
Abstract
Background
microRNAs (miRNAs) are one kind of small non-coding RNAs widely distributed in insects. Many studies have shown that miRNAs play critical roles in development, differentiation, apoptosis, and innate immunity. However, there are a few reports describing miRNAs in Anopheles sinensis, the most common, and one of the dominant malaria mosquito in China. Here, we investigated the global miRNA expression profile across four different developmental stages including embryo, larval, pupal, and adult stages using Illumina Hiseq 2500 sequencing.
Results
In total, 164 miRNAs were obtained out of 107.46 million raw sequencing reads. 99 of them identified as known miRNAs, and the remaining 65 miRNAs were considered as novel. By analyzing the read counts of miRNAs in all developmental stages, 95 miRNAs showed stage-specific expression (q < 0.01 and |log2 (fold change)| > 1) in consecutive stages, indicating that these miRNAs may be involved in critical physiological activity during development. Sixteen miRNAs were identified to be commonly dysregulated throughout four developmental stages. Many miRNAs showed stage-specific expression, such as asi-miR-2943 was exclusively expressed in the embryo stage, and asi-miR-1891 could not be detected in larval stage. The expression of six selected differentially expressed miRNAs identified by qRT-PCR were consistent with our sequencing results. Furthermore, 5296 and 1902 target genes were identified for the dysregulated 68 known and 27 novel miRNAs respectively by combining miRanda and RNAhybrid prediction. GO annotation and KEGG pathway analysis for the predicted genes of dysregulated miRNAs revealed that they might be involved in a broad range of biological processes related with the development, such as membrane, organic substance transport and several key pathways including protein processing in endoplasmic reticulum, propanoate metabolism and folate biosynthesis. Thirty-two key miRNAs were identified by microRNA-gene network analysis.
Conclusion
The present study represents the first global characterization of An. sinensis miRNAs in its four developmental stages. The presence and differential expression of An. sinensis miRNAs imply that such miRNAs may play critical roles in An. sinensis life cycle. A better understanding of the functions of these miRNAs will have great implication for the effective control of vector population and therefore interrupting malaria transmission.
Similar content being viewed by others


Background
Anopheles sinensis is one of the most important disease vectors in China. It is considered as the primary vector of P. vivax malaria due to its wide distribution and high density in central China where several malaria outbreaks occurred in history [1, 2]. Like many mosquitoes, An. sinensis goes through four life stages that include egg, larva, pupa and the adult. During these developmental periods, a fine-tuning complex biological process, such as embryogenesis, organ differentiation, and metamorphosis, etc., is completed.
For the past decades, facilitated by high-throughput sequencing technology, a large number of small non-coding RNAs (ncRNAs) have been discovered in many species [3, 4]. Amongst them, microRNAs (miRNAs) have become the most popular research topic for its critical role in regulating important biological events at the post-transcriptional level. Mature miRNAs are single-stranded, evolutionarily conserved endogenous small ncRNAs of approximately 22 nucleotides (nt) in length, which could lead to transcriptional decay through translational inhibition, transcript degradation or both [5, 6].
The first miRNA repertoire of mosquito species was reported from the African malaria vector Anopheles gambiae [7]. Since then, mosquito miRNAs have been identified and characterized in a number of mosquito species, including Anopheles stephensi [8, 9], Aedes aegypti [10,11,12], Culex fatigans [13,14,15], and many others, but not yet in An. sinensis. Several studies suggested that miRNAs may contribute to an important regulatory role in the post-transcriptional level in developmental stages. For instance, temporal and stage specific expression of miRNAs has been identified in the embryo, larval, pupal and adult stage of Ae. albopictus [16], An. anthropophagus [17], and An. stephensi [9]. Among them, the expression of aal-miR-286b was increased in the embryo, while aal-miR-2942 had the highest level of expression in larvae although it was normally expressed at the embryo, pupal and adult stages [16]. In addition, ast-miR-1891, ast-miR-190-3p, ast-miR-285, ast-miR-988-3p, and ast-miR-989 were absent in the larval stage, while ast-miR-8-3p was the most abundant in both male and female larval stage [9]. These results indicated that dysregulated miRNAs might play pivotal roles in embryogenesis and metamorphosis throughout development. However, the exact functions of these miRNAs remain to be determined.
In this study, we provided a comprehensive repertoire of An. sinensis miRNAs and investigated the differential expression of miRNAs using Solexa sequencing technology, with an aim to identify key regulatory miRNAs through development stages. Selected candidate miRNAs were then validated by qRT-PCR in samples that have been used in sequencing. To explore the potential gene regulatory networks, 32 dysregulated miRNAs were defined as key miRNAs during development and then subjected to miRNA-gene network analysis in terms of developmental pathway analysis. The data described here served as an important step towards the understanding of gene expression modulation by miRNAs in An. sinensis, which will further advance functional studies for novel mosquito control approaches developing and promote elimination campaign in China.
Methods
Mosquito rearing and sample preparation
Anopheles sinensis (China strain) from insectary were reared at 28 ± 2 °C, 70–75% humidity in the Vector Control Reference Laboratory at the National Institute of Parasitic Diseases (NIPD), Shanghai, China. Adult mosquitoes were maintained in screened cages, and provided constant access to water and glucose-soaked sponges.
Sample collections from different developmental stages of An. sinensis for high throughput sequencing are briefly described below. Embryos were collected within 24 h after placing a damp collection filter paper within the cage and were pooled to represent the embryonic stage. In the larval sample collection, we used mixed larval sample (I–IV instars) including early and late larval samples. Pupal samples were collected from a pool of varied ages. Female adults were collected after emerging one to 3 days. All samples were flash frozen in liquid nitrogen immediately following collection, and then stored at − 80 °C until for RNA isolation.
