
Abstract
Background
Five affected individuals with syndromic tremulous dystonia, spasticity, and white matter disease from a consanguineous extended family covering a period of over 24 years are presented. A positional cloning approach utilizing genome-wide linkage, homozygozity mapping and whole exome sequencing was used for genetic characterization. The impact of a calmodulin-binding transcription activator 2, (CAMTA2) isoform 2, hypomorphic mutation on mRNA and protein abundance was studied using fluorescent reporter expression cassettes. Human brain sub-region cDNA libraries were used to study the expression pattern of CAMTA2 transcript variants.
Results
Linkage analysis and homozygozity mapping localized the disease allele to a 2.1 Mb interval on chromosome 17 with a LOD score of 4.58. Whole exome sequencing identified a G>A change in the transcript variant 2 5′UTR of CAMTA2 that was only 6 bases upstream of the translation start site (c.-6G > A) (NM_001171166.1) and segregated with disease in an autosomal recessive manner. Transfection of wild type and mutant 5′UTR-linked fluorescent reporters showed no impact upon mRNA levels but a significant reduction in the protein fluorescent activity implying translation inhibition.
Conclusions
Mutation of CAMTA2 resulting in post-transcriptional inhibition of its own gene activity likely underlies a novel syndromic tremulous dystonia.
Similar content being viewed by others

Background
Currently, classification of dystonia relies upon phenomenology rather than etiology. A natural bifurcation is made based upon whether, with the exception of tremor, it is the sole motor feature (isolated dystonia) or is accompanied by other movement disorders including myoclonus (combined dystonia) [1]. Isolated dystonia may be subdivided into generalized or focal/segmental forms, with combined dystonia further categorized based upon the presence of myoclonus or Parkinsonism [2]. The advent of next generation sequencing has led to rapid expansion of phenotypic and genotypic subsets of syndromes primarily characterized as isolated or combined dystonias with more than 20 loci (DYT1-DYT27; some are redundant) identified to date [2, 3].
Familial-isolated dystonias are predominantly inherited as incompletely penetrant autosomal dominant traits most frequently related to the DYT1 or DYT6 loci with mutations in TOR1A [4, 5] or THAP1 [6, 7], respectively. Additional isolated dystonia loci include DYT23 (CIZ1) [8], DYT24 (ANO3) [9], and DYT25 (GNAL) [10, 11], all of which have recently been extensively reviewed [2]. DYT4 (TUBB4A) may present as an autosomal dominant disorder in adolescence or early adulthood with prominent laryngeal dysphonia and craniocervical, segmental, or generalized dystonia [12, 13]. A rare recessively inherited generalized isolated dystonia results from mutation of PRKRA (DYT16) and is characterized by early onset and frequent association with dystonia-Parkinsonism [2, 14, 15]. Two other recessively inherited isolated dystonias have also been described arising from mutation of DYT2 (HPCA) [16] or DYT27 (COL6A3) [15]. Like these, other dystonia loci such as DYT23 (CIZ1) and DYT24 (ANO3) are yet to be independently confirmed. Dystonic movements are also associated with mutations of ADCY5, identified as the likely cause of a novel movement disorder, familial dyskinesia with facial myokimia (FDFM) [17, 18].
We previously reported a consanguineous extended family having a novel autosomal recessive syndromic tremulous dystonia with spasticity and white matter disease [19]. In this study, we further delineate and genetically characterize the syndrome, identifying CAMTA2 as a novel candidate gene for a syndromic tremulous dystonia, and describe its clinical course and prognosis over a long follow-up period.
Results
The families originated from the Eastern part of the Arabian Peninsula. Five patients were studied (Fig 1a); their current ages ranging from 33 to 46 years. The detailed clinical description was previously reported [19]. In summary, the disease onset was at 7–8 years of age with tremulous movement in fingers and arms that progressed to affect the face, neck, and trunk. Speech became dysarthric and tremulous. The volitional movement was exacerbated by posture and action. Initially, gait was not impaired. Intelligence, personality, and memory were not affected apart from paranoia and depression in two individuals. Patients exhibited a coarse generalized tremor of 4–5 Hz, present at rest, waxing, and waning in amplitude with side-to-side (“no no” type tremor). In addition, there was variable focal or segmental dystonia with retrocollis shoulder elevation and hyperextension of the fingers. There was generalized hyperreflexia with extensor planter response noted around the age of 15 or 16 years. Sensory examination was intact and there was no truncal ataxia. Patients were followed for up to 24 years with no noticeable increase in their movement disorder. The patients were cognitively intact, they were able to finish college, hold independent jobs, get married, and have children. However, all of them exhibited variable degrees of spasticity affecting their legs more than arms (Additional file 1) and two of them required walking aids in their 30’s. Biochemical, organic acid, and lysosomal enzyme studies were normal. Visual and somatosensory evoke potentials showed evidence of prolonged central latencies. Accelerometric recording from the outstretched hand showed 4-Hz tremors. MRI of the brain showed similar abnormalities in all. There was mild symmetrical and confluent white-matter abnormalities sparing the U-fibers and involving the white matter of centrum semiovale, internal capsule, and ventral pons. These findings are not characteristic in any of the classic leukodystrophies. No change was observed in subsequent MRI scans with more than 20 years of follow-up. MR spectroscopy showed no lactate peak or other abnormalities. Patients failed to respond to therapeutic trials of ethanol or anticholinergics. However, some symptomatic benefits were noted with high-dose propranolol, gabapentin, and clonazepam.
