
Abstract
Background
Methanogens are crucial to global methane budget and carbon cycling. Methanogens from the phylum Euryarchaeota are currently classified into one class and seven orders, including two novel methanogen taxa, Methanofastidiosa and Methanomassiliicoccales. The relative importance of the novel methanogens to methane production in the natural environment is poorly understood.
Results
Here, we used a combined metagenomic and metatranscriptomic approach to investigate the metabolic activity of methanogens in mangrove sediments in Futian Nature Reserve, Shenzhen. We obtained 13 metagenome-assembled genomes (MAGs) representing one class (Methanofastidiosa) and five orders (Methanomassiliicoccales, Methanomicrobiales, Methanobacteriales, Methanocellales, and Methanosarcinales) of methanogens, including the two novel methanogens. Comprehensive annotation indicated the presence of an H2–dependent methylotrophic methanogenesis pathway in Methanofastidiosa and Methanomassiliicoccales. Based on the functional gene analysis, hydrogenotrophic and methylotrophic methanogenesis are the dominant pathways in mangrove sediments. MAG mapping revealed that hydrogenotrophic Methanomicrobiales were the most abundant methanogens and that methylotrophic Methanomassiliicoccales were the most active methanogens in the analyzed sediment profile, suggesting their important roles in methane production.
Conclusions
Partial or near-complete genomes of two novel methanogen taxa, Methanofastidiosa and Methanomassiliicoccales, in natural environments were recovered and analyzed here for the first time. The presented findings highlight the ecological importance of the two novel methanogens and complement knowledge of how methane is produced in mangrove ecosystem. This study implies that two novel methanogens play a vital role in carbon cycle.
Video Abstract
Similar content being viewed by others


Background
Methane is the second most important greenhouse gas after CO2. Methanogenesis is conducted by methanogens that thrive in strictly anoxic habitats. Methanogens are considered to play important roles in the global carbon cycle and climate change.
Traditionally, methanogens have been assigned to the phylum Euryarchaeota, and divided into two classes: class I, containing Methanococcales, Methanopyrales, and Methanobacteriales and class II, containing Methanomicrobiales, Methanocellales, and Methanosarcinales [1]. The development of high-throughput sequencing technologies has expanded the knowledge of methanogen diversity. Methanofastidiosa and Methanomassiliicoccales, two newly described novel methanogens, also fall within the phylum Euryarchaeota but do not belong to class I or class II methanogens. Beyond that, Bathyarchaeota, Verstraetearchaeota, Hadesarchaea, Korarchaeota, Nezhaarchaeota, Thaumarchaeota, and Helarchaeota also contain methyl-CoM reductase (MCR)-like enzymes, the key enzyme for methane metabolism, indicating that these archaea might have potentials for methane or short-chain alkane metabolisms [2,3,4,5,6,7]. Further physiological evidence is needed to confirm that notion. No pure cultures of Methanofastidiosa (formerly WSA2/Arc1) have been obtained but genomic data from an anaerobic wastewater treatment bioreactor have been recovered [8]. However, no genome of Methanofastidiosa obtained from the natural environment is available. By contrast, isolates and cultures of Methanomassiliicoccales (formerly RCIII) have been obtained from the human feces, termite gut, and water treatment sludge [9,10,11,12]. Metagenome-assembled genomes (MAGs) of Methanomassiliicoccales have been recently recovered from diverse natural environments [4, 13], yet the details of their metabolism are limited.
Methanogens utilize three pathways, i.e., hydrogenotrophic, acetoclastic, and methylotrophic pathways, to produce methane [14, 15]. Generally, Methanococcales, Methanopyrales, Methanobacteriales, Methanomicrobiales, Methanocellales, and Methanosarcinales are hydrogenotrophs [16]. They utilize H2 and CO2 to produce methane via the methyl branch of the archaeal type Wood–Ljungdahl pathway (WLP) plus methyl-CoM reduction. Methanosarcina and Methanosaeta (Methanosarcinales) are the only acetoclastic methanogens known to date, which dismutate acetate to CH4 and CO2 [17]. Further, methylotrophic methanogens can be classified in two groups: Methanosarcinales that possess cytochromes and other methylotrophs that lack cytochromes [18]. The latter lack the N5-methyl-tetrahydromethanopterin-coenzyme M methyltransterase (MTR) and use H2 reduce methyl-compounds for methane production. They include Methanosphaera (Methanobacteriales), Methanofastidiosa, and Methanomassiliicoccales [19]. Analysis of Methanofastidiosa MAGs revealed that they possess specific methyltransferases to reduce methylated thiol for methanogenesis [8].
In different ecological environments, methanogen communities and the accompanying metabolic pathways are different. For example, hydrogenotrophic Methanococcales and Methanomicrobiales, and acetoclastic Methanosaeta and methylotrophic Methanosarcinales contribute the most to methane emission in marine sediments [20, 21]. In freshwater sediments, acetoclastic Methanosarcinales contribute more to methanogenesis than hydrogenotrophic Methanobacteriales and Methanomicrobiales [22]. Further, hydrogenotrophic and acetoclastic methanogens dominated the total methanogenic community in saline lake sediments with salinity less than 3.5 g L−1 [23]. The relative contribution of hydrogenotrophic methanogens to total methane production increases with depth in lake sediments [24]. However, little is known about the metabolic pathways and ecological roles of Methanofastidiosa and Methanomassiliicoccales in natural environments.
Mangroves are important constituents of the coastal wetlands. They could store atmospheric CO2 as organic matter, so-called “blue carbon,” inhabiting approximately 0.5% of the coast and contributing 10–15% to the global carbon storage [25]. However, mangrove sediment carbon does not remain stored in perpetuity. Some of organic matter are transformed to CH4 and returned to the atmosphere, which has the potential to partially offset blue carbon storage in mangrove sediments [26]. Previous studies using 16S rRNA gene and metagenomics demonstrated that multiple microorganisms including methanogens are widely spread across mangrove sediments [27,28,29,30,31]. However, the metabolic activity and relative contributions to methane production of diverse methanogens in mangroves remain unclear, especially those of Methanofastidiosa and Methanomassiliicoccales.