RNA extraction and small RNA sequencing
Briefly, total RNAs were extracted separately from the four different life cycle stages of An. sinensis (eggs, larvae, pupae and unfed adult females) using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). RNA degradation and contamination were monitored on 1% agarose gels. RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). RNA quality assessment was determined by using Agilent 2100 Bioanalyzer RNA Nano 6000 kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). No rRNA depletion was performed. RNA extracts of the four life stages were sent to Newgenes Inc (Shanghai, China) for small RNA sequencing. A total amount of 3 µg of total RNA was ligated with 3′ and 5′ adaptors followed by reverse transcription using RT primers. The libraries were size-selected for sequencing of RNA fragments of 18–30 nucleotides. These small RNA libraries were then sequenced using Illumina Hiseq 2500 platform (Illumina Inc., USA).
Small RNA sequencing data processing and analysis
Following sequencing, raw data (raw reads) of fastq format were firstly processed through custom perl and python scripts. In this step, clean data (clean reads) were obtained by removing reads containing ploy-N, with 5′ adapter contaminants, without 3′ adapter or the insert tag, containing ploy A or T or G or C and low quality reads from raw data. At the same time, Q20, Q30, and GC-content of the raw data were calculated. Then, a certain range of clean reads from each stage was combined to do all the downstream analyses. All reads were mapped to the An. sinensis strain China genome [GenBank: GCA_000441895.2] by Bowtie [18] without mismatch to analyze their expression and distribution on the reference.
For identification of known and novel miRNA sequences present in the four life stage datasets. Firstly, after removing tags originating from protein-coding genes, repeat sequences, rRNA, tRNA, snRNA, and snoRNA, small RNA tags were mapped to RepeatMasker, Rfam database or those types of data from Anopheles gambiae. Mapped small RNA tags were used to look for known miRNA using miRBase21 [19] as reference. Custom scripts adapted from quantitative script of miRDeep2 were used to obtain the miRNA counts as well as base bias on the first position of identified miRNAs with certain length and on each position of all identified miRNAs. The reads that did not yield a match were used to predict novel miRNAs. The available software miREvo [20] and mirdeep2 [21] were integrated to predict novel miRNA from the small RNA tags unannotated in the former steps. A number of stringent criterions were used for evaluating whether novel miRNA candidates were genuine miRNAs, such as exploring the secondary structure by RNAfold, calculating minimum free energy (MFE) and avoiding isolated base pairs. In each library, same sequences were pooled together to generate expression files (fasta) with their read count and unique ID used for further analysis.
All identified known miRNAs were named according to their most similar miRNAs in the miRNA database (miRBase) release 21 (June 2017). All identified novel miRNAs were named sequentially following the asi-mir-nov1. Raw sequencing data were prepared to submit to the National Center for Biotechnology Information (NCBI).
Quantification of miRNA and differential expression analysis
In order to analyze the relative abundance and expression level of miRNAs, tags per million of total RNA reads (TPM) for each miRNA in all four libraries were calculated. TPM was compared between the libraries to identify miRNAs differentially expressed between two consecutive life stages in the mosquito. Differential expression analysis of two samples was performed using the DEGseq [22] R package. p value was adjusted using q value [23]. q < 0.01 and |log2 (fold change)| > 1 was set as the threshold for significantly differential expression by default.
miRNA target prediction, GO and KEGG enrichment analysis
To predict the potential targeted genes of miRNA, the targeted mRNAs of differentially expressed known and novel miRNAs were predicted by combining miRanda [24] and RNAhybrid [25] to select and assess overlapping predicted targets between each program. Briefly, 3′UTR of An. gambiae downloaded from vectorbase were used for the prediction. The targets showing complementarity with miRNA seed region and binding energy ≤ 20 kcal/mol were selected. The predicted targets were used for further GO and KEGG enrichment analysis.
Gene ontology (GO) enrichment analysis was used on the target gene candidates of differentially expressed miRNAs (“target gene candidates” in the following). GOseq based Wallenius non-central hyper-geometric distribution [26], which could adjust for gene length bias, was implemented for GO enrichment analysis. GO terms with corrected p value less than 0.05 were considered significantly enriched by target genes. KEGG pathway analyses were performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.ad.jp/kegg/) database [27]. In this study, we used KOBAS [28] software to test the statistical enrichment of the target gene candidates in KEGG pathways, with the threshold of corrected p < 0.05.
miRNA-gene network analysis
To identify the key miRNAs in An. sinensis development, microRNA-gene network analysis was conducted for the commonly dysregulated miRNAs identified from the microRNA expression files, the exclusively expressed miRNAs in certain stages (TPM > 10) and the most significantly over/under-expressed miRNAs (>tenfold) and their predicted target developmental genes. Developmental genes of An. gambiae were accessed based on gene annotation in VectorBase [29] and through intensive literature surveys [30,31,32,33]. Interactive networks between key miRNAs and their target developmental genes were constructed by using Cytoscape [34].
miRNA expression validated by quantitative RT-PCR
To validate the presence and expression level of the identified miRNAs, nine miRNAs with differential expression were selected for qRT-PCR as described previously [35]. The total RNA was extracted using TRIzol Reagent (Invitrogen, USA). The reverse transcription reaction was performed with RevertAid First Strand cDNA Synthesis Kit (Fermentas, USA), and the reverse-transcribed products were used as the template for qRT-PCR with miRNA-specific primers (Additional file 1: Table S1). All reactions were assayed in three biological and technical replications, and performed in an ABI-7300 (Applied Biosystems, USA) using SYBR Green qPCR kit (Thermo, USA). Quantitative measurements were normalized to the internal control of U6 small nuclear (U6), and relative expression was calculated by using the 2−ΔΔCt method [14].