Patient video. Patients exhibited a coarse generalized tremor of 4–5 Hz, present at rest, waxing and waning in amplitude with side-to-side (“no no” type tremor). In addition, there was variable focal or segmental dystonia with retrocollis shoulder elevation and hyperextension of the fingers. Patients exhibited generalized hyperreflexia with an extensor planter response being noted. (MOV 39038 kb).

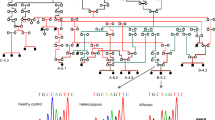
Identification of a dystonia-associated locus on chromosome 17. a Pedigree of an extended family with novel syndromic familial tremulous dystonia. b Genome-wide linkage analysis revealed a maximal peak with a LOD score of 4.58 on chromosome 17. c AutoSNPa output for chromosome 17 reveals an ROH (boxed in red) shared among affected members (IV:1, IV:2, IV:3, IV:7, and IV:8) and not present in unaffected individuals (IV:4, IV:5, IV:6, and IV:9)
Linkage analysis of the nuclear families with three and two affected individuals (Fig. 1a) resulted in independent LOD scores of 2.53 and 2.05, respectively, at rs11651665. Combined analysis (Fig. 1b) localized the disease to a region on chromosome 17 defined proximally by rs743646 and distally by rs34811366 (chr17:4,536,241-6,670,705) with a maximum LOD score of 4.58. Based upon runs of homozygosity shared by all the affected and absent in unaffected individuals (Fig. 1c; red rectangle), we refined the critical interval to a 2.1 Mb region (chr17:4,231,133-6,018,243) containing approximately 60 RefSeq genes (hg19).
Whole exome sequencing of individual IV:7 (Fig. 1a) identified 25,926 variants relative to the hg19 reference sequence, with 1498 variants on chr 17. These were further filtered to exclude all variants present outside our defined ROH (chr 17: 4,231,133-6,018,243) within which we identified 60 variants (Additional file 2). Within the critical interval (chr 17: 4,231,133-6,018,243), 99.42% of bases in the CDS and flanking regions of genes were covered at 30× or better with an average coverage of 425×. By excluding previously reported variants (present in dbSNP with incidence > 0.02; present in 1000 genomes with incidence > 0.02) and retaining those present at a frequency < 2% in more than 1000 Arab exomes held in an in-house database, the list was narrowed to 8. By only focusing on homozygous changes annotated as non-synonymous, splicing variants, frameshift insertions or deletions, and nonsense variants, we decreased the number of candidate variants to 1. It was a G>A change in the 5′UTR of transcript variant 2 of CAMTA2 (c.-6G>A) (NM_001171166.1) (Fig. 2a). This homozygous variant was identified in 56 reads, was confirmed by Sanger sequencing (Fig. 2b), and segregated with disease in the extended family. CAMTA2 has many transcript variants only six (1, 2, 3, 4, 7, and 10) of which are protein coding (https://www.proteinatlas.org/ENSG00000108509-CAMTA2/tissue). The aligned cDNA sequence of the translation start site and adjacent 5′UTR of these six variants show considerable variation (Fig. 3a). The resulting amino terminus amino acid sequences of these transcript variants are aligned and show that the first 12 amino acids of transcript variant 2 are not shared by the other protein coding transcript variants (Fig. 3b). Despite the absence of linkage or homozygozity mapping data to suggest involvement of any known dystonia loci, WES of all affected individuals, parents, and unaffected siblings from both nuclear families was undertaken. There were no variants in any of the known dystonia genes (TOR1A, TUBB4A, THAP1, CIZ1, ANO3, GNAL. SCGE, GCH1, TH, TITF1, TAF1, PRKRA, ATP1A3, SLC63, ADCY5) that survived filtration that removed alleles present in public databases at a frequency > 2%, in any affected individual.