Mangroves are one of the major sources of CH4. We collected CH4 efflux data from literatures for 20 sites of mangrove ecosystems worldwide including Futian Mangrove Nature Reserve (FT) (Additional file 1: Table S1). FT is located in Shenzhen Special Economic Zone. While methanogenesis was not measured in this study, a previous study has reported that methane emission rates in FT range from 242 μmol m−2 day−1 or 0.242 mmol m−2 day−1 to 124 mmol m−2 day−1 [32]. In the current study, we first conducted an overall analysis based on 16S rRNA gene sequences across six mangrove ecosystems to investigate the distribution of methanogens, and the potential interactions between methanogens and other microbial lineages [33]. Then we sampled five sediment layers from FT. Previous 16S rRNA gene analysis of samples collected at the same site revealed the presence and high relative abundance of novel methanogens, including Methanofastidiosa and Methanomassiliicoccales, indicating that methanogens are a dominant archaeal group in FT mangroves [34]. We combined metagenomic and metatranscriptomic analyses to investigate the metabolic activity and relative contributions of diverse methanogens to methane production in a vertical sediment profile in mangrove ecosystem. We recovered and annotated 13 methanogen MAGs to investigate their adaption strategies to the environment. Based on the metabolic analysis and mapping results, we proposed that two novel methanogens were active for methane production and played a vital role in global carbon cycle.
Results
Methanogen diversity and co-occurrence network
The 16S rRNA genes sequence analysis of 78 mangrove sediment samples revealed a variety of methanogens in mangrove wetlands in southeastern China. Among the six sampling sites, methanogen abundance was the highest in Shenzhen, where they accounted for approximately 1.5% of prokaryotes (Additional file 2: Fig. S1a). The community compositions of methanogens at each site are presented in Additional file 2: Fig. S1b. Methanofastidiosa, Methanosarcinales, and Methanomicrobiales were widely distributed in mangrove sediments. Further, Methanomassiliicoccales, Methanofastidiosa, Methanosarcinales, and Methanomicrobiales were the four dominant methanogens in Shenzhen mangroves.
Co-occurrence network analysis based on 16S rRNA gene sequences of 78 mangrove sediment samples revealed interesting potential interactions between methanogens and other microbial taxa (Fig. 1). According to the determined O/R (observed/random incidence) ratios, Micrarchaeota showed the highest non-random association with methanogens. Woesearchaeota, Sedimenticola, Desulfobacca, and Sulfurovum also showed significant non-random association with methanogens.

Co-occurrence network reconstructed based on 16S rRNA gene sequencing data. Different color nodes represent OTUs affiliated with different lineages. Lines (edges) connecting the nodes represent strong (r > 0.6) and significant (P < 0.01) positive correlations. O/R value is the ratio of observed to random co-occurrence incidence between two lineages (methanogens and archaeal phyla or bacterial genera). O/R values of more than 1 indicate a non-random association between two lineages
Genome reconstruction
De novo genomic assembly and binning of metagenome sequencing data from five layers of mangrove sediments in Futian Nature Reserve resulted in the reconstruction of 13 MAGs representing Euryarchaeota methanogens (Table 1). Genome size, GC content, completeness, and contamination are summarized in Table 1. The completeness of 12 MAGs ranged from 69.8 to 99.4%, while one MAG was only 50.3% complete. The contamination degree ranged from 0 to 9.6%, as assessed using CheckM (as described in “Methods” section). MAG genome sizes ranged from 0.61 to 2.33 Mbp.
Phylogeny and relative abundances of methanogens in mangrove sediments
To identify MAG lineages, we constructed phylogenetic trees based on a concatenated set of 16 ribosomal proteins (Fig. 2a), McrA (methyl-coenzyme M reductase alpha subunit) protein sequences (Fig. 2b), and 16S rRNA genes (Additional file 3: Fig. S2). Four MAGs represented two newly described lineages, MF1 and MF2, clustered within the class Methanofastidiosa, a class distinct from other Euryarchaeota and MMA1 and MMA2, clustered with Methanomassiliicoccales. The remaining 9 MAGs corresponded to the traditional methanogen lineages within class I or class II methanogens: MB, belonging to the order Methanobacteriales; MC from the order Methanocellales; MM1, MM2, MM3, and MM4 from the order Methanomicrobiales; and MS1, MS2, and MS3 from the order Methanosarcinales.

Phylogenetic trees of recovered MAGs constructed using inferred 16 ribosomal protein genes (a) and McrA protein sequences (b) using the Bathyarchaeota as the out-group. Names in bold represent MAGs in the current study. Bootstrap values were calculated via non-parametric bootstrapping with 100 replicates, and are represented by gray circles in different sizes. The scale bar indicates 10% estimated phylogenetic divergence. c The relative abundances (metagenome, RPKM, blue) and expression levels (metatranscriptome, RPKM, red) of MAGs affiliated with four dominant methanogen lineages (MF, MMA, MM, and MS)
Nine mcrA gene sequences were retrieved from 13 MAGs (Fig. 2b). A phylogenetic tree constructed using the corresponding McrA sequences was consistent with the tree constructed using conserved ribosomal proteins. The mcrA gene sequences were not retrieved from the MB and MC genome bins, probably because of the incompleteness of the genomes. McrA from MM was affiliated with the sequences from the genus Methanolinea; McrA from MS1 clustered with the sequences from the genus Methanococcoides; and McrA from MS2 clustered with the sequences from the genus Methanosaeta.
To reveal the relative importance of different methanogens in a vertical mangrove sediment profile, the relative abundance and activity of methanogens were next evaluated (Fig. 2c). Members of MM were the most abundant group of methanogens in all layers. Although MMA MAGs were not abundant compared to MM MAGs, they were the most active group in all layers according to the metatranscriptomic analysis. MF and MS coexisted with other methanogens in all layers, but their relative abundance and activity were relatively low.
Relative importance of the three major metabolic pathways for methanogenesis in mangrove sediments
Quantitative PCR analysis revealed a dramatically different abundance of methanogens in the five sampled sediment layers. Indeed, the copy numbers of the mcrA gene ranged from 105 to 106 gene copies per gram sediment, with the highest abundance in the 6–8 cm layer (Additional file 4: Fig. S3).
