
Abstract
Background
Influenza viruses are divided into three types, A, B, and C. Human influenza A and B viruses can cause seasonal epidemics, but influenza C causes only a mild respiratory illness. Influenza A virus can infect various host species. In 2013, human-infectious avian influenza A (H7N9) was first reported in China. By the second week of 2014, there were 210 laboratory-confirmed human cases in the country, and the mortality rate eventually reached 22 %. Rapid and accurate diagnosis of influenza viruses is important for clinical management and epidemiology.
Methods
In this assay, a cost-effective chemiluminescence (CL) detection oligonucleotide microarray was developed to genotype and detect avian influenza A (H7N9), avian influenza A (H5N1), 2009 influenza A (H1N1), seasonal influenza A (H1N1), and seasonal influenza A (H3N2). Influenza A viruses and influenza B viruses were also generally detected using this microarray.
Results
The results of detection of 40 cultivated influenza virus strains showed that the microarray was able to distinguish the subtypes of these influenza viruses very well. The microarray possessed similar or 10 fold higher limit of detection than the real-time RT-PCR method. Sixty-six clinical swab samples were detected using this microarray and verified with real time RT-PCR to evaluate the efficiency of this microarray for clinical testing.
Conclusions
A reliable CL detection oligonucleotide microarray had been developed to genotype and detected these influenza viruses.
Similar content being viewed by others


Background
Influenza viruses are divided into three types, A, B, and C (WHO 1980). Human influenza A and B viruses always cause seasonal epidemics, but influenza C causes only a mild respiratory illness. Influenza A viruses are further divided into various subtypes based on hemagglutinin (HA) and neuraminidase (NA). Currently, seventeen serotypes of hemagglutinin (H1–H17) and ten serotypes of neuraminidase (N1–N10) of influenza A virus have been identified in mammalian and avian species (Fouchier et al. 2005; Zhu et al. 2013). Influenza B viruses are broken down into B/Yamagata and B/Victoria lineages (Zhang et al. 2012). They are not divided into subtypes.
Influenza A virus can infect various host species. The greatest diversity of subtypes is found in wildfowl. The ease with which influenza A can recombine causes viral epidemics to spread across different species (dos Reis et al. 2011). There have been several influenza A pandemics during the past 100 years: influenza A (H1N1) caused Spanish flu in 1918, causing 50 million deaths (Taubenberger and Morens 2006). Another influenza A (H2N2), caused Asian flu in 1957, with one million deaths (Xu et al. 2010). Influenza A (H3N2) caused Hong Kong flu in 1968, with another one million deaths (Viboud et al. 2005). A novel influenza A first noted in 2009, H1N1 caused the swine flu in 2009, with 0.28 million deaths (Dawood et al. 2012). There have also been small-scale epidemics, such as the avian influenza virus A (H5N1) outbreak in Hong Kong in 1997, in which 18 individuals were infected and 6 died (WHO 2004). In addition to the avian influenza virus A H5N1, highly pathogenic avian influenza virus A H5N2 (Yang et al. 2015; Zhan et al. 2012), avian influenza virus A H5N5 (Gu et al. 2011), H5N6 (Li et al. 2016; Qi et al. 2014), H5N8 (Wang et al. 2016; Zhao et al. 2013), and H5N9 (Yu et al. 2015) have also been isolated or confirmed from avian, mice, dog, or humans in China since 2000. It is noteworthy that the first human avian influenza A H5N6 infections were reported in Sichuan province (Nations and F.a.A.O.o.t.U. 2014) and caused 14 laboratory-confirmed human cases (including six deaths) by 9 May 2016 (Organization 2016). In 2013, human-infectious avian influenza A (H7N9) was reported in China (Gao et al. 2013b). By the second week of March 2016, there had been 752 laboratory-confirmed human cases in China, including at least 295 deaths (WHO 2016a, b). The clinical manifestations, severity, and mortality rates of human-infectious avian influenza A (H7N9) were similar to those of avian influenza A (H5N1) and 2009 influenza A (H1N1) but quite different from seasonal influenza (Gao et al. 2013a). Many studies have shown that it is more effective to reduce the severity of the infection when administration of NA inhibitor within 48 h of the onset of influenza symptoms than after 48 h (Hayden et al. 1997; McGeer et al. 2007; Monto et al. 1999). For these reasons, rapid and accurate diagnosis influenza viruses are important to clinical management and epidemiological.
Some molecular diagnostic technologies have been used to diagnose and subtype influenza viruses: antigens and antibody detection (Duman et al. 2013; He et al. 2013), real-time PCR (Choi et al. 2013; Dawood et al. 2009; Gao et al. 2013a; Hackett et al. 2014; Poon et al. 2009; Templeton et al. 2004), sequencing (Deng et al. 2011; Ghedin et al. 2011; Rutvisuttinunt et al. 2013), and microarray (Gall et al. 2009; Han et al. 2008; Heil et al. 2010; Ryabinin et al. 2011). In the assay described here, a chemiluminescence (CL) detection oligonucleotide microarray was developed and used to detect avian influenza A (H7N9), avian influenza A (H5N1), 2009 influenza A (H1N1), seasonal influenza A (H1N1), and seasonal influenza A (H3N2) by genotyping. The microarray also detected influenza A and influenza B viruses in all cases. The efficiency of this CL detection strategy was investigated in actual samples and the results were compared to those of real-time PCR methods.
