
Abstract
Improving the predication efficiency of porcine production performance at early stage will contribute to reducing the breeding and production costs. The intestinal microbiota had received plenty of attention in recent years due to their influence on host health and performance. The purpose of this study was to investigate the relationship between the fecal microbiota at early growth period and porcine feed efficiency (FE) under a commercial feeding environment. Ninety-one pigs were reordered according to the residual feed intake (RFI) values between day 90 on test and day 160 off test, 9 lowest RFI pigs and 9 highest RFI pigs were selected as the LRFI group and the HRFI group, respectively. Fecal samples from pigs in the early grower phase (day 80) were performed for microbial diversity, composition, and predicted functionality by using 16S rRNA sequencing. The results showed that no significant differences in microbial alpha diversity were observed between two RFI groups, whereas, some RFI-associated compositional differences were revealed. In particular, the microbiota of the LRFI group (more feed-efficient) had significantly higher levels of some members of Clostridiales and Bacteroidales (e.g., g_1_68 and g_norank_f_p_2534_18B5), which may promoted FE through protecting gut barrier function, compared with those of the HRFI pigs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis found that the LRFI pigs were likely have microbiota with higher levels of amino acid metabolism. Moreover, redundancy analysis (RDA) showed that litter size, parity, and date of birth had significant effects on the bacterial community structure. These results improved our knowledge of the porcine early-life fecal microbiota and its potential link underlying RFI, which would be useful for future development of microbial biomarkers for predicting and improving porcine FE as well as investigation of targets for dietary strategies.
Similar content being viewed by others

Introduction
Feed accounts for more than 60% of total production costs in growing pigs. Therefore, improving FE has been an important part of the breeding goal in commercial pig production for both economic and environmental reasons. A variety of factors that can influence porcine FE, such as genetics (Do et al. 2014; Onteru et al. 2013; Reyer et al. 2017), diseases (Patience et al. 2015), environment (Chatelet et al. 2018), and diets (Collins et al. 2017; Gilbert et al. 2018). RFI, defined as the difference between the observed feed intake and predicted feed requirements based on average daily gain and backfat, is a useful criterion to measure net FE based on the biological mechanisms that influence FE (Gilbert et al. 2017; Herd and Arthur 2009). Intestinal microbiota is considered as a major “factor”, which plays an important role in the processing of nutrients and the acquisition of energy (Fouhse et al. 2016; Ramayo-Caldas et al. 2016; Xiao et al. 2016). The contribution of intestinal microbiota to pig health and performance, including digestion and metabolism of nutrients, stimulation of immune response, protection from pathogens and stimulation of epithelium cell proliferation is becoming increasingly apparent (Katouli et al. 1997; Konstantinov et al. 2006; Mann et al. 2014; Spreeuwenberg et al. 2001; Thompson et al. 2008). So, the intestinal microbiota could potentially be used to predict or improve porcine FE.
In recent years, the correlations between microbial community and phenotypic traits have become the focus of much attention (Buzoianu et al. 2012; Frese et al. 2015; Pedersen et al. 2013; Vigors et al. 2016). Many researchers have found FE-related bacterial groups in pigs (Quan et al. 2018; Tan et al. 2018; Yang et al. 2017), chickens (Siegerstetter et al. 2017; Yan et al. 2017), and cattle (Li et al. 2019). In pigs, the early colonization and succession of intestinal microbiome has been found to be important for the formation of host phenotypes and specific microbial composition (Collins et al. 2017; Han et al. 2017; Tian et al. 2017). However, little is known about whether the composition of fecal microbiome in young pigs might be used to predict the phenotype of grower-finisher hosts. It had been reported that the increase of Firmicutes with pigs’ growth was consistent with the significantly increased fat deposition in older pigs, compared to 1 month old piglets (Zhao et al. 2015). A large-scale study showed that the fecal microbiota diversity at week 15 and end-of-feeding stage were strongly correlated with back fat and average daily gain of crossbred pigs, whereas very low correlation were detected at weaning (Lu et al. 2018). In the current study, we aimed to investigate the possible relationship between the fecal microbiota and RFI trait of growing stage pigs under a commercial environment. For the purpose, we compared the composition and potential functionality of the fecal microbiota in LRFI and HRFI Duroc pigs using 16S rRNA gene sequencing, and analyzed the effects of host and environmental factors on the microbial community structure.