a Filtering strategy used for identification of a causative mutation in the syndromic familial tremulous dystonia patient. b DNA electrophoregram with the G>A change in 5′UTR (chr17:4890930) of the CAMTA2 gene (reverse strand)

a Alignment of the nucleotide sequences of the six protein coding transcript variants of CAMTA2. b Amino acid alignment of the six isoforms of CAMTA2
The wild-type CAMTA2 5′UTR 35 nt translation initiation region was subjected to analysis by a RNAdraw secondary structure program [20]. The structure obtained is a hairpin with three stems that is weakly stable (ΔG=11.1 kcal). The G>A variant 6 bases upstream of the CAMTA2 transcript variant 2 initiation AUG changed this structure to a more relaxed form (ΔG=9.5 kcal) compared to the wild type. More importantly, the mutant variant secondary structure has only two stems and a wider loop as opposed to the three stem-tight loop of the wild type (Fig. 4a).

SNPs nucleotide sequence variation-mediated translational inhibition of CAMTA2. a Predicted secondary structures for wild type and mutant initiation region of CAMTA2. b Schematic diagram showing the wild type and mutant containing 5′UTR reporter constructs. c HEK293 cells (3 × 104 cells/well) in 96-well plates were transfected with 75 ng of purified wild-type and mutant CAMTA2 5′UTR SGFP-expressing constructs along with control RFP expressing constructs (30 ng). After 20 h, fluorescence was quantified using automated image segmentation and quantification as described in the “Methods” section. The GFP/RFP expression ratio of WT and Mut of 5′UTR are shown as mean ± SD (n = 6). ***p < 0.0001 (Student’s t test). d Real-time qRT-PCR for reporter mRNA levels. HEK293 cells were transfected with wild type and mutant CAMTA2 5′UTR reporters for 24 h. Total RNA was extracted, and qRT-PCR was performed using specific primers for SGFP and RFP as described in the “Methods” section. Data are presented as the mean ± SD of two independent experiments. **p = 0.0014 (Student’s t test)
In order to evaluate the effects of the CAMTA2 transcript variant 2 mutation on 5′UTR-linked activities such as transcription and translation, we constructed a fluorescence reporter cassette that contained a 25 nt initiation region upstream of SGFP (Fig. 4b). We subsequently transfected HEK293 cells with the wild type or mutant CAMTA2 transcript variant 2 5′UTR initiation region containing expression cassettes and measured both the mRNA (qRT-PCR) and protein levels (fluorescence). We observed significant reduction in protein (fluorescence levels; (Fig. 4c)) but not mRNA levels of the mutant CAMTA2 transcript variant 2 expression cassette relative to its wild-type counterpart (Fig. 4d).
PCR amplification of each protein coding isoform of CAMTA2 indicated differential expression of isoform 2 which was only detected in the vermis cerebellum, left and right cerebellum, cerebral cortex, olfactory lobe, and cerebellar peduncles. Expression of this variant was not detected in the corpus callosum, hippocampus, occipital, or frontal lobes (Fig. 5a). GAPDH expression was used as an internal control for PCR amplification and specificity for each isoform confirmed by Sanger sequencing of amplicons as shown for transcript variant 2 (Fig. 5b). Other protein coding transcript variants of CAMTA2 (1, 3, 4, 7, and 10) were ubiquitously expressed in most sub-regions of the brain tested (Additional file 3).

Expression of CAMTA2 transcript variant 2 in sub-regions of human brain. a PCR amplification of CAMTA2 transcript variant 2 and GAPDH (housekeeping gene) in (1) vermis cerebella, (2) amygdala, (3) cerebellum left, (4) cerebellum right, (5) cerebral cortex, (6) corpus callosum, (7) hippocampus, (8) occipital lobe, (9) frontal lobe, (10) olfactory lobe, and (11) cerebellar peduncles. b. Sanger sequencing electrophrogram showing cDNA sequence uniquely identifying CAMTA2 transcript variant 2
Discussion
We have described the clinical and genetic characteristics of a novel autosomal recessive familial tremulous dystonia syndrome, characterized by juvenile onset tremulous dystonia with spasticity and white matter disease. Phenotypically, the syndrome was heterogeneous within the families studied with all affected individuals displaying variable forms of both focal or segmental dystonia and coarse, asymmetrical, generalized tremor involving the head, neck and extremities [19]. Isolated dystonia is a classification reserved for exclusive occurrence of dystonia with the exception of tremor, whereas combined dystonia includes additional symptoms such as myoclonus or Parkinsonism by which it is further sub-classified [2].