Methods
Specimen collection and processing
The six clinically isolated strains of avian influenza A (H7N9) were obtained from the Centers for Disease Control (CDC) of Zhejiang Province. The other cultivated influenza virus strains were obtained from the National Institutes for Food and Drug Control. Clinical throat swab samples were collected from patients suspected of having infected influenza in the 307th Hospital of the Chinese People’s Liberation Army for whom it was not possible to determine whether they had taken medication. Non-influenza respiratory viruses were identified and obtained from the National Institute for Viral Disease Control and Prevention and the 302nd Hospital of the Chinese People’s Liberation Army. Lysate had already been added to all these viral samples before they were acquired for the present study. Total RNAs were extracted using the TIANamp Virus RNA Kit as described in the manufacturer’s protocol (TIANGEN Biotech Beijing Co., Ltd.) and stored at −70 °C until use.
Primer and probe design
HA and NA gene FASTA sequences of avian influenza H7N9 virus, NA gene FASTA sequences of avian influenza H5N1 virus, 2009 influenza A (H1N1), seasonal influenza A (H1N1), influenza A (H3N2), matrix protein (M) gene of influenza A virus, and nonstructural protein (NS) gene of influenza B virus were downloaded from NCBI’s influenza nucleotide database. The sequences were then aligned using AlignX (a component of the Vector NTI Advance 10.3.0) to compare homology. Genotyping primers for these subtypes of influenza A were designed using Primer Premier 5 (PREMIER Biosoft International, USA) and they served as the consensus sequences of the HA and NA genes. Universal primers for influenza A virus and influenza B virus were designed and served as the consensus sequences of the relatively well-conserved M and NS genes. Microarray probes ranging from 20 to 40 nt were designed to detect influenza A virus generally, influenza B virus generally, and these subtypes of influenza A virus. Internal standard primers and probes based on the sequences of Homo sapiens ribonuclease P were designed to monitor specimen extraction, RT-PCR amplification, and microarray hybridization. This marker has been described as a reliable internal positive control marker in several publications (Dare et al. 2016; Fan et al. 2014). All the primers and probes were confirmed using BLAST program of NCBI and then synthesized using Sangon Biotech Co., Ltd. (Shanghai). The primers and probes are shown in Tables 1 and 2.
Microarray preparation
A repeat sequence of (T)12 with an amino-labeled 3′-end was connected to the 3′-end of all the probes so that it could be fixed to the aldehyde-chip surface.
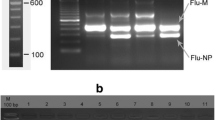
A repeat sequence of (T)20 with a biotin-labeled 5′-end and an amino-labeled 3′-end served as a quality control probe. The probes were used at 50 μM final concentration. They were spotted and repeated three times in the vertical direction on the surface of the aldehyde-chip using uniform proportional printing buffer as described in previous studies (Zhang et al. 2013). The quality control probes were spotted and repeated seven times in the horizontal direction (Fig. 1b).

Microarray layout and CL detection results of parts of cultivated influenza viruses. a Working principle of this CL imaging DNA hybridization method. Steps 1–2 showed that capture probes were fixed to the aldehyde-chip surface. Step 3 showed that the denatured RT-PCR products were hybridized on the capture-chip. Steps 4–5 showed the CL detection principle. Biotin was incorporated into reverse strand on the RT-PCR amplification. Then, HRP modified streptavidin was bound and CL signal was generated by catalysed substrates. b Microarray layout. Capture probes were spotted in triplicate in col. The sequences of (T)20 were repeated seven times for quality control. c CL detection results of parts of cultivated influenza viruses. These cultivated influenza virus strains of influenza virus were derived from the National Institutes for Food and Drug Control and CDC of Zhejiang Province. The results showed that the microarray was able to distinguish the subtypes of avian influenza A (H7N9), avian influenza A (H5N1), 2009 influenza A (H1N1), seasonal influenza A (H1N1), influenza A (H3N2), and influenza B virus very well
RT-PCR amplification
The primers were used in three different RT-PCR systems. Each RT-PCR was performed in a 25 μl reaction volume containing 12.5 μl of 2× One Step Buffer, 1.0 μl of PrimeScript One-step Enzyme Mix (DRR055A, Takara Biotechnology (Dalian) Co., Ltd.), 5 μl of total RNA template, and specific primer mix. RT-PCR system A amplified novel avian influenza A (H7N9) and avian influenza A (H5N1). It contained primer H7F1 0.16 μM, H7R2-B 0.8 μM, NA-1Sa 0.12 μM, NA9r-B-4 0.6 μM, NF11 0.1 μM, NR22B 0.5 μM, RP-F4 0.08 μM, and RP-R4 0.4 μM. RT-PCR system B amplified 2009 influenza A (H1N1), seasonal influenza A (H1N1), and influenza A (H3N2). It contained primer F7 0.1 μM, R4aB 0.5 μM, NF5 0.1 μM, NR5B 0.5 μM, NF12 0.1 μM, NR21B 0.5 μM, RP-F1 0.1 μM, and RP-R1B 0.5 μM. RT-PCR system C amplified influenza A and B viruses. It contained primer MF2 0.1 μM, MR2B 0.5 μM, FBF1 0.1 μM, FBR1B 0.5 μM, PR-F 0.05 μM, and RP-RB 0.25 μM. All the three systems contained a pair of primers that could amplify the gene of Homo sapiens ribonuclease P, which served as an internal standard. Amplifications were performed on a Veriti 96-Well Thermal Cycler PCR system (Applied Biosystems, USA) under the following conditions: 30 min at 50 °C; 2 min at 94 °C; 45 cycles of 20 s at 94 °C, 20 s at 52 °C, and 20 s at 72 °C; and a final extension of 5 min at 72 °C.