Materials and methods
Animals management and sample collection
Ninety-one purebred Duroc pigs used in this study were raised in a commercial farm (Nanning, Guangxi, China). The piglets were weaned at the same age of 28 days and raised under the same nursery conditions. All experimental pigs were moved to an environmentally controlled fattening house (ten pigs in each pen) at the age of day 70, and were fed with the same standard diets without antibiotics or medicines. Daily feed intake (DFI) and individual body weights (BW) data were recorded by Electronic Feed Intake Recording Equipment (FIRE, Osborne, USA) and used to calculate performance indicators, such as average daily feed intake (ADFI), average daily gain (ADG), and feed conversion ratio (FCR). ADFI, individual BW, and FCR data were collected from day 90 to 160. The backfat thickness (BF) was measured using ultrasound measurements (Corometrics Medical Systems, Inc., Wallingford, CT, USA). The equation used to predict RFI has been previously described (Cai et al. 2008). Fresh fecal samples were collected from each individual at 80 ± 1.15 days old and kept frozen in liquid nitrogen for transportation, and then stored at − 80 °C until use.
DNA extraction and sequencing
The DNA extraction, 16S rRNA gene PCR, library preparation and DNA sequencing of the fecal samples were performed by a commercial provider (Shanghai Majorbio Bio-pharm Technology Co.,Ltd, China). The V3–V4 region of the 16S rRNA gene was amplified by polymerase chain reaction (PCR) with universal bacterial 16S rRNA gene PCR amplicon primers (341F-806R) (Kozich et al. 2013). According to the preliminary quantitative results of electrophoresis, the PCR products were detected and quantified by quantifluor™-St blue fluorescence quantitative system (Promega company), and then mixed in corresponding proportion according to the requirements of the sequencing quantity of each sample. All sample libraries sequencing were performed on the Illumina MiSeq platform (Illumina, USA).
Microbial analysis
The 16S rRNA sequencing data were processed using the Quantitative Insights Into Microbial Ecology (QIIME1) (version 1.9.1) platform (Caporaso et al. 2010). The raw sequence reads were filtrated with a minimum overlap of 10 bp and a maximum mismatch ratio 0.2 by using FLASH (version 1.2.11). Operational taxonomic units (OTUs) were picked at 97% similarity cut-off, and the identified taxonomy was then aligned using the Greengenes database (version 13.8). Chimeric sequences were identified and removed in the process of clustering with the software of USEARCH (version 7). OTUs with number of sequences < 20 of the total number of sequences were removed from the OTU table with the software of USEARCH.
The microbial alpha diversity indices of the samples were determined using the Chao1 (richness estimator) (Chao 1984), ACE (abundance-based coverage estimator) (Chao and Yang 1993), Sobs (the observed richness), Shannon (entropy estimator) (Shannon 1948), Simpson (Simpson’s index calculator) (Simpson 1949), Coverage (community coverage), Shen, and PD (Phylogenetic diversity) (Faith 1992), and these indices analyses were also calculated within Mothur (version 1.30.2). Principal component analysis (PCA) was performed at the phylum and the genus level, and the results were visualized using the STAMP program (version 2.1.3) (Parks et al. 2014). Linear discriminant analysis (LDA) effect size (LEfSe) were performed using the LEfSe tool (Segata et al. 2011). The OTU functions of fecal microbial were predicted by the KEGG database, based on performed using phylogenetic investigation of community by reconstruction of unobserved States (PICRUSt) (Langille et al. 2013). RDA was executed, and the significance of total host and environmental factors (including pen, season, birth weight, parity, date of birth, litter size) was tested with Monte Carlo permutations (permu = 999). Host and environmental factors were selected by the functions of envfit (permu = 999) and vif, and the factors with P > 0.05 or vif > 10 were removed from the following analysis.
Result
Basic statistics of porcine performance and RFI
The average RFI value of LRFI and HRFI was − 0.047 ± 0.11 (mean ± SD) (Table 1). Compared with HRFI pigs, ADFI of LRFI pigs was lower 0.45 kg/day (P < 0.01), but showed an improvement in FCR of 0.42 (P < 0.01). However, no remarkable differences between the HRFI and LRFI were observed for ADG (P > 0.05), 100 kg BF (P > 0.05), day 90 BW (P > 0.05), day 160 BW (P > 0.05) and other phenotypes (P > 0.05) (Additional file 1). The phenotypic distributions of 91 pigs including ADFI, ADG, FCR, 90d BW, 160d BW, 100 kg BF and RFI values in the experimental cohort were shown in Additional file 1.