Disease in the families we report is best characterized as a novel syndromic tremulous dystonia. The phenotype that combines dystonia, tremor, spasticity, and white matter disease clearly defines a novel clinical syndrome [19]. The novel nature of this syndrome is highlighted if not confirmed by identification of a previously undescribed single gene disorder segregating in an autosomal recessive manner within the families studied (LOD score 4.58; chr17:4,536,241-6,670,705). Whole exome and Sanger sequencing identified a homoallelic hypomorphic mutation (c.-6G>A) (NM_001171166.1) just upstream of the translation initiation site in the 5′UTR of CAMTA2 transcript variant 2, with which disease segregated in the family studied. However, given the nature of WES where 100% coverage of the target region is almost never achieved and the coverage of only flanking regions (10–20 bp) of coding sequence in this study, and despite supporting functional evidence, we cannot exclude that a coding point mutation, CNV, deep intronic, or other mutation in the critical interval, in tight linkage disequilibrium with the reported variant, is the true causative mutation.
CAMTA proteins include multiple functional amino acid domains that are evolutionarily conserved including nuclear localization signals, nuclear export signals, a unique DNA binding domain CG-1, TIG-1 involved in protein dimerization, and ANK(ankyrin) repeats involved in protein-protein interactions [21,22,23,24]. CAMTA proteins also include a variable number of IQ motifs consisting of a repetitive sequence IQXXXRGXXX known to be associated with binding of Calmodulin (CaM) and CaM-like proteins. Transcription activation domains (TADS) have been reported in Arabadopsis AtCAMTA1, human HsCAMTA2, and functional support derived from reporter genes downstream of TAD’s in plant protoplasts or mammalian cell cultures [25]. Cardiac hypertrophy as a consequence transcriptional reprogramming of cardiac gene expression by CAMTA2 interaction with class II histone deacetylase has been reported [26]. In drosophila, CAMTA is highly expressed in retinal cells and has been shown to induce dFbx14 that deactivates the G-protein coupled receptor (GPCR) rhodopsin. This is essential to maintaining Ca2+ homeostasis of the photo-transduction pathway, a central mechanism of which is light induced Ca2+ influx and efflux regulated through CaM and dmCAMTA [27]. It is not surprising that CAMTA2 is strongly expressed in both developing and adult murine retina; however, its function in this context remains unknown. Given the diverse expression and activity of GPCRs, it is reasonable to speculate that regulation of this class of molecule by CAMTA may modulate cellular growth and differentiation as evidenced in cardiac hypertrophy [26]. Both CAMTA1 and CAMTA2 are highly expressed in the heart and brain [27]. CAMTA2 has six protein coding isoforms (CAMTA2 ENSG00000108509) ranging from 178 to 1241 amino acids. The expression pattern and function of each isoform remains unknown.
In this study, we identified a G>A change in the 5′UTR of CAMTA2 transcript variant 2. The position of the variant, near a splice site (+ 30) and adjacent to the translation initiation site (− 6) suggested that it might impact CAMTA2 transcript variant 2 expression or translation. Our results clearly indicated that the CAMTA2 mutant affected processes downstream of mRNA transcription, most likely translation as protein levels were dramatically reduced while there was no effect on mRNA levels (Fig. 4c–d). As protein expression was severely impacted without any alteration in the coding region (i.e, no amino acid change), the hypomorphic nature of the underlying mutation is clearly evident. It is known that secondary structures in the 5′UTR with stable high-energy forms (ΔG<-50 kcal/mol) inhibit translation if near the mRNA methyl G cap [28]. Hairpin structures of weaker energy coupled with positions far away from the 5’end cap such as in our case, in contrast, enhance translation [28]. The wild-type CAMTA2 5′UTR-predicted structure is near the initiation codon and of weakly stable structure, thus may facilitate translation machinery entry and therefore enhances translation. These activities are lost in the case of the mutant variant. In general, mutations near initiation regions can dramatically affect translation. Context sequence upstream of the initiation codon can modulate the scanning activity of the 40S ribosomal subunit affecting translation; mutations in this region, such as those in the Kozak consensus sequence that direct ribosomes to initiate protein synthesis, negatively affect translation initiation [29].