Results
Small RNAs sequence of the four libraries
We performed small RNA sequencing from eggs, larva, pupa and adult mosquito batches to identify An. sinensis miRNAs expressed during developmental stages. All sequencing data can be found in NCBISRA (PRJNA412443). In total, sequencing of four libraries yielded about 10.75 × 107 reads. After trimming the adaptors, 1.99 × 107 (96.71%), 2.85 × 107 (92.81%), 2.91 × 107 (86.06%) and 2.06 × 107 (92.38%) clean reads from ASE (egg), ASL (larva), ASP (pupa) and ASA (adult) libraries respectively were utilized for further processing (Table 1). The total number of the reads mapped to An. sinensis genome constitutes 82.53, 77.61, 80.55 and 85.75% of the total high-quality sRNA (small RNA) reads from eggs, larvae, pupae, and adult libraries, respectively (Table 1).
Length distribution of small non-coding RNAs in the four libraries
Among the libraries, size distribution of reads varied between 18 and 35 nt. We found that one peak with 21 nt in all libraries, followed by another peak with 20–23 nt which represents miRNAs (Fig. 1). This was consistent with the typical size of miRNAs in other reports. There was also a peak at 24–27 nt in almost all developmental libraries, which corresponds to piRNA-like small RNAs suggesting a possible role in embryogenesis and maintaining genome stability during development.

Length distribution of small RNA reads in a An. sinensis embryo library (ASE), b An. sinensis larval library (ASL), c An. sinensis pupal library (ASP), d An. sinensis female adult library (ASD). X axis represents small RNA read lengths in base pairs while Y axis represents number of reads
Annotation of small non-coding RNAs in the four libraries
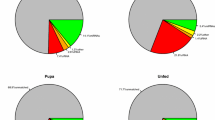
In order to annotate other small non-coding RNAs in the four libraries, all clean reads were aligned against Rfam version 11 [36]. As showed in Fig. 2, the most abundant class of small non-coding RNAs in the eggs’ library was miRNAs (50.10%). However, rRNAs were most abundant in the larvae (6.61%) and pupae library (5.8%), and tRNAs in the adult females’ library (6.87%).

Annotation of small non-coding RNAs from the four life stage libraries of An. sinensis. a An. sinensis embryo library (ASE), b An. sinensis lave library (ASL), c An. sinensis pupa library (ASP). d An. sinensis adult library (ASA). The total reads can be divided into 12 categories; exon−, exon+, intron−, intron+, known miRNA, novel miRNA, others, repeat, rRNA, snoRNA, snRNA and tRNA. The others referred to the other class of small non-coding RNAs and the reads not aligned to RNA families’ database
Identification of miRNAs in the four life stages libraries of An. sinensis
Based on previously known mosquito mature miRNAs in miRBase release 21 (June 2017), we discovered 99 known miRNAs in An. sinensis (Table 2) throughout four development stages. Reads that do not match any known miRNA sequences were further employed by miREvo and mirdeep2 to investigate the folding properties of precursors or hairpins for novel miRNAs. Finally, 65 mature miRNAs were identified as novel (default parameters of miREvo and mirdeep2) (Additional file 2: Table S2). Thus, a total of 164 An. sinensis miRNAs were identified in four life stages libraries of An. sinensis.
miRNA expression pattern during An. sinensis development
The expression patterns of miRNAs in different life stages were examined based on the number of reads revised. The result showed that the expression profiles of miRNAs varied from highly specific to ubiquitous during the four stages. One-hundred and seventeen miRNAs were ubiquitously expressed in all developmental stages, while some miRNAs showed stage-specific patterns (Fig. 3). Among the known miRNAs, asi-miR-276-3p, asi-miR-1, asi-miR-100, asi-miR-8-3p, asi-miR-999, and asi-miR-9a showed constant high expression throughout four development stages. asi-miR-276-3p was the most frequently expressed miRNA in the larvae, the pupae, and the adult females’ library (TPM > 3.17 × 105), whereas asi-miR-1 was the dominant miRNA in the eggs’ library (TPM > 1.87 × 105). Nevertheless, asi-miR-219 had the lowest expression level in all developmental stages. asi-miR-2943 was specifically expressed in the embryo stage, and asi-miR-1891 could not be detected in the larval stage. The highly expressed novel miRNA in the eggs and pupae libraries was asi-miR-nov54 (TPM = 0.45 × 103 and TPM = 1.50 × 103), while in the larvae library and adult females’ library, it was asi-miR-nov55 (TPM = 0.91 × 103) and asi-miR-nov1 (TPM = 0.42 × 103), respectively. However, the expression level of novel miRNAs was generally low throughout all developmental stages when compared with known miRNAs.