Hybridization and signal detection
After amplification, the three reaction products derived from the same template were mixed into one tube (10 µl each). After 5 min of denaturation at 95 °C, the mixture was immediately placed on ice 5 min and then mixed with 5 μl of hybridization buffer (8× SSC, 0.6 % SDS, 10 % formylamine, and 10× Denhardt). A total of 10 μl of hybridization mixture was hybridized on the microarray for 1 h at 45 °C as described (Zhang et al. 2013). Then, the chip was washed for 30 s each with 1× SSC and 0.2 % SDS, 0.2× SSC, and 0.1× SSC at room temperature. Then the chip was incubated with 15 μl of streptavidin-horseradish peroxidase (Str-HRP, SIGMA) for 25 min at 37 °C. Subsequently, the chip was washed with PBST (phosphate buffer, 0.05 % Tween 20) 10 s at room temperature. Finally, 10 μL of pre-mixed CL HRP substrate luminol solution and H2O2 (Millipore Corporation, USA) was added to the chip and immediately detected with a micro-light level imaging system (developed in-house, China Patent Application No. 201310013267.X).
Specificity and sensitivity
The specificity of this microarray was evaluated using 40 cultivated influenza A and B virus strains which were derived from CDC of Zhejiang Province and the National Institutes for Food and Drug Control. The genotype of these cultivated influenza viruses had been determined and cultivated at these two institutions. Influenza A and B Nucleic Acid Detection Kits (PCR-fluorescent probe) (Shenzhen Puruikang Biotech Co., Ltd.) which generally detected influenza A viruses and influenza B viruses, were used to compare results. A panel of non-influenza respiratory viruses, which include parainfluenza virus, adenovirus AD3, AD4, AD30, AD40, measles virus, mumps virus, and respiratory syncytial viruses HK6 and B, were also used to determine the specificity of the microarray.
In order to evaluate the sensitivity of this microarray in the detection of various subtypes of influenza virus, the RNA extraction of cultivated virus strains of 5 subtypes of influenza A virus and influenza B virus were serially diluted in tenfold increments and detected using the microarray. Influenza A and B Nucleic Acid Detection Kits (PCR-fluorescent probe) (Shenzhen Puruikang Biotech Co., Ltd.) were also used to compare results. They amplify the conserved M gene of influenza A viruses and the NS gene region of influenza B viruses.
Clinical samples
To investigate the efficiency of clinical testing, 66 clinical swab samples were collected from the 307th Hospital of the Chinese People’s Liberation Army. Total RNAs were extracted using a TIANamp Virus RNA Kit as described in the manufacturer’s protocol. Then the clinical swab samples were detected using a microarray and verified using four different kinds of real-time RT-PCR kits. These real-time RT-PCR kits included a 2009 Influenza A Virus (H1N1) Nucleic Acid Detection Kit (PCR-fluorescent probe) (Shenzhen Puruikang Biotech Co., Ltd.), 2009 Influenza A Virus (H1N1) Nucleic Acid Detection Kit (PCR-fluorescent probe) (DAAN Gene Co., Ltd.), Seasonal Influenza A Virus (H3) Nucleic Acid Detection Kit (PCR-Fluorescent probe) (DAAN Gene Co., Ltd.), and Diagnosis Kit for H7N9 Avian Influenza Virus RNA (PCR-fluorescent probe) (Shenzhen Puruikang Biotech Co., Ltd.). These real-time RT-PCR kits identified and determined the concentration of HA gene of 2009 influenza A (H1N1), NA gene of 2009 Influenza A Virus (H1N1), influenza A (H3N2), and avian influenza H7N9 virus. They were all used as described in the manufacturer’s protocol.
Results
Primers and probes
Specificity of the microarray
In this assay, a total of 40 cultivated influenza virus strains were tested to determine the specificity of this microarray. The types or subtypes of these strains included 6 avian influenza A (H7N9), 6 influenza A (H3N2), 4 seasonal influenza A (H1N1), 2 2009 influenza A (H1N1), 18 avian influenza A (H5N1), and 4 influenza B virus. The results showed that one strain of H3N2 was mixed with H1N1, one strain of H5N1 was mixed with H1N1, another strain of H5N1 was mixed with H3N2, one strain of influenza B was mixed with H1N1, and one strain of H3N2 did not subtype because of its low concentration. The four mixed samples were confirmed by amplifying the M gene (for influenza A viruses) or NS gene (for influenza B viruses) of influenza using universal primers of influenza A and B viruses (Table 1) respectively and ligating the amplifications into the T-vector which were subsequently Sanger sequenced (data not shown). The Sanger sequencing results were coordinated to that of a microarray (results not shown). The results indicated that the microarray was able to distinguish these subtypes of influenza A and B viruses (Fig. 1c). There were no cross-signals between various influenza A virus subtypes, and the microarray simultaneously detected mixed subtypes in some mixed samples. Furthermore, because we observed a lower signal for H5N1 than the other subtypes with the general influenza A primers targeting the M gene (Fig. 1c), we investigated the number of mismatches of our probes to the H5N1 strains used here. Overall, there were 0, 1 and 1 mismatches in the forward primer, probe and reverse primer regions. In comparison, the suggested universal WHO primers (WHO 2009) also targeting the M gene have 1, 0 and 1 mismatches against the same strains in the forward primer, probe and reverse primer regions, respectively. In addition, the negative microarray results of these common respiratory viruses also demonstrated the specificity of this assay (Fig. 2). The information and detection results of the forty influenza virus strains are shown in Table 3.