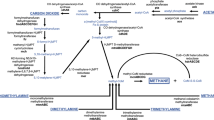
MAG annotation revealed a diverse metabolic potential for methanogenesis. Three complete metabolic pathways (hydrogenotrophic, acetoclastic, and methylotrophic methanogenesis) were identified (Fig. 3). MM and MS MAGs shared the hydrogenotrophic methanogenesis pathway. They possessed genes encoding conserved core enzymes of hydrogenotrophic methanogenesis, including Fwd, Ftr, Mch, Mtd, Mer, Mtr, and Mcr. MS MAGs showed potential for acetoclastic methanogenesis. They contained genes encoding Acs, Cdh, Mtr, and Mcr, enzymes for the utilization of acetate. Further, MF, MMA, and MS MAGs contained genes encoding methyl-compound methyltransferase, i.e., Mts, Mta, Mtm, Mtb, and Mtt. This indicated a potential for methane production via the methylotrophic pathway. Because of the genome incompleteness, no complete methanogenesis pathways were identified in MB and MC MAGs.

Predicted metabolic pathways in the four dominant methanogen lineages (MF, MMA, MM, and MS), based on analysis of 11 MAGs. Genes involved in the metabolism of carbon, nitrogen, and hydrogen; energy conservation; and various transporters are shown in different colors. Predicted proteins in the figures are listed in Additional file 6: Table S2
To evaluate the relative importance of the three metabolic pathways for methane generation in the vertical mangrove sediment profile, the relative abundances and expression of the relevant genes were evaluated (Fig. 4). The functional analysis revealed that the relative abundances of genes associated with autotrophic hydrogenotrophic methanogenesis were higher than those of heterotrophic acetoclastic or methylotrophic methanogenesis pathways in all layers. Further, genes involved in hydrogenotrophic and methylotrophic methanogenesis were highly expressed.

Relative abundances (a) and expression levels (b) of genes involved in three methanogenesis pathways. The relative abundances and expression levels were evaluated based on FPKM values. The three metabolic pathways are represented by different colors (red, hydrogenotrophic; orange, acetoclastic; and blue, methylotrophic)
Next, the relative abundances and expression of genes from different MAGs were evaluated (Additional file 5: Fig. S4). Genes encoding enzymes from the hydrogenotrophic pathway in MM were highly abundant and expressed in all layers. Although the H2–dependent methylotrophic pathway in MF was expressed, the metabolic activity was relatively low. The MMA genes mts, mta, mtb, mtt, and mcr, encoding enzymes involved in the reduction of methyl compounds, were highly expressed.
Discussion
Methane emission in mangroves is strongly affected by anthropogenic activities including aquaculture and sewage [35]. Most pristine mangroves showed low CH4 efflux rate, while mangroves with human disturbances showed significantly higher CH4 efflux rate including FT mangroves (Additional file 1: Table S1). In the current study, we recovered and annotated 13 methanogen MAGs including two novel methanogen taxa, Methanofastidiosa and Methanomassiliicoccales from FT mangrove sediments. We aimed to show the relative importance of the novel methanogens to methane production. Our results showed that, according to transcript levels, Methanomassiliicoccales were the most active methanogens. These observations implied that two novel methanogens make contributions for methane production and play a vital role in carbon cycle.
In the current study, we identified and analyzed diverse methanogens in mangrove sediments. Copies of the mcrA gene ranged from 105 to 106 per gram sediment. The low gene copies might be due to primer pair we used for quantitative PCR. Since there were no generally recognized primers for novel methanogens, we used the primers mlas-mod-F and mcrA-rev-R [36]. This primer pair targets traditional methanogens and has low specificity to the novel methanogens including Methanofastidiosa and Methanomassiliicoccales, potentially underestimate the real abundance of methanogens. Therefore, the mcrA gene abundance could be higher if Methanofastidiosa and Methanomassiliicoccales were taken into account. The highest copy number was observed in the 6–8-cm layer (Additional file 4: Fig. S3). Previous studies similarly reported the highest methane production at the sediment surface [37]. The vertical variations of methanogen abundance are thought to be associated with the changing physiochemical factors in the sediment [24, 38]. The total organic carbon (TOC) concentration in the 0–2-cm layer was approximately 1.78 mg/g and decreased to approximately 0.96 mg/g in the 28–30-cm layer (Additional file 4: Fig. S3). The total nitrogen (TN) concentration also decreased, from 1.61 mg/g at the surface to 0.63 mg/g in deeper layers (Additional file 4: Fig. S3). Since large amounts of terrestrial and riverine nutrients reach and accumulate in the estuary environment, the top layers of the intertidal sediment are characterized by high organic matter content, which provides a suitable environment for the growth of methanogens [25].
We observed that the expression of genes involved in hydrogenotrophic and methylotrophic methanogenesis in the top three layers was high (Fig. 4), indicating that these two pathways are metabolically active and greatly contribute to the methane production in mangrove sediments. This was in agreement with a report that hydrogenotrophic and methylotrophic methanogenesis dominate in coastal sediments [37]. Methylotrophic methanogens might coexist with sulfate-reducing bacteria (SRB) in sulfate-rich environments because of their exclusive utilization of and stronger affinity for methyl compounds. Co-occurrence of methanogens and Sedimenticola, Desulfobacca, and Sulfurovum, which belong to SRB, detected in the current study supports this notion [39,40,41,42]. Methyl compounds such as trimethylamine (TMA) contribute 35–90% of the methane production in coastal sediments [43], which could explain why methylotrophic methanogens play an important role in methane production in mangroves. We also showed that hydrogenotrophic methanogenesis is more abundant and active than methylotrophic methanogenesis. Since mangrove sediments are rich in organic carbon, hydrogenotrophic methanogens could consume H2 and cooperate with syntrophic microbes to degrade short-chain fatty acids [44, 45]. Co-occurrence analysis also revealed a significant non-random association of methanogens and Woesearchaeota. The H2 production and consumption by Woesearchaeota and methanogens might explain this co-occurrence pattern, indicating a high possibility of a syntrophic relationship [46, 47].
We observed a low relative abundance and expression of genes involved in the acetoclastic pathway. This might be explained by that acetate is used by SRB. Salinity plays an important role in regulating methanogenic community [48]. Sulfate concentration is 1.29–2.77 g per kilogram dry weight in Futian mangroves [49]. High levels of salinity/sulfate favor SRB. Due to higher affinity for acetate, SRB have a thermodynamic advantage over acetotrophic methanogens. Acetoclastic methanogens usually dominate methane production in freshwater environments, such as anaerobic digesters, rice fields, and freshwater wetlands [50, 51]. Although 20–22 cm and 28–30 cm were sampled in the current study, we were unable to obtain transcriptomic data for these two layers because of the low quality of RNA extracted for sequencing (data not shown). This might be associated with the slow growth rates of microorganisms in undisturbed deep environments [52].
