
Abstract
Background
Hepatitis B virus (HBV) is highly contagious and causes liver diseases. Globally more than 350 million people are chronically infected and among them above 80 % are from developing countries like Bangladesh. Resistance to existing drugs and vaccines are common phenomenon due to mutations in HBsAg ‘a’ determinant. Due to lack of data about mutations and subtypes of HBV in Bangladesh, this study strongly demands to be documented. Here, we determined the genotypes and subtypes of HBV prevalent in Bangladesh, and their genomic mutations associated with vaccine and drug resistance.
Results
Among 385 samples, a total of 54 (14 %) were found HBV positive, of which 19 samples were subjected to be sequenced. After bioinformatic analysis, we found Genotype D as predominant genotype (73.7 %) with subtypes ayw3 (64.3 %) and ayw2 (35.7 %), followed by genotype A with subtype adw2 (15.8 %), and then genotype C with subtype adr (10.5 %). A significant number of mutations (Thr118Val, Thr125Met, Thr126Ile, Pro127Thr, Ala128Val, Thr131Asn/Ser, Thr/Ser143Leu/Met) were found in ‘a’ determinant region which may admit resistance to the available vaccines and failure of HBsAg detection.
Conclusions
This comprehensive study have clinical importance like disease diagnosis and treatment. It emphasizes HBV infected patients to do molecular diagnosis for choice of anti-viral drugs and effectiveness of vaccines for proper treatment.
Similar content being viewed by others


Background
Hepatitis B virus (HBV) infects over 350 million people globally per year and causes serious acute and chronic liver diseases (Mast et al. 2005; Norder et al. 1993, 1994). Globally, more than 300 million people have chronic HBV infection and approximately two billion are infected (Scheiblauer et al. 2010; Shaha et al. 2015). HBV integrates its genome into liver cell and can reside there for long time which is seem to develop HBV carrier state and thereby, may cause damage to the liver permanently (Suppiah et al. 2014). In Bangladesh, HBV is one of the major causes of hepatocellular carcinoma (HCC), which is known to be a leading cause of death in the world (Suppiah et al. 2014).
Genomic variation of HBV genotypes is determined by genomic relatedness, where the standard cut-off of divergence is about 8 % (Bowyer and Sim 2000). In Bangladesh, the most prevalent genotype is genotype C among the chronic infection of HBV in one study (Rahman et al. 2016) and genotype D among the patients with chronic infection in a tertiary-care hospital in another study (Mamun Al et al. 2006); however the data on subtypes and mutations in surface (S) gene that contains ‘a’ determinant region are not available. The HBsAg ‘a’ determinant region is located between amino acids 124–147 within the major hydrophilic region (MHR amino acids 99–169) of this S gene encoded protein (Velu et al. 2008). Genotyping and Subtyping of HBV is totally based on increasing of variety of antibodies to the ‘a’ determinant (Moradi et al. 2012). Mutations in this region causes vaccine escape as well as HBsAg detection failure mutants through alteration of antigenicity of the protein (Carman et al. 1990).
On the other hand, drug resistance can be caused due to high mutation rate of polymerase gene, which made HBV infection unpreventable even after vaccination. These mutations can be raised mostly among chronic patients who take anti-viral drugs regularly. Vaccine and anti-viral drug resistance have been described as significant factors to treatment failure of hepatitis B as stated by Locarnini and Mason (Locarnini and Mason 2006).
Sequencing part of surface gene from 370 to 861 nt (491 bp) can be termed as partial S gene sequencing that can reveal genotyping, subtyping and common mutations as efficiently as total S gene sequencing (Wang et al. 2013). This small part of the S gene is conserved and contains ‘a’ determinant region as well as many drug resistant and vaccine mutant sites (Wang et al. 2013). Recent reports in different areas of the world, showing the anti-viral drugs resistant strains of HBV, strongly alert the need for monitoring drug resistant and vaccine mutant sites along with mutations in other genes of the genome of HBV (Sayan et al. 2010; Pastor et al. 2009; Han et al. 2009). Herein, we analyzed the partial S gene to determine the genotypes, subtypes and possible mutations in partial S gene of HBV genome through polymerase chain reaction (PCR) and sequencing.