Detection of non-influenza respiratory viruses. The negative microarray results of these common respiratory viruses also demonstrated the specificity of this assay
The limit of detection of the microarray
To assess sensitivity, the microarray detection methods were compared to real-time RT-PCR. Results showed that the microarray have similar (for PH1N1) or tenfold higher limit of detection (LOD) (to the other subtypes of influenza A virus and influenza B virus) than the referenced real-time RT-PCR method. The LOD of general influenza A virus detection was lower than the LOD of genotype detection. The LOD for the two comparator real-time PCR assays was 1 × 103 PFU/ml, so the LOD of the microarray was 1 × 103–10 × 103 PFU/ml. The comparative results of LOD of avian influenza A (H7N9) are shown in Fig. 3. The comparative results of LOD of other influenza viruses are shown in Table 4.

LOD of detection of avian influenza A (H7N9). The RNA extraction of avian influenza (H7N9) was serially diluted in tenfold increments and detected using this microarray and influenza A virus Nucleic Acid Detection Kits (PCR-fluorescent probe) (Shenzhen Puruikang Biotech Co., Ltd.). The microarray possessed a tenfold higher LOD to avian influenza A (H7N9) than the real-time RT-PCR method
Detection of subtypes of cultivated virus and clinical positive swab samples
Sixty-six clinical swab samples were detected by microarray. Results indicated that 25 of the samples were 2009 influenza A (H1N1), 3 were influenza A (H3N2), 26 were influenza A, 1 contained both 2009 influenza A (H1N1) and influenza A (H3N2), and 11 were negative. Four different real-time PCR influenza genotyping kits served as reference methods. Results are shown in Table 5. Some samples that were only shown to contain only influenza A by microarray were confirmed to have specific subtypes using real-time PCR kits. This may be because the microarrays are less sensitive than real-time PCR kits. The results showed that microarray always failed to determine the subtypes of samples when the CT value of real-time RT-PCR exceeded 31. However, general detection of influenza A virus is possible even when the CT values were as high as 39. Consequently, general detection of influenza A virus was more sensitive than genotyping and detection of influenza A virus subtypes.
The statistical data of 66 clinical throat swab samples are shown in Table 6. The sensitivity, specificity, positive predictive values, and negative predicative values of microarray were 91.1, 60.0, 92.7, and 54.5 % those of the corresponding PCR kits. Furthermore, the value of genotyping for positive samples using this microarray was 53.6 %. The low specificity was attributable to the fact that there were four samples shown to contain influenza A by microarray and whose subtypes were not confirmed using real-time PCR kits. The results for these four specimens may be attributable to the low viral load or perhaps the influenza viruses were from subtypes not included in the microarray or real-time PCR kits. The low negative predicative values were attributable to the low sensitivity of the microarray relative to real-time PCR kits, which has been confirmed in cultivated influenza virus strains.
Discussion
Influenza A virus can infect various host species, and the greatest diversity of subtypes is found in wildfowl. The influenza virus genome comprises 8 independent RNA strands, so it can easily undergo antigenic drift, antigenic shift, and reassortment, which allows it to cause major pandemics (Labella and Merel 2013). Over the past 100 years, four subtypes of influenza A virus have had an enormous impact on human beings, H1N1, H3N2, H2N2, and H5N1. Data from the American Center for Disease Control and Prevention have shown the influenza B virus to account for only 14 % of cases from October 2011 to May 2012 (CDC 2012). In 2013, there was a sudden human avian influenza A (H7N9) outbreak, inducing great panic in China. Clinical data suggested that H7N9 caused pneumonia with various degrees of severity, acute lung injury, acute respiratory distress syndrome, and even multiple organ failure. Increased concentrations of C reactive protein were found to be closely associated with mortality (Lu et al. 2014). According to published reports, the median age of patients who died of H7N9 was significantly higher than that of patients with nonfatal cases. Patients found to have increased risk of death upon admission were subjected to cheat imaging and found to have either bilateral lung inflammation or pulmonary consolidation. There were found to be significant levels of lymphopenia and decreases in oxygenation index in patients who eventually died (Ji et al. 2014).
Since October 2013, the strains of influenza A circulating in China have been completely resistant to amantadine drugs. Some of the 2009 influenza A (H1N1) strains have been found to be associated with lower susceptibility to NA inhibitor as indicated by weekly monitoring. Many studies have shown that the severity of the infection is more likely to be reduced if an NA inhibitor is administered within 48 h of the onset of influenza symptoms than when it is administered later (Hayden et al. 1997; McGeer et al. 2007; Monto et al. 1999). Vaccination is another effective way to prevent the spread of pandemic virus and reduce the severity of the disease (Luke and Subbarao 2006). Synthetic DNA vaccines and virus-like-particle vaccines have been developed for avian influenza A (H7N9) (Dormitzer et al. 2013; Fries et al. 2013; Klausberger et al. 2014; Smith et al. 2013; Yan et al. 2014). Because vaccine strains match circulating strains closely, it is important to rapidly and continuously monitor of epidemic influenza virus strains to determine which strains should be covered by seasonal influenza vaccinations (Bandt et al. 2012; Luke and Subbarao 2006; Subbarao et al. 2006). Rapid and accurate diagnosis of avian influenza A (H7N9) and other common influenza viruses is essential to improving clinical patient management and epidemiological investigation.