MF, MMA, MM, and MS were the four dominant methanogen lineages identified in the current study. This was consistent with previous 16S rRNA gene and mcrA sequencing-based identification of Methanomicrobiales and Methanosarcinales in coastal sediments [53,54,55]. Mangrove sediments in the Futian Natural Reserve are characterized by high sulfate concentrations [49], suggesting that adaptation to high salinity is important for mangrove dwelling. Therefore, methanogens should maintain an osmotic pressure equivalent to that of their surroundings, which might be achieved by the accumulation of organic osmotic solutes [56]. Indeed, genes for an organic solute transporter induced by glycine-betaine were identified in MF, MM, and MS MAGs. MF MAGs also encode an osmoprotectant transporter.
Notably, MM was the most abundant group of methanogens in all layers. Further, MM genes encoding enzymes from the hydrogenotrophic pathway were highly abundant and expressed in all layers (Additional file 5: Fig. S4), suggesting MM is the predominant group of hydrogenotrophic methanogens in mangrove sediments. This might be explained by a large number of electron transporters encoded in their genomes, which is advantageous for microbial adaption to low substrate (H2) environments [57]. In addition, MM encoded multiple membrane-bound hydrogenases, including Ech, Eha, and Ehb (Fig. 3 and Additional file 6: Table S2). Finally, MM possessed a complete gene set for the reductive hexulose-phosphate (RHP) pathway for autotrophic carbon fixation. This pathway has been proposed recently and is expected to be widely distributed among Methanomicrobiales [58].
In the current study, the partial MF genomes from a natural environment were reported for the first time. Members of MF are distinguished from the traditional class I and class II methanogens by the lack of genes encoding enzymes for conventional CO2 reduction. Members of MF lack the MTR complex and use H2 as a reductant for methanogenesis. Specifically, MF MAGs harbor genes encoding substrate-specific methyltransferases for multiple methyl compounds, including Mts, Mta, Mtb, and Mtt (Fig. 3 and Additional file 6: Table S2). They also harbor genes for heterodisulfide reductase (hdrABC)/[Ni-Fe] hydrogenase (mvhADG) complex for heterodisulfide coenzyme B coenzyme M (CoB-S-S-CoM) reduction and H2 oxidation. Further, they contain genes encoding the membrane-bound energy-conserving hydrogenase (Ehb) to generate H2. These two processes could be connected to H2 cycling. The mcr genes from MF were expressed. These observations suggested that MF could produce methane via H2 reduction of multiple methyl compounds in mangrove sediments, as opposed to solely relying on methylated thiols, as described previously [8]. That might be because mangrove plants produce methanol and methylamines that could be utilized by methylotrophic methanogens.
Although the H2-dependent methylotrophic pathway in Methanofastidiosa was expressed at 6–8 cm, the metabolic activity was relatively low in mangrove sediments. This might be due to that Methanofastidiosa are heterotrophic methanogens. They generally dwell in eutrophic environments, such as wastewater treatment sludge and digesters [59, 60]. These microorganisms require exogenous organic carbon (acetate or propionate) as a carbon source for growth as they lack genes encoding the carbon fixation pathway (Fig. 3). Organic matter contents in the sampled environments were not high enough for growth of Methanofastidiosa.
Methanomassiliicoccales have been isolated and enriched from human feces, termite gut, and an anaerobic digester [10,11,12, 61]. Recently, Methanomassiliicoccales MAGs have been recovered from the natural environment [4, 13]. In the current study, near-complete genomes of Methanomassiliicoccales from the mangrove sediments were analyzed for the first time. Similar to MF, MMA MAGs harbor a pathway for H2 reduction of methyl compounds (methylsulfides, methanol, and methylamines) (Fig. 3 and Additional file 6: Table S2), as reported in the previously described Methanomassiliicoccales genomes and validated by physiological experiments [18]. The MAGs also contained genes for the HdrABC/MvhADG complex for H2 oxidation. There is evidence for an additional coupling of ferredoxin and heterodisulfide in Methanomassiliicoccales, operated by the association of F420H2 hydrogenase (Fpo) and a second heterodisulfide reductase (HdrD) [62]. Transcripts of the MMA mts, mta, mtb, mtt, and mcr genes encoding proteins for the reduction of methyl compounds were detected, suggesting that MMA might contribute to methane production via the H2-dependent methylotrophic methanogenesis pathway. This could be explained by the availability of a variety of methylotrophic substrates (methylsulfides, methanol, and methylamines) in mangrove sediments.
According to metatranscriptomic analysis in the current study, MMA were the most active group in all layers, suggesting that MMA are well adapted to the fluctuating environment of mangrove sediments. Indeed, prevalence of Methanomassiliicoccales in coastal sediments has been reported [31, 54], indicating their importance for methane production in natural environments. MMA MAGs contained genes encoding oligo- and monosaccharide transporters, which might illustrate adaptation to a heterotrophic lifestyle (Fig. 3). Nevertheless, isolates from natural environments should be obtained and analyzed to validate their inferred physiological characteristics [63].
Conclusions
In conclusion, Methanofastidiosa, Methanomassiliicoccales, Methanomicrobiales, and Methanosarcinales were identified as the four dominant and potential methanogens in mangrove sediments. To the best of our knowledge, metabolic pathways utilized by the two novel methanogens Methanofastidiosa and Methanomassiliicoccales in the natural environment were here analyzed for the first time. Analysis of genes involved in methanogenesis suggested that the hydrogenotrophic and methylotrophic pathways contributed the most to the methane production in mangrove ecosystems. Based on the metagenomic and metatranscriptomic data, Methanomicrobiales and Methanomassiliicoccales are the most abundant and active methanogens, respectively. Collectively, the current study provides insights into the relative importance of diverse methanogens for methane production and advances the understanding of different methanogenesis pathways in mangrove ecosystems. This study implies that two novel methanogens play important roles in global carbon cycle.