Methods
Sample selection
A total of 385 patients with jaundice like illness were included in this study and blood samples were collected from different medical college hospitals of Dhaka city. This study was ethically cleared by ethical committee of Dhaka Medical College, Dhaka-1000, Bangladesh (reference number: DMC-MEU/ECC/2014/16). The samples were collected after obtaining a written consent from every patient. None of the patients were HBV vaccinated. We selected the patients with jaundice like illness who might have been infected by hepatitis B viruses because jaundice is one of the main symptoms of hepatitis B infection.
Serological analysis
Serum samples were allowed to be clotted for approximately 10 min and centrifuged at 5000 rpm for 5 min to separate serum from blood. The obtained serum samples were subjected to analyze Hepatitis B surface antigen (HBsAg) by Enzyme Linked Immune Sorbent Assay (ELISA) using ELISA kits (JAJ International Inc., CA, USA).
DNA extraction
HBV-DNA of several randomly selected HBsAg positive samples was extracted from 200 µl of plasma using pathogen kit (Stratec molecular, Berlin, Germany) according to manufacturer’s instruction. Extracted DNA was stored at −20 °C until use.
Polymerase chain reaction and DNA amplification
Partial S gene was targeted to be amplified in this study. A product of 491 base pairs of partial S gene was amplified using 5′-TCGCTGGATGTGTCTGCGGCGTTTTAT-3′ and 5′-ACCCCATCTTTTTGTTTTGTTAGG-3′ primer pair as forward and reverse primers respectively corresponding to nucleotide position 370–861 (Wang et al. 2013). Four microliter (µl) of extracted DNA was added into the mixture containing 12.5 µl of 2× MiFi Mix, 1.0 µl of each primer (10 pmol) and 6.5 µl nuclease free water making total volume of 25 µl reaction. Then the mixture was amplified using a PCR protocol as follows: 12 min at 95 °C, 35 cycles of 1 min at 94 °C, 1 min at 52 °C, 1 min at 72 °C, and a final elongation step of 7 min at 72 °C. The amplification products of 491 bp were analyzed by agarose gel electrophoresis (AGE) on 1.5 % agarose gel stained with ethidium bromide to observe HBV-DNA positive and negative samples, and viewed under UV illumination.
Post PCR purification and sequence determination
The corresponding amplicons were purified using the PCR clean-up system (Promega Corporation, Madison, WI, USA) according to the manufacturer’s instruction. Final elution contained 50 µl of purified PCR products from which 10 µl was reanalyzed on 1.5 % agarose gel to make sure that the purification step was performed precisely. Sequencing was done with the same primers used to amplify the 491 bp partial S gene sequences.
Determination of genotypes and subtypes
After obtaining the sequences of partial S gene, NCBI-BLAST (BLASTn) was performed to get the sequence similarity with other sequences deposited in the NCBI site. The sequences were also analyzed using EMBOSS Transeq tool of EMBL-EBI server (McWilliam et al. 2013) to find all six frames of in silico translation of proteins and subjected to protein BLAST (PSI) against NCBI protein database to find the correct open reading frame (ORF) of the translated protein. Experimental sequences were aligned using MEGA version 5 tools (Tamura et al. 2011) and genotypes were determined by constructing phylogenetic tree. Phylogenetic tree was constructed by Maximum Likelihood Statistical Method with bootstrap replication of 100 times and analysed using Kimura 2-parameter model with uniform rates among sites. The obtained sequences were aligned by Clustal Omega method (Larkin et al. 2007). The reference sequences for this alignment were local and foreign S gene sequences collected from NCBI GeneBank.
Analysis of mutations
For analyzing respective mutations that are associated with resistance to vaccine and anti-viral drugs the sequences were aligned with standard hepatitis B sequence (Accession number: AB033559) from GenBank database (Moradi et al. 2012) by MEGA version 5 software (Tamura et al. 2011) and visualized using Jalview software (Waterhouse et al. 2009). Amino acid codons 122, 160, 127, 159 and 140 of S gene were used to predict HBsAg subtypes as described elsewhere (Purdy et al. 2007).
Sequence deposition
Partial CDS of S gene sequences is deposited in GenBank under accession numbers: KP240636–KP240654.