Further support for the pathogenicity or association of the CAMTA2 variant with the phenotype described (for which we propose the name Bohlega Syndrome) in affected members of the family studied comes from the expression pattern of CAMTA2 transcript variant 2 in human brain sub-regions where it is expressed in the vermis cerebellum, left and right cerebellum, cerebral cortex, olfactory lobe, and cerebellar peduncles (Fig. 5a). Expression of this isoform was not detected in the corpus callosum, hippocampus, and occipital or frontal lobes (Fig. 5a). Other CAMTA2 protein coding transcripts were ubiquitously expressed in all brain sub-regions tested. In mouse brain, CAMTA2 is expressed in Purkinje cells and granule cells of the dentate gyrus, pyramidal layer, and cerebellum (Additional file 4), but the distribution of each isoform is not known. Dystonia and Parkinsonism, including tremor, have largely been attributed to dysfunction of the basal ganglia. However, more recently, the role of Purkinje cells, the cerebellum, and their links to the basal ganglia in dystonia and Parkinson’s disease have been questioned [30,31,32]. Anatomical studies have identified connections from the basal ganglia and cerebellum via the thalamus to the cortex [33]. Of note, the ability of CAMTA2 knockout mice to mount a hypertrophic response to cardiac stress such as aortic banding is severely compromised [27]. There was no report of any tremor or dystonia in the CAMTA2 knockout mouse. In the affected individuals from our study, absence of any clinical cardiac symptoms extends the apparent discordance between the human and mouse phenotypes, albeit the mouse phenotype was induced by severe stress (aortic banding) [27]. However, the mouse ENCODE consortium mapped transcription factor binding and other regulatory domains across the mouse genome and compared them with the human genome. While they identified substantial conservation, they also identified a large degree of divergence between mouse and man in transcription and transcription regulation [34]. Therefore, differences in mouse and human phenotypes associated with genes such as CAMTA2 involving transcription or transcription regulation should not be unexpected.
Conclusions
Our study identifies a novel dystonia locus CAMTA2 that opens a new line of investigation associated with the role of CAMTA2 in the growth, differentiation, survival, and function of cells and their cellular mechanisms within the brain as related to movement disorders. It also highlights the value of rare inherited disorders in providing insight into more common disease states such as dystonia and tremor.
Methods
Subjects and nucleic acid extraction
Five patients from a consanguineous extended family (two nuclear families) (Fig. 1a) were examined in the Department of Neurosciences, King Faisal Specialist Hospital and Research Center (KFSHRC). They were diagnosed with familial tremulous dystonia. All subjects were enrolled under a KFSHRC IRB-approved (RAC# 2090011) protocol with full-written informed consent.
Genomic DNA extraction
Genomic DNA was extracted from peripheral blood samples of patients and family members using a Gentra Puregene Kit (QIAGEN GmbH, Hilden, Germany). Purity and quantity of DNA samples were assessed spectrophotometrically and stored at − 20 °C until required.
Clinical data and imaging
Patients were seen and examined in the neurology clinic of King Faisal Specialist Hospital and Research Centre (KFSHRC) by certified neurologists (H.A. and S.B.). Briefly, the clinical data was obtained from the charts of patients at the time of the study and during the 20 years of follow-up. Imaging was performed by magnetic resonance imaging (MRI) 3 T using the following sequences: T1, T2, FLAIR, DWI, ADC, and MR spectroscopy. The other clinical, neurophysiological, and laboratory methodology may be reviewed from the previously published clinical paper [19].
Linkage and Homozygosity mapping
All participating individuals (affected and unaffected) were genotyped using an Affymetrix Axiom Genome-Wide CEU array containing 587,353 markers (Affymetrix, Santa Clara, CA, USA) following the manufacturer’s protocol (http://www.affymetrix.com/support/technical/manuals.affx). Resulting genotypes were analyzed for shared runs of homozygosity (ROH) using autoSNPa (http://dna.leeds.ac.uk/autosnpa/). Linkage analysis was performed using the Allegro module of Easy Linkage assuming autosomal recessive inheritance and 100% penetrance (http://genetik.charite.de/hoffmann/easyLINKAGE/index.html). For linkage analysis, the extended family was broken down to reflect the two consanguineous nuclear families, with both independent LOD scores and a combined total LOD score being calculated, in a single multipoint analysis of both nuclear families.