Heatmaps clustering of miRNAs expressed in the four life stage libraries of An. sinensis. The clustering was performed on all known (a) and novel (b) miRNAs based on TPM (tags per million) and the four life stage samples. Each row represents one miRNA and each column represents a stage. The color scale (shown on the left) illustrates the number of the reads of a miRNA across the life stages
MicroRNA regulation from egg to adult during An. sinensis development
Hierarchical heat map revealed different expression patterns of the miRNAs between the two consecutive developmental stages (Fig. 3). Based on q-value and log fold change, 33 significantly down-regulated miRNAs (26 for known and 7 for novel miRNAs) and 41 significantly up-regulated miRNAs (25 for known and 16 for novel) were identified from the embyro to the larval stage. 20 down-regulated miRNAs (18 for known and 2 for novel miRNAs) and 22 up-regulated miRNAs (18 for known and 4 for novel) were observed form the larval to the pupal stage. 22 significantly down-regulated miRNAs (14 for known and 8 for novel miRNAs) and 15 significantly up-regulated miRNAs (12 for known and 3 for novel) were expressed form the pupal to adult stage (Fig. 4). It is noteworthy that the total number of dysregulated miRNAs decreased from the embryo to the adult stage. We also identified two known miRNAs (asi-miR-2943 and asi-miR-286b) to be significantly over-expressed (>tenfold) form the embryo to the larval stage and one known miRNA (asi-miR-1891) to be significantly under-expressed form the pupal to the adult stage (>tenfold) (Fig. 4). Sixteen miRNAs were differentially expressed in all developmental stages of the mosquito. Based on the analysis of these regulated miRNAs between the two consecutive developmental stages, five miRNAs (asi-miR-1890, asi-miR-193, asi-miR-263a-5p, asi-miR-263b-5p and asi-miR-nov54) were down-regulated in the larval stage whereas get up-regulated in the pupal stage but down-regulated in the adult stage again. Seven miRNAs (asi-miR-1175-3p, asi-miR-1175-5p, asi-miR-281-3p, asi-miR-281-5p, asi-miR-317, asi-miR-34-5p and asi-miR-nov55) were up-regulated in the larval stage whereas get down-regulated in the pupal but up-regulated in the adult stage again. asi-miR-nov23 and asi-miR-2765 were up-regulated in the larval stage whereas get down-regulated in both pupal and adult stage. asi-miR-2944a-5p was down-regulated in the larval and the pupal stages but up-regulated in the adult stage. asi-miR-989 was down-regulated in larval and pupal stages but up-regulated in the adult stages.

Fold-change of miRNAs during the development of An. sinensis. a Log2 fold change of miRNAs between embryo and larval stage, b Log2 fold change of miRNAs between larva and pupal stage, c Log2 fold change of miRNAs between pupal and adult stage. X axis represents miRNAs while Y axis represents fold-change of miRNAs
Validation of differentially expressed miRNA
To validate the small RNA sequencing results, we performed quantitative real-time PCR analysis by selecting nine miRNAs that showed dramatic changes in expression level at different developmental stages. The result (Fig. 5) showed that six miRNAs (asi-miR-276-3p, asi-miR-2943, asi-miR-1891, asi-miR-286b, asi-miR-1175-3p, and asi-miR-nov54) showed similar expression patterns as those revealed by our sequencing result. However, three miRNAs (asi-miR-263a-5p, asi-miR-nov23, and asi-miR-nov55) expression levels detected by qRT-PCR were inconsistent with that of our sequencing results due to some unknown reasons.

Validation of the differentially expressed miRNAs by Quantitative real-time PCR. The transcript levels of six miRNAs at different stages were calculated relative to the amount of U6 small nuclear after normalization. The real time PCR data with bars represent a mean ± SD from three independent experiments
miRNA target gene prediction, GO enrichment, and KEGG pathway analysis
To understand the role of dysregulated miRNAs in physiological functions and biologic processes during development in An. sinensis, miRNA target gene prediction was performed to identify the putative target genes based on miRNA/mRNA complementarity. Multiple/different miRNA target prediction algorithms were applied to minimize the number of putative or false positive target genes. The target gene candidates of the differentially expressed miRNAs between two consecutive developmental stages of An. sinensis were predicted. A total of 7198 annotated mRNA transcripts were predicted as target gene candidates for 95 differentially expressed miRNAs (Additional file 3: Table S3). Among them, 5296 and 1902 target gene candidates were identified for 68 known and 27 novel miRNAs, respectively.
GO term annotation and KEGG Pathway analysis were used to further understand biological functions and signaling pathways that could potentially be regulated by these putative target genes. We found that the putative target genes of the differentially expressed miRNAs between two consecutive developmental stages of An. sinensis were involved in a broad range of biological functions and signaling pathways (Fig. 6). Detailed classifications about the biological process, cellular component, and molecular function of the miRNA targets were shown in Fig. 6a. By GO annotation, targets of miRNAs regulated from the embryo to the larval stage in the mosquito were enriched in intracellular, cytoplasm and cellular process. Targets of miRNAs regulated from larva to pupal stage in the mosquito were enriched in membrane part, membrane, cytoplasm and organic substance transport. Targets of miRNAs regulated from the pupa to the adult stage in the mosquito were enriched in structural molecule activity, membrane part and transport. Similarly, by KEGG pathway analysis, we found that 119 enriched pathways were identified in this study. The most enriched pathway included protein processing in endoplasmic reticulum, propanoate metabolism, folate biosynthesis, Citrate cycle (TCA cycle) and valine, leucine and isoleucine degradation (Fig. 6b).