Results
Genotype D as most prevalent genotype in Bangladesh
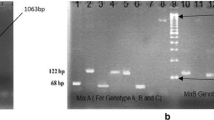
A total of 54 (14 %) samples were observed HBsAg positive. Among them 19 samples were randomly selected to be analyzed by PCR and then nucleotide sequencing. After phylogenetic analysis, 14 (73.7 %) samples were found as genotype D, followed by genotype A (15.8 %) and then genotype C (10.5 %). Phylogenetic tree revealed the similarity of these sequences varying regionally across the world. Furthermore, genotype D was comprised of two subtypes namely, ayw3 (64.3 %) and ayw2 (35.7 %), whereas, subtype adw2 and adr were specific for genotype A and genotype C respectively as shown in Fig. 1.

Distribution of HBV genotypes in Bangladesh
Evolutionary relationship of our HBV with other circulating HBVs
After constructing phylogenetic tree, we observed strong similarity of our partial S protein sequence with other Bangladeshi sequences (up to 97 %) and sequences from India (up to 100 %) and Malaysia (96 %) as shown in Fig. 2. However, sequences comprising genotype D were evolutionarily diverted (Fig. 2). On the other hand, there were several amino acid mutations like K122R (amino acid Lysine at 122 position of S protein instead of Arginine), T131S and I208T in genotype C (Fig. 3). The above mutations might have possible effect on structural and functional activity of S protein which might be a significant cause of resistance. Phylogenetic tree of above alignment revealed that our sequences were closely related with other Bangladeshi existing sequences with a similarity of more than 90 % (Fig. 2a). Phylogenetic analysis revealed the highest similarity of genotype A, C and D sequences with the sequences from India, Malaysia and India respectively (Fig. 2b).

Phylogenetic analysis of HBV sequences with a different existing Bangladeshi strains and b foreign strains collected from NCBI GenBank. Our experimental sequences have been marked with boxes

Alignment of HBV sequences with a different Bangladeshi strains and b foreign strains. Our experimental sequences have been marked with boxes. Matched amino acids were presented as dot and mismatched were shown in symbols. Alignment was done by MEGA v5.0 software and visualized using Jalview v2.8.1 software
Multiple mutations of our sequences in ‘a’ determinant region
Several mutations were observed in ‘a’ determinant region of S gene such as, T118V, T125M, T126I, P127T, A128V and T/S143L/M according to different genotypes of HBV (Table 1). These mutations probably have strong impact on functions of S gene especially resistance to different drugs as well as vaccine.
Discussion
As the severity of HBV is increasing day by day and resistance to the anti-viral drugs and vaccines is raising exponentially, it is essential to demonstrate the mutation status of HBV genome across the world. Hence, the purpose of this study was to determine the genotypes and subtypes of HBV prevailing in Bangladesh and to demonstrate possible mutations in ‘a’ determinant region that might be responsible for resistance to different anti-viral drugs and vaccines.
In this study, we found genotype D (73.7 %) as most prevalent in Bangladesh, followed by genotype A (15.8 %) and then genotype C (10.5 %), which is consistent with other study done in India (Ismail et al. 2012). We used different existing Bangladeshi and foreign sequences of different genotypes as well as a standard GenBank sequence (Accession number: AB033559) (Moradi et al. 2012) from NCBI GenBank to determine the genotypes in Bangladesh and aligned the sequences of those genotypes to observe subtypes. In Bangladesh, the predominant subtypes associated with genotype D are ayw3 (64.3 %) and ayw2 (35.7 %), and subtypes adw2 and adr were specific for genotype A and C respectively. One previous study in Bangladesh documented the predominant genotype in Bangladesh is genotype D which was in accordance with our study (Mahtab et al. 2008; Mamun Al et al. 2006).
We demonstrated several amino acid variations in genotypes A, C and D comparing with the previously documented NCBI sequences. Whereas, genotype A was more conserved than genotypes C and D as shown in Fig. 3. Genotype D was found more susceptible to surface gene mutations (Table 1).
The highest similarity of genotype A and D was found with the sequences from India. However, genotype C was found most similar with the sequences from Malaysia, which are highly communicated countries by Bangladesh.