Whole exome sequencing and analysis
DNA was amplified to obtain a whole exome Ion Proton AmpliSeq library which was further used for emulsion PCR on an Ion One Touch System followed by an enrichment process using an Ion OneTouch ES, both procedures following the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA). The template-positive Ion PI Ion Sphere particles were processed for sequencing on the Ion Proton instrument (Life Technologies, Carlsbad, CA, USA). Approximately 15–17 Gb of sequence was generated per sequencing run. Reads were mapped to UCSC hg19 (http://genome.ucsc.edu/) and variants identified using the Ion Torrent pipeline (Life Technologies, Carlsbad, CA, USA). Based upon the consanguineous inbred pedigree and a clear autosomal recessive inheritance pattern in the family studied, the resultant variant caller file (vcf) was filtered to include only homozygous variants in the critical interval on chromosome identified by linkage and homozygosity mapping, within the pre-determined ROH shared by all affected individuals only. These were further filtered to only retain variants that were present in public databases including NHLBI Exome Sequencing Project, 1000 genomes project (phase 3), Exome Aggregation Consortium (ExAC) (version 0.3.1), dbSNP (build 141), and Kaviar (Known VARiants) (September 2015) databases at an incidence of < 2%. Included variants were further selected based upon pathogenicity predicted by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and CADD (http://cadd.gs.washington.edu/info). Potential causative variant(s) were validated by Sanger sequencing and further vetted for familial segregation based upon autosomal recessive inheritance. For validation, an amplicon incorporating the variant of interest was amplified and sequenced using a BigDye Terminator kit (Applied Biosystems, Foster City, CA) and run on an ABI 3730xl automated sequencer (Applied Biosystems, Foster City, CA). SeqScape v.2.6 software (Applied Biosystems, Foster City, CA) was used to align sequence data and confirm variants. CAMTA2 transcript variant 2 is a human isoform and aligns with the human reference sequence of this gene.
Expression constructs
The CAM2TA 5′UTR reporter expression constructs were generated by using a cloning-free PCR method [35]. The CAM2TA 5′UTR wild type and mutant sequences (underlined) were incorporated in the forward primer: CCTATCAGTGATAGGGCGGCTGGGTATATAATGGAAGGGGGGTCGGTGTGGG/AGTAGAATGGCCAGCAAGG. The sequence also contained a minimal promoter (TATA box in italics), 15 bases of complementary sequence to an expression vector containing strong modified EGFP, UWr-EGFP (SGFP), and a 3’UTR [36]. The reverse primer is complementary to a downstream region of the poly(A) site on the vector. Briefly, PCR was performed with the following conditions: 2.5 units of HotStart Taq(Qiagen) and 0.2 units of Pfx polymerase (Invitrogen, Carlsbad, CA) mix, 100 ng, template and cycle conditions: 95 °C for 12 min, 31 cycles of: 94 °C, 1 min., 55 °C, 1 min., 72 °C, 4 min., and final extension at 72 °C for 7 min. The PCR products were purified using Qiagen PCR purification columns and eluted in sterile water. The PCR products were run on a 1.2% agarose gel and visualized by ethidium bromide under UV light to verify size and quality. The resultant purified constructs contained a minimal promoter, CAM2TA2 5′UTR, SGFP reporter, and 3’UTR, and were used for transfection.
Cells and transfection
HEK293 was obtained from the ATCC (Rockville, MD) and cultured in DMEM medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and antibiotics at 37 °C in 5% CO2. Cells were seeded at 3 × 104 cells per well in 96-well clear-bottom black plates (Matrix technologies, Hudson, NH) and incubated overnight. Cells were transfected with purified reporter constructs. Transfections with CAM2TA2 5′UTR-SGFP reporter along with a normalization control RFP expression plasmid driven by constitutive yet non-inducible RPS30 promoter [37] were performed in serum-free medium for 4 h, using Lipofectinamine 2000 (Invitrogen) according to the manufacturer’s instructions. Subsequently, the cell culture were replaced with completed DMEM medium and incubated further for 16 h. At these conditions, no effect on cell viability was observed. All transfections were performed in several replicates as indicated in the text. Fluorescence were acquired by the BD imaging pathway and quantified using the Proxcell imaging segmentation and quantification program [38]. The variance in GFP fluorescence among replicate microwells was < 10%. Data are presented as mean values ± standard deviation (SD) of total fluorescence intensity in each well, with at least six replicate readings.
Real-time RT-PCR
Quantitative real-time (qRT-PCR) was performed for SGFP and RFP reporter mRNA using SybrGreen Real-Time PCR. Briefly, total RNAs were treated with DNase I, and cDNAs were used with Lightcycler Faststart DNA master SYBR Green I, (Roche, USA). The following primers were used: SGFP forward: GCCAGGTCTTTTCTGCAGTCACCG; reverse: GTAGTGCAGATGAACTTCAGG); human GAPDH forward: GGCAAATTCAACGGCACAGT; reverse: GATGGTGATGGGCTTCCC. In all cases, relative quantification of expression was determined using the standard curve method and the values obtained were within both detectable and linear range. Average concentrations were normalized to the endogenous GAPDH gene. The final results were converted to ratio ± SD of the specific mRNA levels to housekeeping mRNA levels. qRT-PCR was performed in multiplex using the Chroma 4DNA Engine thermocycler (Bio-Rad).
