagulation and fibrinolysis. US Patent 20080268483 A1.
Gonsalves WI, Pruthi RK, Patnaik MM. The new oral anticoagulants in clinial practice. Mayo Clin Proc. 2013;88:495–511.
Gresele P, Fuster V, Lopez J, Page C, Vermylen J. Platelets in hematologic and cardiovascular disorders: a clinical handbook. In: Shantsila E, Watson T, GYH L, editors. Laboratory investigation of platelets. United Kingdom: Cambridge University Press; 2007. p. 129.
Han F, Yao W, Yang X, Liu X. Experimental study on anticoagulant and antiplatelet aggregation activity of a chemically sulfated marine polysaccharide YCP. Int J Biol Macromol. 2005;36:201–7.
Hartert H. Blutgerinnungsstudien mit der thrombelastographie, einem neuen untersuchungsverfahren. Klin Wochenschr. 1948;26:577–83.
Hawkey CM, Stafford JL. A standard clot method for the assay of plasminogen activators, anti-activators, and plasmin. J Clin Pathol. 1964;17:175.
Howell M. A method for assessing clot lysis. J Clin Pathol. 1964;17:310.
Hsieh PW, Hwang TL, Wu CC, Chiang SZ, Wu CI, Wu YC. The evaluation and structure-activity relationships of 2-benzoylaminobenzoic esters and their analogues as anti-inflammatory and antiplatelet aggregation agents. Bioorg Med Chem Lett. 2007;17:1812–7.
Jespersen J, Astrup T. A study of the fibrin plate assay of fibrinolytic agents. Optimal conditions, reproducibility and precision. Haemostasis. 1983;13:301–15.
Johns DG, Cohen RA. Thrombosis and haemorrhage. In: Loscalzo J, Schafer AI, editors. Laboratory methods in haemostasis. Philadelphia: Lippincott Williams & Wilkins; 2003. p. 394.
Ju X, Cao X, Sun Y, Wang Z, Cao C, Liu J, Jiang J. Purification and characterisation of a fibrinolytic enzyme from Streptomyces sp. XZNUM 00004. World J Microbiol Biotechnol. 2012;28:2479–86.
Kamiya S, Hagimori M, Ogasawara M, Arakawa M. In vivo evaluation method of the effect of nattokinase on carrageenan-induced tail thrombosis in a rat model. Acta Haematol. 2010;124:218–24.
Kotb E. Activity assessment of microbial fibrinolytic enzymes. Appl Microbiol Biotechnol. 2013;97:6647–65.
Li W, McIntyre TM, Silverstein RL. Ferric chloride-induced murine carotid arterial injury: a model of redox pathology. Redox Biol. 2013;1:50–5.
Lip G, Andreotti F, Fauchier L, Huber K, Hylek E, Knight E, Lane D, Levi M, Marin F, Palareti G, Kirchhof P. Bleeding risk assessment and management in atrial fibrillation patients. Thromb Haemost. 2011;106:997–1011.
Lisman T, de Groot PG, Meijers CMJ, Rosendaal FR. Reduced plasma fibrinolytic potential is a risk factor for venous thrombosis. Blood. 2005;29:566–70.
Longstaff C, Thelwell C. Understanding the enzymology of fibrinolysis and improving thrombolytic therapy. FEBS Lett. 2005;579:3303–9.
Lu F, Lu Z, Bie X, Yao Z, Wang Y, Lu Y, Guo Y. Purification and characterisation of a novel anticoagulant and fibrinolytic enzyme produced by endophytic bacterium Paenibacillus polymyxa EJS-3. Thromb Res. 2010;126:349–55.
Mahla E, Lang T, Vicenzi MN, Werkgartner G, Maier R, Probst C, Metzler H. Thromboelastography for monitoring prolonged hypercoagulability after major abdominal surgery. Anesth Analg. 2001;92:572–7.
Marieb EN, Hoehn K. Human anatomy and physiology. 8th ed. San Francisco: Benjamin Cummings; 2010.
Marsh NA, Gaffney NJ. The rapid fibrin plate—a method for plasminogen activator assay. Thromb Haemostat. 1977;38:545–51.
Mavromatis BH, Kessler CM. D-Dimer testing: the role of the clinical laboratory in the diagnosis of pulmonary embolism. J Clin Pathol. 2001;54:664–8.
Miesbach W, Seifried E. New direct oral anticoagulants-current therapeutic options and treatment recommendations for bleeding complications. Thromb Haemost. 2012;108:625–32.
Millar WT, Smith JF. The comparison of solid phase and fibrin plate methods for the measurement of plasminogen activators. Thromb Res. 1983;30:431–9.
Narasimhan MK, Chandrasekaran M, Rajesh M. Fibrinolytic enzyme production by newly isolated Bacillus cereus SRM-001 with enhanced in-vitro clot lysis potential. J Gen Appl Microbiol. 2015;61:157–64.
Nieuwenhuizen W, Schielen WJG, Yonekawa O, Tesser GI, Voskuilen M. Fibrinogen, thrombosis, coagulation, and fibrinolysis. In: Liu CY, Chien S, editors. Studies on the localisation and accessibility of sites in fibrin which are involved in the acceleration of the activation of plasminogen by tissue-type plasminogen activator. New York: Springer Science and Business Media; 2012. p. 83–91.
Nordby E, Arnesen H, Andersen P, Godal HC. The euglobulin clot lysis time, a rapid and sensitive method for the assay of fibrinolytic activity after venous stasis. Scand J Haematol. 1980;25:407–11.