Differences in the fecal bacterial alpha diversity of the HRFI and LRFI pigs
A total of 3,254,159 sequence reads were obtained from 91 fecal samples, with an average of 35,759 reads per sample (ranging from 23,641 to 73,941). After subsampling each sample to an equal sequencing depth (23,562 reads per sample) and clustering, 1011 OTUs at 97% identity were obtained. From a taxonomic perspective, 14 phyla, 24 class, 33 order, 60 families, and 114 genera were identified across all pig fecal samples.
Sobs, Shannon index, Simpson index, ACE, Chao 1, Coverage, Shen, PD index values were used as parameters of the alpha diversity of fecal microbiota in our study (Table 2). The Sobs metric was 575.33 for the LRFI group and 549.22 for the HRFI group. The Shannon metric was 4.48 for the LRFI group and 4.32 for the HRFI group. The Simpson metric was 0.03 for the LRFI group and 0.06 for the HRFI group. The ACE metric was 660.52 for the LRFI group and 637.15 for the HRFI group. The Chao 1 metric was 670.15 for the LRFI group and 644.95 for the HRFI group. The Coverage metric was 0.99 for the LRFI group and 0.99 for the HRFI group. The Shen metric was 62.50 for the LRFI group and 64.95 for the HRFI group. The PD metric was 48.09 for the LRFI group and 45.48 for the HRFI group. No significant differences for any of the indices of alpha diversity measured (P > 0.05).
Differences in the fecal microbial taxa represented in the HRFI and LRFI pigs
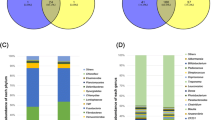
At the phylum level, the microbiota of the LRFI and HRFI groups shared 13 phyla (92.86%, Fig. 1a), whereas Fusobacteria uniquely identified in the LRFI group. The three dominant phyla detected in both groups were Firmicutes (70.41% in the LRFI group and 75.34% in the HRFI group), Bacteroidetes (25.02% in the LRFI group and 21.04% in the HRFI group), and Actinobacteria (1.43% in the LRFI group and 1.13% in the HRFI group) (Fig. 1c).

Compositions of the fecal microbiota of the LRFI and HRFI pigs. The number of phylum (a) and genera (b) shared by the two groups are shown in Venn diagrams. The different colors represent different groups, the numbers in the overlapping part represent the number of species in two groups, and the numbers in the non-overlapping parts represent the number of species unique to the corresponding group. The overall compositions of the fecal microbiota of the LRFI and HRFI groups were represented as bar plots at the phyla (c) and the genus level (d). The different colors represent the columns, and the length of the columns represent the proportion of the species in the two groups
At the genus level, the fecal microbiota of the two groups shared 106 genera (92.98%), with 7 (6.14%) and 1 genus (0.0088%) uniquely identified in the LRFI group and the HRFI group, respectively (see Fig. 1b). Four dominant genera, Prevotella (16.25%), norank_f_Ruminococcaceae (10.25%), Megasphaera (6.34%), and norank_f_Lachnospiraceae (5.31%) were found in the LRFI group, whereas the four dominant genera in the HRFI group were Prevotella (12.48%), Lactobacillus (11.57%), norank_f_Ruminococcaceae (11.18%), and norank_f_S24-7 (4.93%) (Fig. 1d).
PCA was used to compare the total microbial composition of the LRFI and HRFI pigs at the level of phylum and genus. In the PCA diagram, the microbial communities were not separated at phylum (Fig. 2a) and genus (Fig. 2b) levels. Then, we compared the relative abundance of microbial members between the two groups and found some significant different phyla and genera (P < 0.05), although they have a lower abundance. Such as the level of Chlamydiae phylum was significantly higher in the HRFI group than those in the LRFI group (P = 0.037) (Fig. 2c). At the genus level, the levels of Chlamydia (P = 0.0034), norank_f__Erysipelotrichaceae (P = 0.016), g_Kitasatospora (P = 0.0034) were significantly higher in the HRFI group. In contrast, the levels of norank_f_p_2534_18B5 (P = 0.034), g_1_68 (P = 0.034) were significantly higher in the LRFI group than those in the HRFI group (Fig. 2d).