Currently, real-time RT-PCR is the most widely used molecular diagnostic approach to the detection of influenza virus in clinical settings (Choi et al. 2013; Dawood et al. 2009; Gao et al. 2013a; Hackett et al. 2014; Poon et al. 2009; Templeton et al. 2004). Although the real-time RT-PCR approach is generally more sensitive than microarray, only a few subtypes of influenza can be detected in the same reaction (Choi et al. 2013; Kuo et al. 2014). Microarrays are high-throughput and can be used to simultaneously detect several or even all the subtypes of influenza (Gall et al. 2009; Han et al. 2008; Heil et al. 2010; Ryabinin et al. 2011). However, traditional microarray detection requires expensive fluorescence scanners, which limit its use. Analytical CL is a versatile, sensitive method of detection with a wide range of uses, including enzyme-linked immunosorbent assays (Liu et al. 2012; Maiolini et al. 2013), lateral flow immunoassays (Maiolini et al. 2013; Wolter et al. 2008; Wutz et al. 2013), flow-through hybridization assays (Hommatsu et al. 2013), capillary electrophoresis (Jiang et al. 2013), flow injection CL analysis (Tan and Song 2013), and magnetic bead-based DNA hybridization assays (Li and He 2009). This is the first paper to subtype influenza viruses using a chemiluminescent oligonucleotide microarray. In this assay, a sensitive CL method that relies on horseradish peroxidase was used to catalyze the luminol-H2O2 for a conventional oligonucleotide microarray. A proprietary potable CL imaging system was established. The imaging system was based on CCD camera imaging technology and equipped with a power supply suitable for portable use (China Patent Application No. 201310013267.X). Furthermore, other commercial CL imagers based on CCD imaging technology (e.g. Amersham Imager 600, GE Healthcare Life Sciences) could also be used for this CL microarray. The new CL imager had a lower cost and a much faster detection speed than our previous visual microarray system which was based on quantum dot-catalyzed silver deposition. The cost of this CL imaging microarray was also lower than that of our previous fluorescence based microarray system (Zhang et al. 2013).
This assay was designed based on the following ideas: (1) priority was given to the detection avian influenza A (H7N9) (H7 and N9 were detected simultaneously). (2) Other than avian influenza A (H7N9), only the most common subtypes of influenza virus were detected by genotyping in this microarray. (3) Other than genotyping detection, the microarray also universal detected the more conservative M gene of influenza A viruses and NS gene of influenza B viruses to extend the range of the application. (4) One-step RT-PCR amplification was used to reduce the length of the experimental procedure and so reduce the risk of contamination. (5) Human-derived primers and probes served as internal standards; they were added to all the amplification reactions to monitor extraction of clinical samples, RT-PCR amplification, and microarray hybridization. (6) Low-cost CL imaging technology was used for detection in the microarray. This may allow this system to be used on a large scale.
A low-density CL oligonucleotide microarray was developed for genotyping detection of the newest avian influenza A (H7N9), avian influenza A (H5N1), 2009 influenza A (H1N1), seasonal influenza A (H1N1), and influenza A (H3N2). Influenza A viruses and influenza B viruses were also generally detected using this microarray. The microarray kits used here were capable of rapid, high-throughput, highly accurate readings, and the entire detection time from sample extraction to the production of genotyping results was 6–8 h. Six avian influenza A (H7N9) culture viruses and thirty-four other influenza culture viruses served as positive references to confirm the specificity and sensitivity of the microarray. Some non-influenza respiratory viruses were also tested using the microarray to further confirm this specificity. All the results showed that the microarray accurately detected the genotypes of these influenza viruses and of some mixed influenza strains. The LOD of genotyping and universal detection of influenza viruses were similar to or ten-fold higher than reference values for influenza real-time RT-PCR kits. Sixty-six clinical swab samples were assessed using microarray to determine the efficiency of this type of clinical testing and epidemiology. Results showed that the specificity and sensitivity of the microarray met the needs of clinical and epidemiological studies of the influenza virus.
This method has some limitations. RT-PCR amplification was here divided into three groups to amplify different subtypes of influenza viruses. This complicated the operation. However, reducing the number of amplification systems (increasing the number of primers in one amplification system) may have decreased sensitivity. Here, 11 pairs of primers were divided into three groups of amplification systems, and the LOD of this microarray was still ten times higher than that of the real-time PCR kits used for reference (the sensitivity of 2009 influenza A virus was similar to that of real-time RT-PCR kit). The 66 clinical swab samples were detected and microarray failed to show subtypes of samples when the CT value of real-time RT-PCR more than 32. However, general detection of influenza A virus was still positive even when the CT values were as high as 39. Consequently, the sensitivity of this CL detection strategy and microarray could be improved by further optimization. Moreover, avian influenza A (H7N9) and (H5N1) were not actually comparatively tested using the developed microarray and PCR assays due to the deficiency of clinical specimens.
Furthermore, the use of this strategy was restricted because of the limitations of detection for only novel avian influenza A (H7N9) and some of the most common human influenza viruses. The other influenza A subtypes such as H5N6 and H9N2, can not be typed. Thus, sequencing or other diagnostic methods are still needed to determine the exact subtypes of those influenza A viruses.
Conclusions
A reliable CL detection oligonucleotide microarray was developed to genotype and detected avian influenza A (H7N9), avian influenza A (H5N1), 2009 influenza A (H1N1), seasonal influenza A (H1N1), and seasonal influenza A (H3N2). Influenza A viruses and influenza B viruses were also generally detected using this microarray. The results of detection of 40 cultivated influenza virus strains showed the microarray to be able to distinguish the subtypes of these influenza viruses very well. The microarray possessed similar or tenfold higher LOD than the real-time RT-PCR method. Sixty-six clinical swab samples were detected by microarray and verified using real-time RT-PCR to evaluate the efficiency of this microarray for clinical testing.