Gene ontology annotation by Goseq and KEGG pathway analysis by KOBAS for target genes of differentially expressed miRNAs. a Shows partial GO enrichment for the predicted target genes in ontologies of biological processes, cellular component, and molecular function. b Shows the enriched pathways for the predicted target genes during different stages of mosquito development
miRNA-gene network analyses identified key miRNAs and developmental genes
Through analysis, 16 commonly dysregulated miRNAs identified from the miRNA expression files, 15 exclusively expressed miRNAs in certain stages (TPM > 10), and three the most significantly over/under-expressed miRNAs (>tenfold) were defined as the key miRNAs in An. sinensis development. A total of 546 developmental genes were obtained based on gene annotation in VectorBase and intensive literature surveys. After removing duplicates and invalid target genes, miRNA-gene network was built based on 32 key miRNAs and 104 developmental genes (Additional file 4: Table S4). We developed a network to demonstrate the overlapping miRNA targets for miRNAs since miRNAs usually have many mRNA targets. The number of targets for asi-miR-317, asi-miR-2944a-5p, asi-miR-263b-5p, asi-miR-263a-5p, and asi-miR-2943 was 10, 7, 6, 6 and 5, respectively (Fig. 7). This network also indicated that several mRNAs were predicted to be targets of one miRNAs. For example, AGAP001999, AGAP007696, AGAP005245, and AGAP004050 were targeted by two to five miRNAs, respectively (Fig. 7). As expected, the overall pattern suggests a positive correlation between the miRNA expression and developmental genes, such as AGAP000048 (APC), AGAP005449 (Cbl), AGAP001867 (hep), which encoding important components of major developmental signaling pathways.

miRNA-mRNA interaction network predicted from 32 key miRNAs and 104 developmental genes. miRNAs are in green while mRNAs are in pink
Discussion
In the present study, the miRNA expression profile of An. sinensis in four different life stages was analyzed by NGS technology, and approximately 100 million high-quality reads were obtained. The mapping rates of high-quality reads to the An. sinensis genome were between 77.61 and 85.75%. This might be subject to incomplete reference genome. The size distribution of small RNA sequence reads showed peaks between 20 AND 30 nucleotides comparing to the average miRNA length. Annotation of the sRNAs in the four libraries indicated that the most abundant class of sRNAs in the eggs, larvae, pupae and adult females’ libraries was miRNAs, rRNAs (larvae and pupae) and tRNAs respectively. This result is consistent with previous research in Anopheles funestus [37], indicating that protein synthesis during development is of great importance.
In total, 99 known miRNAs and 65 novel miRNAs expressed in the four life stages of the An. sinensis were identified. As expected, most of the known miRNAs identified in An. sinensis were highly conserved across diverse mosquito species, suggesting that the conserved miRNAs may involve in many critically fundamental regulatory pathways. Novel miRNAs found in this study are not conserved in the Anopheles genus, and we cannot find homologs in other insect species by searching the miRBase, suggesting that these might be species-specific miRNAs. However, the predicted novel miRNAs exhibited much lower expression levels when compared with conserved miRNAs, which is also consistent with previous reports [9, 37]. Previously, Dritsou and his colleagues [6] computationally predicted 115 miRNAs (after removing 33 duplicates and 11 putatively new miRNA, 71 left) in An. sinensis from the genome level. We compared our miRNA dataset with theirs and found that 33 miRNAs overlap with our predictions. Among these miRNAs, 19 miRNAs could be found in Ae. aegypti, An. gambiae, and Cu. quinquefasciatus. Six miRNAs (miR-193, miR-2765, miR-277-5p miR-2b, miR-315-5p and miR-9b) were only found in Ae. aegypti. In contrast, we validate more miRNAs existence and detect their expression pattern at different developmental stages.
To obtain insight into possible stage-specific miRNAs and their roles in An. sinensis, we investigated the expression profile of the miRNAs identified in the four life stage libraries. One-hundred and seventeen miRNAs were ubiquitously expressed in all developmental stages. The top five most highly expressed miRNA across four developmental stages were asi-miR-276-3p, asi-miR-1, asi-miR-100, asi-miR-9a and miR-8, which is basically in line with previous report in other mosquito species [15, 38]. The most highly expressed miRNA in female An. sinensis was asi-miR-276-3p. In contrast, Skalsky et al. [15] reported that miR-184 was the most highly expressed miRNA in C7/10 Ae. albopictus cells and blood-fed female Cx. quinquefasciatus mosquitoes [15]. afu-miR-8 was the dominant miRNA in the unfed adult An. funestus females [37]. In addition, miR-1, miR-184, and miR-263 ranked among the most highly expressed miRNAs in both Ae. aegypti and An. stephensi [27]. aga-bantam, aga-miR-263a, aga-miR-8, aga-miR-10, aga-miR-184 and aga-miR-281 were the most abundantly expressed miRNAs in An. gambiae [38]. Thus, despite the differences in expression level, most of these reported abundant miRNAs in different mosquito lineages are still among the top 10 most highly expressed miRNA in An. sinensis.
Comparisons between egg and larva, larva and pupa, and pupa and unfed adult female are shown in Figs. 3 and 4. A majority of miRNAs were differentially expressed in the four stages. For example, 41 miRNAs were up-regulated and another 33 down-regulated between the embryo and larva stage. Especially, asi-miR-2943 and asi-miR-286b were up-regulated more than tenfold change. While between the larva and the pupa, 22 miRNAs were up-regulated and 20 other miRNAs were down-regulated. We also identified 15 significantly up-regulated miRNAs and 22 significantly down-regulated miRNAs from the pupal to the adult stage. And asi-miR-1891 was significantly down-regulated in this process. Dramatic alternation of miRNA expression during these three stages signifies that these miRNAs may be involved in the regulation of metamorphosis during development. What’s more, asi-miR-2943 was exclusively expressed in the embryo stage, which is also highly expressed in the embryos in both Ae. aegypti, An. stephensi [39] and An. anthropophagus [17], indicating its important roles in embryogenesis across many mosquito species. However, miR-2943 was present at low levels in both Cx. quinquefasciatus and Ae. albopictus C7/10 cells [15]. Similarly, miR-1891 was most abundantly expressed in Ae. aegypti and An. stephensi pupal stage [39], however, asi-miR-276-3p was the highest expressed miRNAs in An. sinensis pupae, although asi-miR-1891 also exhibited a high expression. These results indicate that, on the one hand, miRNA profiles of mosquito lineage may differ from each other, one the other hand, specifically expressed miRNAs or regulatory miRNAs during developmental stages may also be different. In addition, sample preparation, detection methods and sensitivity may also lead to inconsistency between different studies.