The genomic mutation in partial S region of HBV have clinical importance. Mutations in ‘a’ determinant region are associated with functional properties of HBV as described above. Different mutations in ‘a’ determinant were found in our sequences as shown in Table 1. Among which, Thr118Val, Thr125Met, Pro127Thr, Ala128Val and Thr/Ser143Leu/Met were observed in genotype D sequences, which is thought to have strong impacts on functions of S gene. For example, substitution Pro127Thr has been alarmed as great public health significance. Patients with this mutation lack to exhibit quantifiable HBsAg as declared in different studies (Simon et al. 2013; Scheiblauer et al. 2010). Furthermore, Sequences of genotype C were found to have Thr126Ile and Thr131Ser mutations, and genotype A sequences have only Thr131Asn mutation in ‘a’ determinant region. Although activity of these mutations was not documented, they might lead to the conformational change of ‘a’ determinant region and as a result, vaccine would be less immunogenic to induce an antibody response even after complete vaccination. Hence, this study recommend to demonstrate experimentally whether these mutations have any resistant property to anti-viral drug or vaccine.
Conclusions
This study is unique for three reasons (1) a detail mutation profile of ‘a’ determinant region of S gene was delineated (2) amino acid comparison of our sequences with different existing Bangladeshi and foreign sequences was described and lastly (3) this is the first comprehensive study detecting the mutations and subtypes of HBV in Bangladesh. This study has the importance to direct clinicians to diagnose the disease properly and decide the choice of drugs or vaccines for the treatment of hepatitis B and HBV carriers.
Abbreviations
- AGE:
-
agarose gel electrophoresis
- bp:
-
basepair
- ELISA:
-
Enzyme Linked Immune Sorbent Assay
- HBV:
-
hepatitis B virus
- HBsAg:
-
hepatitis B surface antigen
- HCC:
-
hepatocellular carcinoma
- MHR:
-
major hydrophilic region
- nt:
-
nucleotide
- ORF:
-
open reading frame
References
Bowyer SM, Sim JG (2000) Relationships within and between genotypes of hepatitis B virus at points across the genome: footprints of recombination in certain isolates. J Gen Virol 81(Pt 2):379–392
Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G, Tanzi E, Zuckerman AJ, Thomas HC (1990) Vaccine-induced escape mutant of hepatitis B virus. Lancet 336(8711):325–329
Han Y, Huang LH, Liu CM, Yang S, Li J, Lin ZM, Kong XF, de Yu M, Zhang DH, Jin GD, Lu ZM, Gong QM, Zhang XX (2009) Characterization of hepatitis B virus reverse transcriptase sequences in Chinese treatment naive patients. J Gastroenterol Hepatol 24(8):1417–1423. doi:10.1111/j.1440-1746.2009.05864.x
Ismail AM, Samuel P, Eapen CE, Kannangai R, Abraham P (2012) Antiviral resistance mutations and genotype-associated amino acid substitutions in treatment-naive hepatitis B virus-infected individuals from the Indian subcontinent. Intervirology 55(1):36–44. doi:10.1159/000323521
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. doi:10.1093/bioinformatics/btm404
Locarnini S, Mason WS (2006) Cellular and virological mechanisms of HBV drug resistance. J Hepatol 44(2):422–431. doi:10.1016/j.jhep.2005.11.036
Mahtab MA, Rahman S, Khan M (2008) Occult hepatitis B virus related decompensated cirrhosis of liver in young males: first report of two cases from Bangladesh. Hepat Mon 8(2):147–150
Mamun Al M, Kumar SI, Rahman S, Kamal M, Khan M, Aggarwal R (2006) Hepatitis B genotypes virus among chronically infected patients in a tertiary-care hospital in Bangladesh. Indian J Gastroenterol 25(4):219–221
Mast EE, Margolis HS, Fiore AE, Brink EW, Goldstein ST, Wang SA, Moyer LA, Bell BP, Alter MJ (2005) A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 54(RR-16):1–31
McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R (2013) Analysis tool Web services from the EMBL-EBI. Nucl Acids Res 41(Web Server issue):W597–W600. doi:10.1093/nar/gkt376