Methods
Diversity and community network analysis of methanogens in mangrove sediments
Prokaryotic 16S rRNA genes in 78 sediment samples from 6 mangrove ecosystems across southern China were previously sequenced using the primer pair 515F/806R, and raw reads were processed as previously described [33]. Operational taxonomic units (OTUs) were picked at 97% cutoff using QIIME scripts [64]. Representative sequences of each OTU were assigned according to the SILVA SSUPara database (v132) [65]. Sequences belonging to methanogens were extracted to make “Methanogens_OTU table.” Diversity of methanogens in mangrove sediments was calculated based on the “Methanogen_OTU table.” Abundant and ubiquitous OTUs whose abundance > 0.001% of total sequences and occurrence in more than one sample were selected to make “Core_OTU table.” To explore the co-occurrence patterns between methanogens and other microbes, network analysis was conducted by calculating the correlations based on the “Core_OTU table” [66]. The obtained network reflected positive correlations (edges) among OTUs (nodes) with Spearman’s ρ > 0.6 and FDR-adjusted p value < 0.01. The network contained 3548 nodes and 36,248 edges. The random and observed incidence of co-occurrence patterns between methanogens and archaeal phyla or bacterial genera were calculated [67]. Only OTUs of methanogens and taxa with the O/R (observed/random incidence) ratio above 1 were retained for visualization. The network was constructed, characterized, and visualized using R packages (vegan and igraph) and the software gephi [68, 69].
Sample collection, nucleic acid extraction, and metagenome/metatranscriptome sequencing
A 32-cm sediment core, vertically stratified at 2-cm–depth intervals (32 cm in total), was collected for DNA extraction at an intertidal mudflat in the Futian Nature Reserve of Shenzhen (22.53°N, 114°E) in April 2017. A description of physicochemical parameters’ measurement can be found in previous study [34]. Samples from the 0–2, 6–8, 12–14, 20–22, and 28–30-cm layers were selected for metagenomic analysis. For the above fiver layers, genomic DNA was extracted from 5 g of wet sediment using DNeasy PowerSoil kit (Qiagen, Germany), according to the manufacturer’s instructions, and stored at – 20 °C. Metagenomic sequence data were generated using Illumina Hiseq 2000 instrument at Novogene Bioinformatics Technology Co., Ltd. (Tianjin, China). Approximately 110 Gbp (2 × 150 bp paired-end reads) of raw sequence data were generated for each sample.
The samples for metatranscriptomic analysis were collected at the same site as that used for metagenomic analysis from the 0–2, 6–8, and 12–14-cm layers in April 2018. The sediments were preserved immediately after collection in the LifeGuard Soil Preservation Solution (Qiagen) to prevent RNA degradation. Total RNA was isolated from wet sediment (4–20 g) using an RNeasy PowerSoil Total RNA kit (Qiagen), according to the manufacturer’s protocol. Genomic DNA was removed by using TURBO DNA-free kit (Ambion, USA), and the remaining RNA was concentrated and purified by using the RNeasy MinElute Kit (Qiagen). The extracted RNA (approximately 3 μg per sample) was paired-end sequenced using Illumina Hiseq 2000 instrument at Novogene (Tianjin, China). Approximately 8 Gbp (2 × 150 bp paired-end reads) of raw sequence data were generated for each sample.
Determination of methanogen abundances in the sediment layers
Abundances of the mcrA gene in the five sediment layers were determined by quantitative PCR using the primers mlas-mod-F and mcrA-rev-R [36], and an iCycler iQ 5 thermocycler (Bio-Rad, USA). The reaction volume was 25 μl. Each reaction contained 12.5 μl of 2 × SYBR® Premix Ex TaqTM (Takara Biotechnology, Japan), 0.5 μl of each primer (10 μM), and 2 μl of diluted DNA template (1–10 ng). The amplification program consisted of 30 cycles of 30 s at 95 °C, 45 s at 55 °C, and 30 s at 72 °C. Standard curves were generated using 10-fold serial dilutions of a plasmid containing the mcrA gene fragments. The PCR efficiency ranged between 90 and 100%, with R2 value of 0.99.
De novo assembly, binning, and annotation
Metagenomic datasets generated for the five layers of mudflat sediments were used in a combined assembly to recover genomes. The raw reads were dereplicated and trimmed using Sickle (https://github.com/najoshi/sickle). High-quality metagenomic sequences were de novo assembled using IDBA-UD [70] with the following parameters: -mink 65, -maxk 145, and -step 10. Genome binning of the assembled fragments was done using MetaBAT [71]. Partial and near-complete genomes were recovered after binning. Manual refining of MAGs was performed using Anvi’o to remove contaminating contigs [72]. The completeness, contamination, and strain heterogeneity of MAGs were evaluated by using CheckM (version 1.0.5) [73]. Thirteen MAGs representing one class (Methanofastidiosa) and five orders (Methanomassiliicoccales, Methanobacteriales, Methanocellales, Methanomicrobiales, and Methanosarcinales) of methanogens were selected for further analysis. The MAGs were translated by Prodigal using the “-p meta” parameters [74]. For each predicted coding sequence (CDS), protein function was annotated using the KEGG server (BlastKOALA) and eggNOG-mapper [75, 76].
Phylogenetic analysis
The concatenated ribosomal protein tree was generated as described elsewhere [77]. Briefly, several reference genomes of methanogens from the phylum Euryarchaeota were downloaded from the NCBI (https://www.ncbi.nlm.nih.gov/) and IMG-M (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) databases. The information for reference genomes is provided in Additional file 7: Table S3. For the analysis, genes for 16 ribosomal proteins (ribosomal proteins L2–L6, L14–L16, L18, L22, L24, S3, S8, S10, S17, and S19) were used [46]. The 16 ribosomal proteins were aligned independently using MUSCLE [78]. The amino acid alignments were then used for phylogenetic tree construction using FastTree with default parameters [79].
Genes annotated as 16S rRNA and mcrA were extracted against the SILVA SSU132 [65] and mcrA database downloaded from FunGene (http://fungene.cme.msu.edu/index.spr), respectively. Reference sequences of the 16S rRNA and mcrA genes of methanogens from the phylum Euryarchaeota were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) and FunGene, respectively. The 16S rRNA gene sequences were aligned using SINA [80]. The amino acid sequences of predicted McrA proteins were aligned using ClustalW [81]. Phylogenetic trees for the 16S rRNA gene and McrA protein sequence were constructed in FastTree using default parameters [79]. The trees were visualized by using iTOL [82], and rooted using the Bathyarchaeota as an out-group.
Gene abundance and expression
Raw reads generated after metatranscriptomic sequencing were filtered using SortMeRna to remove tRNA and rRNA sequences [83]. Reads per kilobase of transcript per million mapped reads (RPKM) for genomic and transcriptomic reads were calculated to determine the relative abundance and expression activity of MAGs, respectively. Relative abundances were determined by mapping to methanogen MAGs using Bowtie [84]. Expression activity was determined by mapping non-rRNA transcripts to methanogen MAGs using BWA-MEM [85]. The abundance and expression of key genes encoding enzymes involved in methanogenesis were calculated by mapping raw metagenome and metatranscriptome reads to each CDS of MAGs using fragments per kilobase of transcript per million mapped reads (FPKM), respectively.