Moreover, we observed that the expression level of miRNAs did not correlate with the fold change. Some miRNAs showed higher expression but with low fold changes. For instance, asi-miR-276-3p, asi-miR-1, asi-miR-100 and asi-miR-7 were among the top 10 most highly expressed miRNAs in all developmental stages but showed low log2-fold changes. However, asi-miR-193 and asi-miR-nov54 were low expressed miRNAs in An. sinensis developmental stages but showed high log2-fold changes. Additionally, asi-miR-309a, asi-miR-2943, asi-miR-315-3p and asi-miR-iab-4-3p could not be detected in the pupae and larvae libraries, asi-miR-275-5p and asi-miR-2491-3p was not identified in the pupae and adult females’ libraries, and asi-miR-980-3p was not found from the pupae library. These results indicate that the expressions of some miRNAs are probably stage specific. We assumed that stage-specific miRNAs might be involved in regulation of embryogenesis, organ differentiation, growth, and reproduction during a specific developmental stage. Investigation on stage-specific miRNAs may detail understanding of mosquito biology and could provide mosquito-specific targets for disease control. However, the function of majority of the miRNAs remains to be determined.
To confirm our sequencing results, we performed quantitative real-time PCR analysis using the RNA samples used for small RNA NGS. Thus, nine miRNAs that showed dramatic changes in expression level at different developmental stages were selected for validation. All nine miRNAs (asi-miR-276-3p, asi-miR-2943, asi-miR-1891, asi-miR-286b, asi-miR-1175-3p, asi-miR-263a-5p, asi-miR-nov23, asi-miR-nov54, and asi-miR-nov55) could be detected in the sample. Six miRNAs showed similar expression patterns as those revealed by our sequencing analysis, which indicated a low false discovery rate of our sequencing data and supported the validity of the profiling. However, the expression levels of asi-miR-263a-5p, asi-miR-nov23, and asi-miR-nov55 detected by qRT-PCR were inconsistent with that of our sequencing results. We inferred that this might be caused by the experimental conditions, such as base bias, amplification efficiency, and analytical error in both small RNA NGS and the qRT-PCR, but some other unknown reasons cannot be excluded.
Mosquitoes grow through four phases of metamorphosis: egg, larva, pupa and adult. Knowing the different stages of the mosquito’s life will help us to prevent mosquitoes and disease transmission. To further understand the role of miRNAs in An. sinensis, miRNA target gene prediction was performed based on miRNA/mRNA interactions to provide molecular insight into their function by using two computational methods. A total of 7198 annotated mRNA transcripts were predicted as putative target genes for 95 differentially expressed miRNAs. The enriched biological functions and signaling pathways with significant role at a specific stage of development were predicted by GO term annotation and KEGG Pathway analysis. Metamorphosis from embryo to larval and from pupal to adult stage involves biological functions such as cellular component, membrane, cytoplasm and organic substance transport. In addition, enriched pathways involved in Protein processing in endoplasmic reticulum, Propanoate metabolism, Folate biosynthesis, Citrate cycle (TCA cycle) and Valine, leucine and isoleucine degradation were found. These biological functions and pathways might be important for degeneration and formation of tissue/organ, which happens during immature stages to complete its transition from egg to adult.
Network of miRNA-mRNA interactions was predicted by using results from miRNA analysis and mRNA retrieved from developmental genes of An. gambiae and then visualized by Cytoscape. Further analyses indicated that a number of miRNAs (asi-miR-317, asi-miR-2944a-5p, asi-miR-263b-5p, asi-miR-263a-5p, and asi-miR-2943) target a cluster of mRNA and vice versa. The candidate targets mentioned here have been reported to be key components in developmental pathways [31]. For example, AGAP002352, one of the targets of asi-miR-263b-5p, is an annotated apoptosis-related gene (p53) in Ae. aegypti and many other mosquitoes. p53 plays an important role in programmed cell death in relation to development [40]. Also, AGAP003108 (htl), AGAP001867 (hep) and AGAP005245 (lola) were the common target of asi-miR-281-5p. htl is one of two FGF (Fibroblast Growth Factor) receptors, and FGF signaling regulates a variety of biological processes during development, such as cell differentiation and cell proliferation [41]. hep is one component of Ras signaling pathways which also could regulate many developmental processes [42]. lola, which is called axon guidance gene that regulate wiring of the olfactory system in D. melanogaster [43], have been reported to have many orthologs in mosquito [31]. In addition, knock-in and knock-down of specifically and temporally expressed miRNAs in Ae. albopictus by microinjection have improved our understanding of regulated miRNAs in mosquito development [16]. Intervention on aal-miR-286b led to a profound decrease in the hatching rate of embryos. In addition, eclosion rate of larvae in aal-miR-2942 knocked-down group showed a significant decrease compared to control group. A marked reduction in longevity and fecundity was also observed in the miR-1891 inhibitor group. These research findings have established the exact functional roles these developmental genes and miRNAs in regulating the stage-specific development. However, additional extensive experimental analyses are required to confirm the interactions of miRNA-gene regulatory role in many phenotypic changes during developmental stages in further ongoing research work.