Composition of the fecal microbiota of the LRFI and HRFI pigs at the phylum and genera level. Principal component analysis (PCA) plot at phylum (a) and genera (b) level. The X-axis and Y-axis represent the principal component axes, and the percentage represents the value of the component’s interpretation of the different sample composition. The points of different colors or shapes represent samples of different groups, and the closer the two sample points, the more similar the two species composition. The phylum (c) and genera (d) represented at significantly different levels in the microbiota of two groups were shown in an extended error bar plot. The X-axis represents different groups, different colored boxes represent different groups, and the Y-axis represents the average relative abundance of a species in different groups. The differences in the compositions were tested using a two-sided Welch’s test, and P < 0.05 was marked with “*”
The LEfSe tool was used to identify specialized microbial communities in the HRFI and LRFI groups. The results of LEfSe analysis showed that 5 (3 genera unique to the HRFI group and 2 genera unique to the LRFI group) genera were potential biomarkers for distinguishing between high and low RFI groups (Fig. 3). These results confirmed the significant of enrichment of p_Chlamydiae, c_Chlamydiia, g_Chlamydia, f_Chlamydiaceae, o_Chlamydiales, f_Streptomycetaceae, g_Kitasatospora and norank_f__Erysipelotrichaceae in the HRFI group. We also found that, o_Burkholderiales, f_Tissierellaceae_, g_1_68, p_2534_18B5, and norank_f_p_2534_18B5 were higher relative abundances in the LRFI group compared to those in the HRFI group (Fig. 3).

Indicator bacteria with LDA scores of 2 or greater in bacterial communities associated with LRFI and HRFI groups. The X-axis represents the LDA score, different colored boxes represent different groups, and the higher the LDA score, the greater the impact of species abundance on the difference effect
Predicted KEGG pathways differences of fecal microbiota between the HRFI and LRFI pigs
Several of the predicted KEGG pathways were significantly differentially identified in the fecal microbiota of the HRFI and LRFI pigs (Fig. 4). “Cysteine and methionine metabolism” pathway was predicted at significantly a higher level in the microbiota of the LRFI group (1.00%) than in that of the HRFI group (0.95%). In contrast, “fructose and mannose metabolism”, “penicillin and cephalosporin biosynthesis”, “phosphotransferase system (PTS)”, “beta-Lactam resistance”, and “base excision repair” pathways which were involved in “carbohydrate metabolism”, “biosynthesis of other secondary metabolites”, “membrane transport”, “drug resistance: antimicrobial”, and “replication and repair” at the higher KEGG pathway hierarchical levels, were predicted at significantly higher levels in the microbiota of the HRFI group (0.99, 0.02, 0.55, and 0.02%, respectively) than those in that of the LRFI group (0.92, 0.02, 0.43, and 0.02%, respectively).

The different functions of the fecal microbiota of the LRFI and HRFI groups. The microbial functions were predicted using PICRUSt at the third level of the KEGG pathway and were expressed as relative. The differences in the compositions were tested using a two-sided Welch’s test, permutation test replicates 1000, and P < 0.05 was considered significant
Host and environmental factors affecting porcine fecal microbial community structure
Host and environmental factors may influence microbial community structures. Therefore, we investigated the possible correlations between the microbial community structure and the variables of environmental and host characteristics. After removal of the redundant variables, five host and environmental characteristics (including pen, birth weight, parity, date of birth, and litter size) were chosen for RDA. As a result, litter size was found to have significant (P = 0.007) effect on the fecal bacterial community structure at phylum level (Fig. 5a), which the first axis and second axis explained 21.15% and 0.56% of total microbial variance, respectively. Besides, at genera level, date of birth and parity had significant (P = 0.019 and 0.024) effects on the fecal microbial composition (Fig. 5b), which the first axis and second axis explained 21.17% and 8.32% of total microbial variance, respectively.