Availability of data and materials
The genome bins generated and analyzed during the current study are available in the NCBI and can be viewed under Project PRJNA587831. The metagenome and transcriptome data can be viewed in NODE (http://www.biosino.org/node) by pasting the accession (OEP000712) into the text search box or through the URL: http://www.biosino.org/node/project/detail/OEP000712.
Abbreviations
- MAGs:
-
Metagenome-assembled genomes
- MCR:
-
Methyl-CoM reductase
- WLP:
-
Wood–Ljungdahl pathway
- MTR:
-
N5-methyl-tetrahydromethanopterin-coenzyme M methyltransterase
- mcrA :
-
Methyl-coenzyme M reductase alpha subunit
- FT:
-
Futian Mangrove Nature Reserve
- O/R:
-
Observed/random incidence
- TOC:
-
Total organic carbon
- TN:
-
Total nitrogen
- RHP:
-
Reductive hexulose-phosphate
- Hdr:
-
Heterodisulfide reductase
- Mvh:
-
[Ni-Fe] hydrogenase
- CoB-S-S-CoM:
-
Heterodisulfide coenzyme B coenzyme M
- Ehb:
-
Energy-conserving hydrogenase
- Fpo:
-
F420H2 hydrogenase
- Acs:
-
Acetyl-CoA synthetase
- Cdh:
-
Carbon monoxide dehydrogenase
- CDS:
-
Coding sequence
- RPKM:
-
Reads per kilobase of transcript per million mapped reads
- FPKM:
-
Fragments per kilobase of transcript per million mapped reads
References
Borrel G, Adam PS, Gribaldo S. Methanogenesis and the Wood-Ljungdahl pathway: an ancient, versatile, and fragile association. Genome Biol Evol. 2016;8:1706–11.
Seitz KW, Dombrowski N, Eme L, Spang A, Lombard J, Sieber JR, Teske AP, Ettema TJG, Baker BJ. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat Commun. 2019;10:1822.
Baker BJ, Saw JH, Lind AE, Lazar CS, Hinrichs KU, Teske AP, Ettema TJ. Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat Microbiol. 2016;1:16002.
Hua Z-S, Wang Y-L, Evans PN, Qu Y-N, Goh KM, Rao Y-Z, Qi Y-L, Li Y-X, Huang M-J, Jiao J-Y, Chen Y-T, Mao Y-P, Shu W-S, Hozzein W, Hedlund BP, Tyson GW, Zhang T, Li W-J. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nature Commun. 2019;10.
Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, Tyson GW. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science. 2015;350:434–8.
Vanwonterghem I, Evans PN, Parks DH, Jensen PD, Woodcroft BJ, Hugenholtz P, Tyson GW. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat Microbiol. 2016;1:16170.
Wang Y, Wegener G, Hou J, Wang F, Xiao X. Expanding anaerobic alkane metabolism in the domain of archaea. Nat Microbiol. 2019;4:595–602.
Nobu MK, Narihiro T, Kuroda K, Mei R, Liu WT. Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 2016;10:2478–87.
Iino T, Tamaki H, Tamazawa S, Ueno Y, Ohkuma M, Suzuki K-I, Igarashi Y, Haruta S. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. Nov. and Methanomassiliicoccales ord. Nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ. 2013;28:244–50.
Paul K, Nonoh JO, Mikulski L, Brune A. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol. 2012;78:8245–53.
Borrel G, Harris HMB, Parisot N, Gaci N, Tottey W, Mihajlovski A, Deane J, Gribaldo S, Bardot O, Peyretaillade E, Peyret P, O’Toole PW, Brugèrea J-F. Genome sequence of “Candidatus Methanomassiliicoccus intestinalis” issoire-mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announcements. 2013;1:e00453–13.
Borrel G, Harris HMB, Tottey W, Mihajlovski A, Parisot N, Peyretaillade E, Peyret P, Gribaldo S, O’Toole PW, Brugèrea J-F. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J Bacteriol. 2012;194:6944–5.
Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2017;2:1533–42.
Welte CU. Revival of archaeal methane microbiology. mSystems. 2018;3:e00181–17.
Borrel G, Adam PS, McKay LJ, Chen LX, Sierra-Garcia IN, Sieber CMK, Letourneur Q, Ghozlane A, Andersen GL, Li WJ, Hallam SJ, Muyzer G, de Oliveira VM, Inskeep WP, Banfield JF, Gribaldo S. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat Microbiol. 2019;4:603–13.
Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–89.
Ferry JG. Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Curr Opin Biotechnol. 2011;22:351–7.
Lang K, Schuldes J, Klingl A, Poehlein A, Daniel R, Brune A. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of “Candidatus Methanoplasma termitum”. Appl Environ Microbiol. 2015;81:1338–52.
Borrel G, Gaci N, Peyret P, O'Toole PW, Gribaldo S, Brugere JF. Unique characteristics of the pyrrolysine system in the 7th order of methanogens: implications for the evolution of a genetic code expansion cassette. Archaea. 2014;2014:374146.
Carr SA, Schubotz F, Dunbar RB, Mills CT, Dias R, Summons RE, Mandernack KW. Acetoclastic methanosaeta are dominant methanogens in organic-rich antarctic marine sediments. ISME J. 2018;12:330–42.
Strapoc D, Picardal FW, Turich C, Schaperdoth I, Macalady JL, Lipp JS, Lin YS, Ertefai TF, Schubotz F, Hinrichs KU, Mastalerz M, Schimmelmann A. Methane-producing microbial community in a coal bed of the illinois basin. Appl Environ Microbiol. 2008;74:2424–32.
Conrad R. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol Ecol. 1999;28:193–202.
Liu Y, Priscu JC, Xiong J, Conrad R, Vick-Majors T, Chu H, Hou J. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol Ecol. 2016;92.
Chan OC, Claus P, Casper P, Ulrich A, Lueders T, Conrad R. Vertical distribution of structure and function of the methanogenic archaeal community in lake dagow sediment. Environ Microbiol. 2005;7:1139–49.