Although miRNAs are reported to play a role in development in C. elegans, fruit flies and other insects, and significant stage-specific expression was observed for a number of miRNAs in various mosquito species. However, the role of miRNAs in metamorphosis and development is far less understood and remains an enigma. For some specific or novel miRNAs, their functional roles still need to be confirmed by biological experiments. Furthermore, only very limited specific target genes have been identified for small part of miRNAs. It is important to note that the result in this work is a key step towards improving our understanding of the complexity and regulation mode of miRNAs in An. sinensis embryogenesis and metamorphosis. Further analysis of stage-specific miRNA expression and functions would be helpful to decipher the complex genetic network that controls mosquito at a crucial stage.
Anopheles sinensis is the most common and wide-distributed malaria vector exhibiting a variety of ecological behaviors and feeding habits throughout the China. It is also of great medical and veterinary importance. Thus, identification of key miRNAs required for gene regulation throughout the life cycle will not only help to a better understanding of its developmental biology but also may help to conceive novel approaches to control this vector. However, the function and molecular mechanism of these miRNAs are still need to be further investigated.
Conclusions
In conclusion, this study provides a comprehensive account of miRNA profile and its dysregulated expression patterns across four different stages of An. sinensis development using the high throughput sequencing. Several miRNAs show stage-specific expression pattern in particular developmental stages. We also investigate targets of these dysregulated miRNAs and their related functions in important life activities in order to provide a better understanding of mosquito development. In turn, such information would provide the basis to conceive novel approaches to control this vector in future.
Abbreviations
- An. sinensis :
-
Anopheles sinensis
- P. vivax :
-
Plasmodium vivax
- An. gambiae :
-
Anopheles gambiae
- An. stephensi :
-
Anopheles stephensi
- Ae. aegypti :
-
Aedes aegypti
- Cu. fatigans :
-
Culex fatigans
- Ae. albopictus :
-
Aedes albopictus
- An. anthropophagus :
-
Anopheles anthropophagus
- miRNA:
-
microRNA
- Pre-miRNA:
-
precursor microRNA
- ASE:
-
An. sinensis egg
- ASL:
-
An. sinensis larvae
- ASP:
-
An. sinensis pupa
- ASA:
-
An. sinensis adult female
- piRNA:
-
Piwi-interacting RNA
- nt:
-
Nucleotide
- GO:
-
gene ontology
- KEGG:
-
kyoto encyclopedia of genes and genomes
- UTR:
-
untranslated region
- NGS:
-
next generation sequencing
- TPM:
-
tags per million
- mRNA:
-
messenger RNA
- rRNA:
-
ribosomal RNA
- tRNA:
-
transfer RNA
- small ncRNAs:
-
small non-coding RNAs
- snoRNA:
-
small nucleolar RNA
- qRT-PCR:
-
reverse transcription quantitative real-time polymerase chain reaction
References
Feng X, Zhang S, Huang F, Zhang L, Feng J, Xia Z, et al. Biology, bionomics and molecular biology of Anopheles sinensis Wiedemann 1828 (Diptera: Culicidae), main malaria vector in China. Front Microbiol. 2017;8:1473.
Feng X, Huang L, Lin L, Yang M, Ma Y. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera: Culicidae) in China inferred by cox1 gene. Parasit Vectors. 2017;10(1):75.
Lyu Y, Shen Y, Li H, Chen Y, Guo L, Zhao Y, et al. New microRNAs in Drosophila–birth, death and cycles of adaptive evolution. PLoS Genet. 2014;10(1):e1004096.
Zhang G, Hussain M, O’Neill SL, Asgari S. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proc Natl Acad Sci USA. 2013;110(25):10276–81.
Lucas KJ, Myles KM, Raikhel AS. Small RNAs: a new frontier in mosquito biology. Trends Parasitol. 2013;29(6):295.
Dritsou V, Deligianni E, Dialynas E, Allen J, Poulakakis N, Louis C, et al. Non-coding RNA gene families in the genomes of anopheline mosquitoes. BMC Genom. 2014;15:1038.
Wang X, Zhang J, Li F, Gu J, He T, Zhang X, et al. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21(18):3610–4.
Krishnan R, Kumar V, Ananth V, Singh S, Nair AS, Dhar PK. Computational identification of novel microRNAs and their targets in the malarial vector, Anopheles stephensi. Syst Synth Biol. 2015;9(1–2):11–7.
Jain S, Rana V, Tridibes A, Sunil S, Bhatnagar RK. Dynamic expression of miRNAs across immature and adult stages of the malaria mosquito Anopheles stephensi. Parasit Vectors. 2015;8:179.
Zhang X, Aksoy E, Girke T, Raikhel AS, Karginov FV. Transcriptome-wide microRNA and target dynamics in the fat body during the gonadotrophic cycle of Aedes aegypti. Proc Natl Acad Sci USA. 2017;114(10):1895–903.
Hussain M, Walker T, O’Neill SL, Asgari S. Blood meal induced microRNA regulates development and immune associated genes in the Dengue mosquito vector, Aedes aegypti. Insect Biochem Mol Biol. 2013;43(2):146–52.
Akbari OS, Antoshechkin I, Amrhein H, Williams B, Diloreto R, Sandler J, et al. The developmental transcriptome of the mosquito Aedes aegypti, an invasive species and major arbovirus vector. G3 (Bethesda). 2013;3(9):1493–509.
Zotzmann S, Steinbrink A, Schleich K, Frantzmann F, Xoumpholphakdy C, Spaeth M, et al. Bacterial diversity of cosmopolitan Culex pipiens and invasive Aedes japonicus from Germany. Parasitol Res. 2017;116(7):1899–906.