Moradi A, Zhand S, Ghaemi A, Javid N, Tabarraei A (2012) Mutations in the S gene region of hepatitis B virus genotype D in Golestan Province-Iran. Virus Genes 44(3):382–387. doi:10.1007/s11262-012-0715-z
Norder H, Hammas B, Lee SD, Bile K, Courouce AM, Mushahwar IK, Magnius LO (1993) Genetic relatedness of hepatitis B viral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen. J Gen Virol 74(Pt 7):1341–1348
Norder H, Courouce AM, Magnius LO (1994) Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 198(2):489–503. doi:10.1006/viro.1994.1060
Pastor R, Habersetzer F, Fafi-Kremer S, Doffoel M, Baumert TF, Gut JP, Stoll-Keller F, Schvoerer E (2009) Hepatitis B virus mutations potentially conferring adefovir/tenofovir resistance in treatment-naive patients. World J Gastroenterol 15(6):753–755
Purdy MA, Talekar G, Swenson P, Araujo A, Fields H (2007) A new algorithm for deduction of hepatitis B surface antigen subtype determinants from the amino acid sequence. Intervirology 50(1):45–51. doi:10.1159/000096312
Rahman MA, Hakim F, Ahmed M, Ahsan CR, Nessa J, Yasmin M (2016) Prevalence of genotypes and subtypes of hepatitis B viruses in Bangladeshi population. SpringerPlus 5:278. doi:10.1186/s40064-016-1840-2
Sayan M, Akhan SC, Meric M (2010) Naturally occurring amino-acid substitutions to nucleos(t)ide analogues in treatment naive Turkish patients with chronic hepatitis B. J Viral Hepat 17(1):23–27. doi:10.1111/j.1365-2893.2009.01149.x
Scheiblauer H, El-Nageh M, Diaz S, Nick S, Zeichhardt H, Grunert HP, Prince A (2010) Performance evaluation of 70 hepatitis B virus (HBV) surface antigen (HBsAg) assays from around the world by a geographically diverse panel with an array of HBV genotypes and HBsAg subtypes. Vox Sang 98(3 Pt 2):403–414. doi:10.1111/j.1423-0410.2009.01272.x
Shaha M, Hoque SA, Ahmed MF, Rahman SR (2015) Effects of risk factors on anti-HBs development in hepatitis B vaccinated and nonvaccinated populations. Viral Immunol 28(4):217–221. doi:10.1089/vim.2014.0147
Simon B, Kundi M, Puchhammer E (2013) Analysis of mutations in the S gene of hepatitis B virus strains in patients with chronic infection by online bioinformatics tools. J Clin Microbiol 51(1):163–168. doi:10.1128/JCM.01630-12
Suppiah J, Mohd Zain R, Haji Nawi S, Bahari N, Saat Z (2014) Drug-resistance associated mutations in polymerase (p) gene of hepatitis B virus isolated from malaysian HBV carriers. Hepat Mon 14(1):e13173. doi:10.5812/hepatmon.13173
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121
Velu V, Saravanan S, Nandakumar S, Dhevahi E, Shankar EM, Murugavel KG, Kumarasamy T, Thyagarajan SP (2008) Transmission of “a” determinant variants of hepatitis B virus in immunized babies born to HBsAg carrier mothers. Jpn J Infect Dis 61(1):73–76
Wang F, Lu L, Yu C, Lv Z, Luo X, Wan C, Hu Z, Zhu Q, Deng Y, Zhang C (2013) Development of a novel DNA sequencing method not only for hepatitis B virus genotyping but also for drug resistant mutation detection. BMC Med Genomics 6(Suppl 1):S15. doi:10.1186/1755-8794-6-S1-S15
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–1191. doi:10.1093/bioinformatics/btp033
Authors’ contributions
MS was involved in designing the study and prepared the manuscript; SRR supervised the study group, involved in designing the study and revised the manuscript critically; SAH was involved in conception and design, drafting the manuscript or revising it critically for important intellectual content. All authors contributed equally to this work. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to the participated patients for their cooperation with this study.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shaha, M., Hoque, S.A. & Rahman, S.R. Molecular epidemiology of hepatitis B virus isolated from Bangladesh. SpringerPlus 5, 1513 (2016). https://doi.org/10.1186/s40064-016-3174-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-3174-5