Alongi DM. Carbon cycling and storage in mangrove forests. Ann Rev Mar Sci. 2014;6:195–219.
Rosentreter JA, Maher DT, Erler DV, Murray RH, Eyre BD. Methane emissions partially offset “blue carbon” burial in mangroves. Sci Adv. 2018;4:eaao4985.
Cai M, Liu Y, Yin X, Zhou Z, Friedrich MW, Richter-Heitmann T, Nimzyk R, Kulkarni A, Wang X, Li W, Pan J, Yang Y, Gu J-D, Li M. Diverse asgard archaea including the novel phylum Gerdarchaeota participate in organic matter degradation. Sci China Life Sci. 2020;63.
Liu Y, Zhou Z, Pan J, Baker BJ, Gu JD, Li M. Comparative genomic inference suggests mixotrophic lifestyle for Thorarchaeota. ISME J. 2018;12:1021–31.
Pan J, Zhou Z, Beja O, Cai M, Yang Y, Liu Y, Gu JD, Li M. Genomic and transcriptomic evidence of light-sensing, porphyrin biosynthesis, calvin-benson-bassham cycle, and urea production in Bathyarchaeota. Microbiome. 2020;8:43.
Zhou Z, Liu Y, Lloyd KG, Pan J, Yang Y, Gu JD, Li M. Genomic and transcriptomic insights into the ecology and metabolism of benthic archaeal cosmopolitan, Thermoprofundales (MBG-D archaea). ISME J. 2019;13:885–901.
Zhou Z, Chen J, Cao H, Han P, Gu JD. Analysis of methane-producing and metabolizing archaeal and bacterial communities in sediments of the northern south china sea and coastal mai po nature reserve revealed by PCR amplification of mcrA and pmoA genes. Front Microbiol. 2014;5:789.
Chen GC, Tam NF, Ye Y. Summer fluxes of atmospheric greenhouse gases N2O, CH4 and CO2 from mangrove soil in South China. Sci Total Environ. 2010;408:2761–7.
Zhang C-J, Pan J, Duan C-H, Wang Y-M, Liu Y, Sun J, Zhou H-C, Song X, Li M. Prokaryotic diversity in mangrove sediments across southeastern China fundamentally differs from that in other biomes. mSystems. 2019;4:e00442–19.
Pan J, Chen Y, Wang Y, Zhou Z, Li M. Vertical distribution of Bathyarchaeotal communities in mangrove wetlands suggests distinct niche preference of Bathyarchaeota subgroup 6. Microb Ecol. 2019;77:417–28.
Zheng X, Guo J, Song W, Feng J, Lin G. Methane emission from mangrove wetland soils is marginal but can be stimulated significantly by anthropogenic activities. Forests. 2018;9:738.
Steinberg LM, Regan JM. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl Environ Microbiol. 2008;74:6663–71.
Xiao KQ, Beulig F, Kjeldsen KU, Jorgensen BB, Risgaard-Petersen N. Concurrent methane production and oxidation in surface sediment from Aarhus Bay, Denmark. Front Microbiol. 2017;8:1198.
Chen J, Wang F, Jiang L, Yin X, Xiao X. Stratified communities of active archaea in shallow sediments of the Pearl River Estuary, Southern China. Curr Microbiol. 2013;67:41–50.
Mitterer RM. Methanogenesis and sulfate reduction in marine sediments: a new model. Earth and Planetary Science Letters. 2010;295:358–66.
Wasmund K, Mussmann M, Loy A. The life sulfuric: microbial ecology of sulfur cycling in marine sediments. Environ Microbiol Rep. 2017;9:323–44.
Flood BE, Jones DS, Bailey JV. Complete genome sequence of Sedimenticola thiotaurini strain SIP-G1, a polyphosphate- and polyhydroxyalkanoate-accumulating sulfur-oxidizing Gammaproteobacterium isolated from salt marsh sediments. Genome Announcements. 2015;3:e00671–15.
Goker M, Teshima H, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, Han C, Goodwin L, Pitluck S, Huntemann M, Liolios K, Ivanova N, Pagani I, Mavromatis K, Ovchinikova G, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Brambilla EM, Rohde M, Spring S, Detter JC, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kyrpides NC, Klenk HP. Complete genome sequence of the acetate-degrading sulfate reducer Desulfobacca acetoxidans type strain (ASRB2). Stand Genomic Sci. 2011;4:393–401.
Jameson E, Stephenson J, Jones H, Millard A, Kaster AK, Purdy KJ, Airs R, Murrell JC, Chen Y. Deltaproteobacteria (Pelobacter) and Methanococcoides are responsible for choline-dependent methanogenesis in a coastal saltmarsh sediment. ISME J. 2019;13:277–89.
Liu P, Lu Y. Concerted metabolic shifts give new insights into the syntrophic mechanism between propionate-fermenting Pelotomaculum thermopropionicum and hydrogenotrophic Methanocella conradii. Front Microbiol. 2018;9:1551.
Xia X, Zhang J, Song T, Lu Y. Stimulation of Smithella-dominating propionate oxidation in a sediment enrichment by magnetite and carbon nanotubes. Environ Microbiol Rep. 2019;11:236–48.
Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, Frischkorn KR, Tringe SG, Singh A, Markillie LM, Taylor RC, Williams KH, Banfield JF. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol. 2015;25:690–701.
Liu X, Li M, Castelle CJ, Probst AJ, Zhou Z, Pan J, Liu Y, Banfield JF, Gu JD. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome. 2018;6:102.
Zhang CJ, Chen YL, Pan J, Wang YM, Li M. Spatial and seasonal variation of methanogenic community in a river-bay system in South China. Appl Microbiol Biotechnol. 2020;104:4593–603.
Wu S, Li R, Xie S, Shi C. Depth-related change of sulfate-reducing bacteria community in mangrove sediments: the influence of heavy metal contamination. Mar Pollut Bull. 2019;140:443–50.
Narrowe AB, Angle JC, Daly RA, Stefanik KC, Wrighton KC, Miller CS. High-resolution sequencing reveals unexplored archaeal diversity in freshwater wetland soils. Environ Microbiol. 2017;19:2192–209.
Lyu Z, Shao N, Akinyemi T, Whitman WB. Methanogenesis. Curr Biol. 2018;28:727–32.