Hong S, Guo Q, Wang W, Hu S, Fang F, Lv Y, et al. Identification of differentially expressed microRNAs in Culex pipiens and their potential roles in pyrethroid resistance. Insect Biochem Mol Biol. 2014;55C:39–50.
Skalsky RL, Vanlandingham DL, Scholle F, Higgs S, Cullen BR. Identification of microRNAs expressed in two mosquito vectors, Aedes albopictus and Culex quinquefasciatus. BMC Genom. 2010;11:119.
Puthiyakunnon S, Yao Y, Li Y, Gu J, Peng H, Chen X. Functional characterization of three MicroRNAs of the Asian tiger mosquito, Aedes albopictus. Parasit Vectors. 2013;6(1):230.
Liu W, Huang H, Xing C, Li C, Tan F, Liang S. Identification and characterization of the expression profile of microRNAs in Anopheles anthropophagus. Parasit Vectors. 2014;7:159.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–4.
Wen M, Shen Y, Shi S, Tang T. miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012;13(1):1–10.
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136.
Storey JD. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat. 2003;31(6):2013–35.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(Web Server issue):W451–4.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–93.
Giraldo-Calderon GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas E, Topalis P, et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2015;43(Database issue):D707–13.
Duman-Scheel M, Flannery E, Behura SK, Haugen M, Clemons A, Severson DW. Functional genetic and comparative genomic analysis of vector mosquito development. Dev Biol. 2011;356(1):243.
Behura SK, Haugen M, Flannery E, Sarro J, Tessier CR, Severson DW, et al. Comparative genomic analysis of drosophila melanogaster and vector mosquito developmental genes. PLoS ONE. 2011;6(7):e21504.
Costa-da-Silva AL, Marinotti O, Ribeiro JM, Silva MC, Lopes AR, Barros MS, et al. Transcriptome sequencing and developmental regulation of gene expression in Anopheles aquasalis. PLoS Negl Trop Dis. 2014;8(7):e3005.
Akbari OS, Antoshechkin I, Amrhein H, Williams B, Diloreto R, Sandler J, et al. The developmental transcriptome of the mosquito Aedes aegypti, an invasive species and major arbovirus vector. G3 (Bethesda, Md). 2013;3(9):1493–509.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Lei Z, Lv Y, Wang W, Guo Q, Zou F, Hu S, et al. MiR-278-3p regulates pyrethroid resistance in Culex pipiens pallens. Parasitol Res. 2015;114(2):699–706.
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–41.
Allam M, Spillings BL, Abdalla H, Mapiye D, Koekemoer LL, Christoffels A. Identification and characterization of microRNAs expressed in the African malaria vector Anopheles funestus life stages using high throughput sequencing. Malar J. 2016;15(1):542.
Biryukova I, Ye T, Levashina E. Transcriptome-wide analysis of microRNA expression in the malaria mosquito Anopheles gambiae. BMC Genom. 2014;15:557.
Hu W, Criscione F, Liang S, Tu Z. MicroRNAs of two medically important mosquito species: Aedes aegypti and Anopheles stephensi. Insect Mol Biol. 2015;24(2):240–52.
Waskar M, Landis GN, Shen J, Curtis C, Tozer K, Abdueva D, et al. Drosophila melanogaster p53 has developmental stage-specific and sex-specific effects on adult life span indicative of sexual antagonistic pleiotropy. Aging. 2009;1(11):903.
Woodcock SA, Hughes DA. p120 Ras GTPase-activating protein associates with fibroblast growth factor receptors in Drosophila. Biochem J. 2004;380(Pt 3):767.
Morey M, Serras F, Baguñà J, Hafen E, Corominas M. Modulation of the Ras/MAPK signalling pathway by the redox function of selenoproteins in Drosophila melanogaster. Dev Biol. 2001;238(1):145–56.
Goeke S, Greene EA, Grant PK, Gates MA, Crowner D, Aigaki T, et al. Alternative splicing of lola generates 19 transcription factors controlling axon guidance in Drosophila. Nat Neurosci. 2003;6(9):917.
Authors’ contributions
XYF conceived and designed the study, XYF and WH performed the experiments. XYF and HW analyzed the data and drafted the manuscript. JWW and SSZ critically revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Dongxiao Yang from the School of Bioinformatics, Nanjing Agricultural University for providing the computational algorithm involved in the work. This work was supported by the National Natural Science Foundation of China (Nos. 81271867 and 91431104), International Science and Technology Cooperation Program of China (No. 2014DFA31130) and National Science and Technology Major Program (No. 2009ZX10004-302). This work was also supported by Shanghai Municipal Commission of Health and Family Planning Grant (No. 20174Y0089).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI BioProject repository, [PRJNA412443, this Sequence Read Archive will be released on 2018-10-01 or upon publication].
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Primers used in qRT-PCR validation.
Additional file 2: Table S2.
Novel miRNAs identified in four life stages libraries of An. sinensis.
Additional file 3: Table S3.
Target gene candidates for known and novel miRNAs in An. sinensis.
Additional file 4: Table S4.
Developmental target gene candidates for 32 key miRNAs during development in An. sinensis.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Feng, X., Wu, J., Zhou, S. et al. Characterization and potential role of microRNA in the Chinese dominant malaria mosquito Anopheles sinensis (Diptera: Culicidae) throughout four different life stages. Cell Biosci 8, 29 (2018). https://doi.org/10.1186/s13578-018-0227-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-018-0227-1