Abbreviations
- CDC:
-
the Centers for Disease Control
- CL:
-
chemiluminescence
- HA:
-
hemagglutinin
- LOD:
-
limit of detection
- M gene:
-
matrix protein gene
- NA:
-
neuraminidase
- NS gene:
-
nonstructural protein gene
- Str-HRP:
-
streptavidin-horseradish peroxidase
References
Bandt D, Monecke S, Scott C, Gall A, Hoffmann B, Ehricht R (2012) Economic high-throughput-identification of influenza A subtypes from clinical specimens with a DNA-oligonucleotide microarray in an outbreak situation. Mol Cell Probes 26:6–10
CDC (2012) Update: influenza activity - United States, 2011–12 season and composition of the 2012–13 influenza vaccine. MMWR Morb Mortal Wkly Rep 61:414–420
Choi JH, Kim MS, Lee JY, Lee NJ, Kwon D, Kang MG, Kang C (2013) Development and evaluation of multiplex real-time RT-PCR assays for seasonal, pandemic A/H1pdm09 and avian A/H5 influenza viruses detection. J Microbiol 51:252–257
Dare R, Zhu Y, Williams JV, Griffin M, Talbot HK (2016) Detection of influenza by real time RT-PCR is not affected by delays in respiratory specimen processing. J Med Virol 88:1891–1895
Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360:2605–2615
Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P et al (2012) Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis 12:687–695
Deng YM, Caldwell N, Barr IG (2011) Rapid detection and subtyping of human influenza A viruses and reassortants by pyrosequencing. PLoS ONE 6:e23400
Dormitzer PR, Suphaphiphat P, Gibson DG, DE Wentworth, Stockwell TB, Algire MA, Alperovich N, Barro M, Brown DM, Craig S et al (2013) Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci Transl Med 5:185ra168
dos Reis M, Tamuri AU, Hay AJ, Goldstein RA (2011) Charting the host adaptation of influenza viruses. Mol Biol Evol 28:1755–1767
Duman M, Gencpnar P, Ozbek OA, Ozdemir D, Sayner AA (2013) Value of rapid antigen test for pandemic influenza A (H1N1) 2009 in the pediatric emergency department. Pediatr Emerg Care 29:612–616
Fan J, Cui D, Lau S, Xie G, Guo X, Zheng S, Huang X, Yang S, Yang X, Huo Z et al (2014) Detection of a novel avian influenza A (H7N9) virus in humans by multiplex one-step real-time RT-PCR assay. BMC Infect Dis 14:541
Fouchier RA, Munster V, Wallensten A, Bestebroer TM, Herfst S, Smith D, Rimmelzwaan GF, Olsen B, Osterhaus AD (2005) Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol 79:2814–2822
Fries LF, Smith GE, Glenn GM (2013) A recombinant viruslike particle influenza A (H7N9) vaccine. N Engl J Med 369:2564–2566
Gall A, Hoffmann B, Harder T, Grund C, Ehricht R, Beer M (2009) Rapid and highly sensitive neuraminidase subtyping of avian influenza viruses by use of a diagnostic DNA microarray. J Clin Microbiol 47:2985–2988
Gao HN, Lu HZ, Cao B, Du B, Shang H, Gan JH, Lu SH, Yang YD, Fang Q, Shen YZ et al (2013a) Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med 368:2277–2285
Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K et al (2013b) Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368:1888–1897
Ghedin E, Laplante J, DePasse J, Wentworth DE, Santos RP, Lepow ML, Porter J, Stellrecht K, Lin X, Operario D et al (2011) Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis 203:168–174
Gu M, Liu W, Cao Y, Peng D, Wang X, Wan H, Zhao G, Xu Q, Zhang W, Song Q et al (2011) Novel reassortant highly pathogenic avian influenza (H5N5) viruses in domestic ducks, China. Emerg Infect Dis 17:1060–1063
Hackett H, Bialasiewicz S, Jacob K, Bletchly C, Harrower B, Nimmo GR, Nissen MD, Sloots TP, Whiley DM (2014) Screening for H7N9 influenza A by matrix gene-based real-time reverse-transcription PCR. J Virol Methods 195:123–125
Han X, Lin X, Liu B, Hou Y, Huang J, Wu S, Liu J, Mei L, Jia G, Zhu Q (2008) Simultaneously subtyping of all influenza A viruses using DNA microarrays. J Virol Methods 152:117–121
Hayden FG, Osterhaus AD, Treanor JJ, Fleming DM, Aoki FY, Nicholson KG, Bohnen AM, Hirst HM, Keene O, Wightman K (1997) Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenzavirus infections. GG167 Influenza Study Group. N Engl J Med 337:874–880
He F, Prabakaran M, Tan Y, Indira K, Kumar SR, Kwang J (2013) Development of dual-function ELISA for effective antigen and antibody detection against H7 avian influenza virus. BMC Microbiol 13:219
Heil GL, McCarthy T, Yoon KJ, Liu S, Saad MD, Smith CB, Houck JA, Dawson ED, Rowlen KL, Gray GC (2010) MChip, a low density microarray, differentiates among seasonal human H1N1, North American swine H1N1, and the 2009 pandemic H1N1. Influenza Other Respir Viruses 4:411–416
Hommatsu M, Okahashi H, Ohta K, Tamai Y, Tsukagoshi K, Hashimoto M (2013) Development of a PCR/LDR/flow-through hybridization assay using a capillary tube, probe DNA-immobilized magnetic beads and chemiluminescence detection. Anal Sci 29:689–695
Ji H, Gu Q, Chen LL, Xu K, Ling X, Bao CJ, Tang FY, Qi X, Wu YQ, Ai J et al (2014) Epidemiological and Clinical Characteristics and Risk Factors for Death of Patients with Avian Influenza A H7N9 Virus Infection from Jiangsu Province, Eastern China. PLoS ONE 9:e89581