Borton MA, Daly RA, O'Banion B, Hoyt DW, Marcus DN, Welch S, Hastings SS, Meulia T, Wolfe RA, Booker AE, Sharma S, Cole DR, Wunch K, Moore JD, Darrah TH, Wilkins MJ, Wrighton KC. Comparative genomics and physiology of the genus Methanohalophilus, a prevalent methanogen in hydraulically fractured shale. Environ Microbiol. 2018;20:4596–611.
Webster G, Sass H, Cragg BA, Gorra R, Knab NJ, Green CJ, Mathes F, Fry JC, Weightman AJ, Parkes RJ. Enrichment and cultivation of prokaryotes associated with the sulphate-methane transition zone of diffusion-controlled sediments of Aarhus Bay, Denmark, under heterotrophic conditions. FEMS Microbiol Ecol. 2011;77:248–63.
Li W, Guan W, Chen H, Liao B, Hu J, Peng C, Rui J, Tian J, Zhu D, He Y. Archaeal communities in the sediments of different mangrove stands at Dongzhaigang. China. J Soil Sediment. 2016;16:1995–2004.
Hu A, Hou L, Yu CP. Biogeography of planktonic and benthic archaeal communities in a subtropical eutrophic estuary of China. Microb Ecol. 2015;70:322–35.
Roessler M, Pfluger K, Flach H, Lienard T, Gottschalk G, Muller V. Identification of a salt-induced primary transporter for glycine betaine in the methanogen Methanosarcina mazei Go1. Appl Environ Microbiol. 2002;68:2133–9.
Browne P, Tamaki H, Kyrpides N, Woyke T, Goodwin L, Imachi H, Brauer S, Yavitt JB, Liu WT, Zinder S, Cadillo-Quiroz H. Genomic composition and dynamics among Methanomicrobiales predict adaptation to contrasting environments. ISME J. 2017;11:87–99.
Kono T, Mehrotra S, Endo C, Kizu N, Matusda M, Kimura H, Mizohata E, Inoue T, Hasunuma T, Yokota A, Matsumura H, Ashida H. A RuBisCO-mediated carbon metabolic pathway in methanogenic archaea. Nat Commun. 2017;8:14007.
Kuroda K, Hatamoto M, Nakahara N, Abe K, Takahashi M, Araki N, Yamaguchi T. Community composition of known and uncultured archaeal lineages in anaerobic or anoxic wastewater treatment sludge. Microb Ecol. 2015;69:586–96.
Wilkins D, Lu XY, Shen Z, Chen J, Lee PK. Pyrosequencing of mcrA and archaeal 16S rRNA genes reveals diversity and substrate preferences of methanogen communities in anaerobic digesters. Appl Environ Microbiol. 2015;81:604–13.
Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. Nov., sp. Nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. 2012;62:1902–7.
Kroninger L, Berger S, Welte C, Deppenmeier U. Evidence for the involvement of two heterodisulfide reductases in the energy-conserving system of Methanomassiliicoccus luminyensis. FEBS J. 2016;283:472–83.
Sun Y, Liu Y, Pan J, Wang F, Li M. Perspectives on cultivation strategies of archaea. Microb Ecol. 2019;79:770–84.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:590–6.
Shetty SA, Hugenholtz F, Lahti L, Smidt H, de Vos WM. Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol Rev. 2017;41:182–99.
Ju F, Zhang T. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J. 2015;9:683–95.
Bastian M, Heymann S, Jacomy M. Gephi : An open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media 2009 San Jose, CA, USA.
Ju F, Xia Y, Guo F, Wang Z, Zhang T. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ Microbiol. 2014;16:2421–32.
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Eren AM, Esen OC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ. 2015;3:e1319.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. Checkm: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31.
Huerta-Cepas J, Forslund K, Szklarczyk D, Jensen LJ, von Mering C, Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 2016;34:2115–22.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, Suzuki Y, Dudek N, Relman DA, Finstad KM, Amundson R, Thomas BC, Banfield JF. A new view of the tree of life. Nat Microbiol. 2016;1:16048.
Edgar RC. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Price MN, Dehal PS, Arkin AP. Fasttree 2 – approximately maximum-likelihood trees for large alignments. Plos One. 2010;5:e9490.
Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947-8.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Acknowledgements
We thank Joanna Mackie for providing professional writing services. We acknowledge the editor and anonymous reviewers for reviewing our manuscript. They made good comments to help us improve the paper.
Funding
This work was financially supported by the National Natural Science Foundation of China (grant no. 91851105, 31970105, and 31622002), the Shenzhen Science and Technology Program (grant no. JCYJ20170818091727570 and KQTD20180412181334790), the Key Project of Department of Education of Guangdong Province (grant no. 2017KZDXM071), China Postdoctoral Science Foundation (no. 2018 M630977), and the CAS Interdisciplinary Innovation Team (grant no. JCTD-2018-16).
Author information
Authors and Affiliations
Contributions
CJZ and ML conceived the study. CJZ, JP, and YL sampled sediments in the field. CJZ carried out laboratory work with the help from CHD and analyzed the data with the help from JP, YL, and CHD. CJZ wrote the paper with the help from all co-authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Range of CH4 emission from different mangrove ecosystems across the world.
Additional file 2: Figure S1.
Relative abundance of methanogens in prokaryotes (a) and community composition of methanogens (b) among 6 mangroves across southeastern China.
Additional file 3: Figure S2.
Phylogenetic trees of recovered MAGs using 16S rRNA gene sequences. Each name in bold represent a MAG. Bootstrap values were calculated via non-parametric bootstrapping with 100 replicates, and are represented by grey circles in different sizes. The scale bar indicates 10% estimated phylogenetic divergence.
Additional file 4: Figure S3.
Physiochemical properties and mcrA gene copies in five layers of mangrove sediments.
Additional file 5: Figure S4.
The relative abundances (metagenome, FPKM, blue) and expression levels (metatranscriptome, FPKM, red) of genes involved for methanogenesis with affiliation to four dominant methanogens (MF, MMA, MM and MS) at five layers in mangrove sediments.
Additional file 6: Table S2.
Genes of the recovered MAGs related to methanogenesis, energy conservation, carbon, nitrogen, sulfate, amino acid and ABC transporter.
Additional file 7: Table S3.
Reference genomes used to build the methanogens ribosomal protein tree (Fig. 1a).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, CJ., Pan, J., Liu, Y. et al. Genomic and transcriptomic insights into methanogenesis potential of novel methanogens from mangrove sediments. Microbiome 8, 94 (2020). https://doi.org/10.1186/s40168-020-00876-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-020-00876-z