Amplification of CAMTA2 protein coding isoforms from human cDNA libraries
We used cDNA libraries prepared from total RNA isolated from normal human adult brain tissues: vermis cerebelli, amygdala, cerebellum left, cerebellum right, cerebral cortex, corpus callosum, hippocampus, occipital lobe, frontal lobe, olfactory, and cerebellar peduncles (BioChain Institute, Newark, CA) to determine the expression pattern of CAMTA2 isoforms in human brain sub-regions, skeletal muscle, and cardiac muscle. The cDNA libraries were used as template for PCR amplification of protein coding CAMTA2 isoforms (isoform 1, isoform 2, isoform 3, isoform 4, isoform 7, and isoform 10). Briefly, PCR mixtures containing 2.5 ng cDNA, 1× PCR buffer, 2.5 mM each dNTP, 0.5 μM each primer, and 0.25 U HotStar Taq polymerase (Qiagen) were cycled 30 times at 95 °C for 15 min, 95 °C for 45 s, 64 °C for 45 s, and 72 °C for 60 s. GAPDH was amplified in a multiplex reaction as an internal PCR control. Primer information is available on request. The resulting amplicons were evaluated by electrophoresis on a 2% agarose gel. Representative bands were sequenced to confirm amplicon specificity.
References
Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Movement disorders : official journal of the Movement Disorder Society. 2013;28(7):863–73.
Balint B, Bhatia KP. Isolated and combined dystonia syndromes—an update on new genes and their phenotypes. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2015;22(4):610–7.
Charlesworth G, Bhatia KP, Wood NW. The genetics of dystonia: new twists in an old tale. Brain : a journal of neurology. 2013;136(Pt 7):2017–37.
Ozelius LJ, Hewett J, Kramer P, et al. Fine localization of the torsion dystonia gene (DYT1) on human chromosome 9q34: YAC map and linkage disequilibrium. Genome Res. 1997;7(5):483–94.
Zirn B, Grundmann K, Huppke P, et al. Novel TOR1A mutation p.Arg288Gln in early-onset dystonia (DYT1). J Neurol Neurosurg Psychiatry. 2008;79(12):1327–30.
Clot F, Grabli D, Burbaud P, et al. Screening of the THAP1 gene in patients with early-onset dystonia: myoclonic jerks are part of the dystonia 6 phenotype. Neurogenetics. 2011;12(1):87–9.
Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41(3):286–8.
Xiao J, Uitti RJ, Zhao Y, et al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann Neurol. 2012;71(4):458–69.
Charlesworth G, Plagnol V, Holmstrom KM, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91(6):1041–50.
Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet. 2013;45(1):88–92.
Saunders-Pullman R, Fuchs T, San Luciano M, et al. Heterogeneity in primary dystonia: lessons from THAP1, GNAL, and TOR1A in Amish-Mennonites. Movement disorders : official journal of the Movement Disorder Society. 2014;29(6):812–8.
Hersheson J, Mencacci NE, Davis M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol. 2013;73(4):546–53.
Wilcox RA, Winkler S, Lohmann K, Klein C. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Movement disorders : official journal of the Movement Disorder Society. 2011;26(13):2404–8.
Camargos S, Scholz S, Simon-Sanchez J, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7(3):207–15.
Zech M, Lam DD, Francescatto L, et al. Recessive mutations in the alpha3 (VI) collagen gene COL6A3 cause early-onset isolated dystonia. Am J Hum Genet. 2015;96(6):883–93.
Charlesworth G, Angelova PR, Bartolome-Robledo F, et al. Mutations in HPCA cause autosomal-recessive primary isolated dystonia. Am J Hum Genet. 2015;96(4):657–65.
Chen YZ, Friedman JR, Chen DH, et al. Gain-of-function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol. 2014;75(4):542–9.
Chen YZ, Matsushita MM, Robertson P, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol. 2012;69(5):630–5.
Bohlega S, Stigsby B, al-Kawi MZ, et al. Familial tremulous and myoclonic dystonia with white matter changes in brain magnetic resonance imaging. Movement disorders : official journal of the Movement Disorder Society. 1995;10(4):513–7.
Matzura O, Wennborg A. RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Computer applications in the biosciences : CABIOS. 1996;12(3):247–9.
Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J Mol Biol. 1999;287(5):1023–40.
Bouche N, Scharlat A, Snedden W, Bouchez D, Fromm H. A novel family of calmodulin-binding transcription activators in multicellular organisms. J Biol Chem. 2002;277(24):21851–61.