Jiang J, Zhao S, Huang Y, Qin G, Ye F (2013) Highly sensitive immunoassay of carcinoembryonic antigen by capillary electrophoresis with gold nanoparticles amplified chemiluminescence detection. J Chromatogr A 1282:161–166
Klausberger M, Wilde M, Palmberger D, Hai R, Albrecht RA, Margine I, Hirsh A, Garcia-Sastre A, Grabherr R, Krammer F (2014) One-shot vaccination with an insect cell-derived low-dose influenza A H7 virus-like particle preparation protects mice against H7N9 challenge. Vaccine 32:355–362
Kuo RL, Yang SL, Liu YC, Chen LT, Mok CK, Kuo SM, Shih SR, Tsao KC (2014) Influenza A/B virus detection and influenza A virus subtyping with emphasis on the novel H7N9 virus by using multiplex real-time RT-PCR. J Virol Methods 208:41–46
Labella AM, Merel SE (2013). Influenza. Med Clin North Am 97:621–645, x
Li H, He Z (2009) Magnetic bead-based DNA hybridization assay with chemiluminescence and chemiluminescent imaging detection. Analyst 134:800–804
Li K, Liu H, Yang Z, Li T, Di B, Chen Z, Lu J, Chen G, Zhao P, Yang L et al (2016) Clinical and epidemiological characteristics of a patient infected with H5N6 avian influenza A virus. J Clin Virol 82:20–26
Liu Z, Song C, Li Y, Liu F, Zhang K, Sun Y, Li H, Wei Y, Xu Z, Zhang C et al (2012) Development of highly sensitive chemiluminescence enzyme immunoassay based on the anti-recombinant H(C) subunit of botulinum neurotoxin type A monoclonal antibodies. Anal Chim Acta 735:23–30
Lu S, Li T, Xi X, Chen Q, Liu X, Zhang B, Ou J, Liu J, Wang Q, Zhu B et al (2014) Prognosis of 18 H7N9 avian influenza patients in Shanghai. PLoS ONE 9:e88728
Luke CJ, Subbarao K (2006) Vaccines for pandemic influenza. Emerg Infect Dis 12:66–72
Maiolini E, Ferri E, Pitasi AL, Montoya A, Di Giovanni M, Errani E, Girotti S (2013) Bisphenol a determination in baby bottles by chemiluminescence enzyme-linked immunosorbent assay, lateral flow immunoassay and liquid chromatography tandem mass spectrometry. Analyst. doi:10.1039/c3an00552f
McGeer A, Green KA, Plevneshi A, Shigayeva A, Siddiqi N, Raboud J, Low DE (2007) Antiviral therapy and outcomes of influenza requiring hospitalization in Ontario, Canada. Clin Infect Dis 45:1568–1575
Monto AS, Fleming DM, Henry D, de Groot R, Makela M, Klein T, Elliott M, Keene ON, Man CY (1999) Efficacy and safety of the neuraminidase inhibitor zanamivirin the treatment of influenza A and B virus infections. J Infect Dis 180:254–261
Nations, F.a.A.O.o.t.U. (2014) Avian influenza A (H5N6): the latest addition to emerging zoonotic avian influenza threats in East and Southeast Asia. http://www.fao.org/3/a-i4199e.pdf. Accessed 30 Nov 2014
Poon LL, Chan KH, Smith GJ, Leung CS, Guan Y, Yuen KY, Peiris JS (2009) Molecular detection of a novel human influenza (H1N1) of pandemic potential by conventional and real-time quantitative RT-PCR assays. Clin Chem 55:1555–1558
Qi X, Cui L, Yu H, Ge Y, Tang F (2014) Whole-genome sequence of a reassortant H5N6 avian influenza virus isolated from a live poultry market in China, 2013. Genome Announc 2. doi:10.1128/genomeA.00706-14
Rutvisuttinunt W, Chinnawirotpisan P, Simasathien S, Shrestha SK, Yoon IK, Klungthong C, Fernandez S (2013) Simultaneous and complete genome sequencing of influenza A and B with high coverage by Illumina MiSeq Platform. J Virol Methods 193:394–404
Ryabinin VA, Kostina EV, Maksakova GA, Neverov AA, Chumakov KM, Sinyakov AN (2011) Universal oligonucleotide microarray for sub-typing of Influenza A virus. PLoS ONE 6:e17529
Smith GE, Flyer DC, Raghunandan R, Liu Y, Wei Z, Wu Y, Kpamegan E, Courbron D, Fries LF 3rd, Glenn GM (2013) Development of influenza H7N9 virus like particle (VLP) vaccine: homologous A/Anhui/1/2013 (H7N9) protection and heterologous A/chicken/Jalisco/CPA1/2012 (H7N3) cross-protection in vaccinated mice challenged with H7N9 virus. Vaccine 31:4305–4313
Subbarao K, Murphy BR, Fauci AS (2006) Development of effective vaccines against pandemic influenza. Immunity 24:5–9
Tan X, Song Z (2013) Human saliva-based quantitative monitoring of clarithromycin by flow injection chemiluminescence analysis: a pharmacokinetic study. Appl Biochem Biotechnol 172:1320–1331. doi:10.1007/s12010-013-0605-4
Taubenberger JK, Morens DM (2006) 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 12:15–22
Templeton KE, Scheltinga SA, Beersma MF, Kroes AC, Claas EC (2004) Rapid and sensitive method using multiplex real-time PCR for diagnosis of infections by influenza a and influenza B viruses, respiratory syncytial virus, and parainfluenza viruses 1, 2, 3, and 4. J Clin Microbiol 42:1564–1569
Viboud C, Grais RF, Lafont BA, Miller MA, Simonsen L (2005) Multinational impact of the 1968 Hong Kong influenza pandemic: evidence for a smoldering pandemic. J Infect Dis 192:233–248