Olson JD, Cunningham MT, Higgins RA, Eby CS, Brandt JT. D-dimer: simple test, tough problems. Arch Pathol Lab Med. 2013;137:1030–8.
Peng S, Zhao M. Pharmaceutical bioassays: methods and applications, methods and applications of thrombus-related assays. New Jersey: Wiley; 2009. p. 59.
Peng Y, Yang X, Zhang Y. Microbial fibrinolytic enzymes: an overview of source, production, properties, and thrombolytic activity in vivo. Appl Microbiol Biotechnol. 2005;69:126–32.
Porrett PM, Drebin JA, Atluri P, Karakousis GC, Roses RE. The surgical review and subtitle: an integrated basic and clinical science study guide. Philadelphia: Williams & Wilkins; 2005.
Prasad S, Kashyap RS, Deopujari JY, Purohit HJ, Taori GM, Daginawala HF. Development of an in vitro model to study clot lysis activity of thrombolytic drugs. Thromb J. 2006;4:14.
Rooks MD, Rodriguez Jr J, Blechner M, Zusmanis K, Hutton W. Comparative study of intraarterial and intravenous anticoagulants in microvascular anastomoses. Microsurgery. 1994;15:123–9.
Simkhada JR, Cho SS, Mander P, Choi YH, Yoo JC. Purification, biochemical properties and antithrombotic effect of a novel Streptomyces enzyme on carrageenan-induced mice tail thrombosis model. Thromb Res. 2012;129:176–82.
Smith AA, Jacobson LJ, Miller BI, Hathaway WE, Manco-Johnson MJ. A new euglobulin clot lysis assay for global fibrinolysis. Thromb Res. 2003;112:329–37.
Sumi H, Hamada H, Nakanishi K, Hiratani H. Enhancement of the fibrinolytic activity in plasma by oral administration of nattokinase. Acta Haematol. 1990;84:139–43.
Sumi H, Hamada H, Tsushima H, Mihara H, Muraki H. A novel fibrinolytic enzyme (nattokinase) in the vegetable cheese Natto; a typical and popular soybean food in the Japanese diet. Experientia. 1987;43:1110–1.
Surin WR, Prakash P, Barthwal MK, Dikshit M. Optimisation of ferric chloride induced thrombosis model in rats: effect of anti-platelet and anti-coagulant drugs. J Pharmacol Toxicol Methods. 2009;61:287–91.
Tan WT, Green CJ. The rat groin flap as an experimental model in microsurgery. Ann Acad Med Singap. 1987;16:170–4.
Turpie AGG, Chin BSP, Lip GYH. Venous thromboembolism: pathophysiology, clinical features, and prevention. BMJ. 2002;325:887–90.
Verstraete M. Third-generation thrombolytic drugs. Am J Med. 2000;109:52–8.
Wakai A, Gleeson A, Winter D. Role of fibrin D-dimer testing in emergency medicine. Emerg Med J. 2003;20:319–26.
Wallmichrath J, Baumeister RG, Gottschalk O, Giunta RE, Frick A. The free groin flap in the rat: a model for improving microsurgical skills and for microvascular perfusion studies. J Plast Surg Hand Surg. 2014;48:191–6.
Wang CT, Ji BP, Nout BLR, Li PL, Ji H, Chen LF. Purification and characterisation of a fibrinolytic enzyme of Bacillus subtilis DC33, isolated from Chinese traditional Douchi. J Ind Microbiol Biotechnol. 2006;33:750–8.
Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2079–93.
Xiang YZ, Kang LY, Gao XM, Shang HC, Zhang JH, Zhang BL. Strategies for antiplatelet targets and agents. Thromb Res. 2008;123:35–49.
Yan F, Yan J, Sun W, Yao L, Wang J, Qi Y, Xu H. Thrombolytic effect of Subtilisin QK on carrageenan induced thrombosis model in mice. J Thromb Thrombolysis. 2009;28:444–8.
Yogesh D, Halami PM. A fibrin degrading serine metallo protease of Bacillus circulans with α-chain specificity. Food Biosci. 2015;11:72–8.
Yuan J, Yang J, Zhuang Z, Yang Y, Lin L, Wang S. Thrombolytic effects of Douchi fibrinolytic enzyme from Bacillus subtilis LD-8547 in vitro and in vivo. BMC Biotechnol. 2012;12:1–9.
Zdanowicz MM. Essentials of pathophysiology for pharmacy. Florida: CRC Press; 2002.
Acknowledgements
Anusha Krishnamurthy thanks the National Institute of Technology Karnataka (N.I.T.K.), India, for providing a fellowship to pursue doctoral research work.
Funding
No funding sources to declare.
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
AK prepared the manuscript under the guidance of PDB. PDB corrected and edited the manuscript. SBS corrected the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
AK is a doctoral student working under the guidance of PDB on “Fibrinolytic enzyme production from marine Serratia marcescens subsp. Sakuensis”. PDB is an Assistant professor in Chemical Engineering department, National Institute of Technology Karnataka. His research interest includes production of therapeutic enzymes, entomopathogenic nematodes and probiotic organisms and storage stability of edible oils. SBS is an assistant professor in Department of Physiology, United Arab Emirates University. His research interest includes gastrointestinal physiology and intestine electrical activity.
Ethics approval
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krishnamurthy, A., Belur, P.D. & Subramanya, S.B. Methods available to assess therapeutic potential of fibrinolytic enzymes of microbial origin: a review. J Anal Sci Technol 9, 10 (2018). https://doi.org/10.1186/s40543-018-0143-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-018-0143-3