Muller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature. 1995;373(6512):311–7.
Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 1999;24(8):311–6.
Finkler A, Ashery-Padan R, Fromm H. CAMTAs: calmodulin-binding transcription activators from plants to human. FEBS Lett. 2007;581(21):3893–8.
Schwartz RJ, Schneider MD. CAMTA in cardiac hypertrophy. Cell. 2006;125(3):427–9.
Song K, Backs J, McAnally J, et al. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell. 2006;125(3):453–66.
Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA. 2006;12(5):851–61.
Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37.
Babij R, Lee M, Cortes E, Vonsattel JP, Faust PL, Louis ED. Purkinje cell axonal anatomy: quantifying morphometric changes in essential tremor versus control brains. Brain : a journal of neurology. 2013;136(Pt 10):3051–61.
Lewis MM, Galley S, Johnson S, Stevenson J, Huang X, McKeown MJ. The role of the cerebellum in the pathophysiology of Parkinson’s disease. The Canadian journal of neurological sciences. J Can Sci Neurol. 2013;40(3):299–306.
Prudente CN, Hess EJ, Jinnah HA. Dystonia as a network disorder: what is the role of the cerebellum? Neuroscience. 2014;260:23–35.
Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev. 2000;31(2–3):236–50.
Yue F, Cheng Y, Breschi A, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515(7527):355–64.
al-Haj L, Al-Ahmadi W, Al-Saif M, Demirkaya O, Khabar KS. Cloning-free regulated monitoring of reporter and gene expression. BMC Mol Biol. 2009;10:20.
Al-Saif M, Khabar KS. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20(5):954–9.
Hitti E, Al-Yahya S, Al-Saif M, et al. A versatile ribosomal protein promoter-based reporter system for selective assessment of RNA stability and post-transcriptional control. RNA. 2010;16(6):1245–55.
Mahmoud L, Al-Saif M, Amer HM, Sheikh M, Almajhdi FN, Khabar KS. Green fluorescent protein reporter system with transcriptional sequence heterogeneity for monitoring the interferon response. J Virol. 2011;85(18):9268–75.
Acknowledgements
The authors would like to thank the family members for their participation in this study. We thank the Genotyping and Sequencing Core Facilities of King Faisal Specialist Hospital & Research Centre (KFSHRC) for their contributions. Work described was supported by grants from KFSHRC (RAC#2090011) and King Abdulaziz City for Science and Technology (KACST) (KACST-12-BIO2943-20). We would also like to acknowledge the support of KACST through the Saudi Human Genome Project (SHGP).
Funding
This work was funded through grants from King Faisal Specialist Hospital & Research Centre (RAC#2090011) and King Abdulaziz City for Science and Technology (KACST-12-BIO2943-20).
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
DM contributed to the conception and design, acquisition and analysis of data, and drafting of the manuscript and figures. HA participated in the acquisition and analysis of data, and drafting of the manuscript and figures. EN participated in the acquisition and analysis of data and drafting of the manuscript and figures. BA contributed to the acquisition and analysis of data and drafting of the manuscript and figures. MAB contributed to the acquisition and analysis of data. MAS participated in the acquisition and analysis of data. SW contributed to the acquisition and analysis of data and drafting of the manuscript and figures. BM participated in the conception and design, acquisition and analysis of data and drafting of the manuscript and figures. KK participated in the conception and design, acquisition and analysis of data and drafting of the manuscript and figures. SB contributed to the conception and design, acquisition and analysis of data and drafting of the manuscript and figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all enrolled participants in an IRB-approved project (RAC#2090011). The study was carried out in accordance with the declaration of Helsinki under IRB guidelines of the King Faisal Specialist Hospital and Research Centre (KFSHRC), Riyadh, Saudi Arabia.
Consent for publication
Written informed consent was obtained to publish the video included in the supplement to this article.
Competing interests
The authors declare no competing interest regarding the production of this article. The authors have no personal financial or institutional interest in any of the drugs, materials, or devices described in this article.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 2:
Variants in candidate interval. (PDF 217 kb)
Additional file 3:
Expression of CAMTA2 isoforms, 1, 3, 4, 7 and 10 in sub-regions of the brain. (PDF 594 kb)
Additional file 4:
CAMTA2 expression in brain. (PDF 1628 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Monies, D., Abou Al-Shaar, H., Goljan, E.A. et al. Identification of a novel genetic locus underlying tremor and dystonia. Hum Genomics 11, 25 (2017). https://doi.org/10.1186/s40246-017-0123-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-017-0123-5