Wang X, Meng F, Wang D, Liu X, Chen S, Qin T, Peng D, Liu X (2016) Characteristics of two highly pathogenic avian influenza H5N8 viruses with different pathogenicity in mice. Archives Virol 161:3365–3374
WHO (1980) A revision of the system of nomenclature for influenza viruses: a WHO memorandum. Bull World Health Organ 58:585–591
WHO (2004) Avian influenza A (H5N1) in humans and poultry in Viet Nam. http://www.who.int/csr/don/2004_01_13/en/. Accessed 13 Jan 2004
WHO (2009) CDC protocol of realtime RT-PCR for influenza A (H1N1). http://www.who.int/csr/resources/publications/swineflu/CDCRealtimeRTPCR_SwineH1Assay-2009_20090430.pdf?ua=1. Accessed 6 Oct 2009
WHO (2016a) Influenza at the human-animal interface. http://www.who.int/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_05_09_2016.pdf. Accessed 9 May 2016
WHO (2016b) Influenza at the human–animal interface. http://www.who.int/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_04_04_2016.pdf. Accessed 4 Apr 2016
Wolter A, Niessner R, Seidel M (2008) Detection of Escherichia coli O157:H7, Salmonella typhimurium, and Legionella pneumophila in water using a flow-through chemiluminescence microarray readout system. Anal Chem 80:5854–5863
Wutz K, Meyer VK, Wacheck S, Krol P, Gareis M, Nolting C, Struck F, Soutschek E, Bocher O, Niessner R et al (2013) New route for fast detection of antibodies against zoonotic pathogens in sera of slaughtered pigs by means of flow-through chemiluminescence immunochips. Anal Chem 85:5279–5285
Xu R, McBride R, Paulson JC, Basler CF, Wilson IA (2010) Structure, receptor binding, and antigenicity of influenza virus hemagglutinins from the 1957 H2N2 pandemic. J Virol 84:1715–1721
Yan J, Villarreal DO, Racine T, Chu JS, Walters JN, Morrow MP, Khan AS, Sardesai NY, Kim JJ, Kobinger GP et al (2014) Protective immunity to H7N9 influenza viruses elicited by synthetic DNA vaccine. Vaccine 32:2833–2842
Yang S, Chen G, Sun J, Li F, Hua HC (2015) Sequence and phylogenetic analyses of five low pathogenic avian influenza H5N2 viruses isolated in China. Acta Virol 59:140–147
Yu Y, Wang X, Jin T, Wang H, Si W, Yang H, Wu J, Yan Y, Liu G, Sang X et al (2015) Newly emergent highly pathogenic H5N9 subtype avian influenza A virus. J Virol 89:8806–8815
Zhan GJ, Ling ZS, Zhu YL, Jiang SJ, Xie ZJ (2012) Genetic characterization of a novel influenza A virus H5N2 isolated from a dog in China. Vet Microbiol 155:409–416
Zhang N, Fang S, Wang T, Li J, Cheng X, Zhao C, Wang X, Lv X, Wu C, Zhang R et al (2012) Applicability of a sensitive duplex real-time PCR assay for identifying B/Yamagata and B/Victoria lineages of influenza virus from clinical specimens. Appl Microbiol Biotechnol 93:797–805
Zhang Y, Liu Q, Wang D, Chen S, Wang S (2013) Simultaneous detection of oseltamivir- and amantadine-resistant influenza by oligonucleotide microarray visualization. PLoS ONE 8:e57154
Zhao K, Gu M, Zhong L, Duan Z, Zhang Y, Zhu Y, Zhao G, Zhao M, Chen Z, Hu S et al (2013) Characterization of three H5N5 and one H5N8 highly pathogenic avian influenza viruses in China. Vet Microbiol 163:351–357
Zhu X, Yu W, McBride R, Li Y, Chen LM, Donis RO, Tong S, Paulson JC, Wilson IA (2013) Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc Natl Acad Sci USA 110:1458–1463
Authors’ contributions
YZ and QL performed the experiment and wrote the main manuscript text; DW participated in the experiment; SC participated in the experimental results discussed; XW and SW guided and developed the experimental program. All authors read and approved the final version of the manuscript.
Acknowledgements
We wish to thank the National Institutes for Food and Drug Control, the Centers for Disease Control of Zhejiang Province, the 307th Hospital of the Chinese People’s Liberation Army, National Institute for Viral Disease Control and Prevention, and the 302nd Hospital of the Chinese People’s Liberation Army for providing cultivated samples of influenza virus, clinical throat swab samples of influenza A virus, and non-influenza respiratory viruses.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
In this assay, the only human materials used were clinical throat swab samples collected from hospital patients. Informed written consent was obtained from all patients. Ethical approval for this work was obtained from the Research Ethics committee of the Academy of Military Medical Sciences, Beijing.
Funding
This work was financially supported by the National Science and Technology Major Project (No. 2012ZX09301003-005) and the Science and Technology Major of Guangdong Province (2012A080203005). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yingjie Zhang and Qiqi Liu contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, Y., Liu, Q., Wang, D. et al. Genotyping and detection of common avian and human origin-influenza viruses using a portable chemiluminescence imaging microarray. SpringerPlus 5, 1871 (2016). https://doi.org/10.1186/s40064-016-3482-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-3482-9