The relationship of host and environmental factors to the microbial community structure. At phylum (a) and genus (b) level Distance-based Redundancy Analysis (RDA) plot showing the relationship of pen, season, birth weight, parity, date of birth and litter size to the microbial community structure. The red arrow indicates the host and environmental factors. The length of the arrow was representing the degree of interpretation of the factors to the species. The angle between the arrows represents the correlation. The distance from the sample point to the factor represents the relative influence of the factor on the distribution of the microbial community. Correlation heatmap of the top fourteen phyla (c), the top fifty genus (d) and environmental and host factors. The X-axis and Y-axis were host and environmental factors and species, respectively. The R-value was shown in different colors in the figure. The legend on the right was the color range of different R values. The value of P < 0.05 or P < 0.01 was marked with “*” or “**”
The correlation heatmap showed that the relationship between bacterial phyla and environmental factors were different (Fig. 5c). Actinobacteria showed a significant positive correlation with date of birth (Spearman correlation coefficient: 0.50, P = 0.036). Fibrobacteres demonstrated a significant negative correlation with litter size (Spearman correlation coefficient: − 0.59, P = 0.010). The factor of pen was significant positive correlated with Proteobacteria (Spearman correlation coefficient: 0.53, P = 0.020), whereas significant negative correlated with WPS-2 (Spearman correlation coefficient: − 0.49, P = 0.040). The birth weight revealed a significant negative correlation with Firmicutes (Spearman correlation coefficient: − 0.52, P = 0.027), whereas significant positive correlated with Proteobacteria (Spearman correlation coefficient: 0.54, P = 0.021) and Bacteroidetes (Spearman correlation coefficient: 0.51, P = 0.032). Similarly, result of heatmap showed the relationship between environmental factors and bacterial genera at the genus level were different (Fig. 5d). The factor of date of birth was significant positive correlated with [Eubacterium] (Spearman correlation coefficient: 0.47, P = 0.049), Catenibacterium (Spearman correlation coefficient: 0.55, P = 0.018), Collinsella (Spearman correlation coefficient: 0.54, P = 0.021) and g_Oscillospira (Spearman correlation coefficient: 0.48, P = 0.044), whereas significant negatively correlated with Clostridium (Spearman correlation coefficient: − 0.66, P = 0.003), g_norank_o_Bacteroidales (Spearman correlation coefficient: − 0.49, P = 0.038), g_norank_o_Clostridiales (Spearman correlation coefficient: − 0.47, P = 0.049). The birth weight revealed a significant positive correlation with [Prevotella] (Spearman correlation coefficient: 0.49, P = 0.036), Enterobacteriaceae (Spearman correlation coefficient: 0.49, P = 0.039) and Phascolarctobacterium (Spearman correlation coefficient: 0.58, P = 0.012), nevertheless, significant negatively correlated with g_norank_f_Coriobacteriaceae (Spearman correlation coefficient: − 0.49, P = 0.041). The factor of parity was significant positive correlated with [Mogibacteriaceae] (Spearman correlation coefficient: 0.53, P = 0.024) and g_norank_f_S24-7 (Spearman correlation coefficient: 0.63, P = 0.005), however, significant negatively correlated with Roseburia (Spearman correlation coefficient: − 0.58, P = 0.012), Anaerovibrio (Spearman correlation coefficient: − 0.55, P = 0.017), Faecalibacterium (Spearman correlation coefficient: − 0.47, P = 0.049). Megamonas (Spearman correlation coefficient: − 0.53, P = 0.024) and Megasphaera (Spearman correlation coefficient: − 0.52, P = 0.026). The factor of pen was significant positive correlated with Clostridium (Spearman correlation coefficient: − 0.48, P = 0.044).
Discussion
Recent studies in porcine intestinal microbiome using next-generation sequencing have greatly expanded our understanding on the role of the gut microbiota in different phenotypes (Han et al. 2017; McCormack et al. 2017; Tan et al. 2018), and demonstrated associations between porcine intestinal microbes composition and growth traits. Several studies indicated that the differences within the gut microbiome explained the variability of FE in pigs. For example, Oscilibacter, Christensenellaceae, and Cellulosilyticum were more abundant in high FE pigs (McCormack et al. 2017). In another study, Ruminococcaceae, Christensenellaceae, Akkermansia, and Lachnospiraceae were reported to have a positive relationship to porcine FE, nevertheless, Faecalibacterium has a negative association with porcine FE (Yang et al. 2017). However, most of these studies were focused on the grower-finisher pigs (Fang et al. 2017; Tan et al. 2018), with very few studies involving the FE of pigs through differences in microbiome comparison of early-life pigs. Bacterial colonization in the gastrointestinal tract of early-life pigs has been suggested to be crucial for the formation of host phenotypes, and concurrently helpful for early selection of pigs (Mach et al. 2015). Early selection of breeding stock can expand the scale of testing and increase selection intensity, especially for low-heritability traits (such as RFI), and can achieve rapid genetic progress.
Consistent with a previous report (Yang et al. 2018), the LRFI pigs tended to have higher α variety (Chao 1, Sob, Shannon, ACE, Coverage, and PD) than the HRFI pigs, albeit no significant difference in richness or evenness were found between the two groups. The composition of intestinal microorganisms can be influenced by diet, age, host genetics and environmental factors (Benson et al. 2010; Buzoianu et al. 2012; Kumar et al. 2019). Previous studies indicated that there were significant differences in intestinal microbial composition among pigs at different grow stages (Ke et al. 2019; Kumar et al. 2019). In this study, the dominant phyla within the fecal microbiota were Firmicutes, Bacteroidetes and Actinobacteria, and the six most abundant genera were Prevotella, Ruminococcaceae_f_norank, Lactobacillus, Megasphaera, Lachnospiraceae_f_norank and S24-7_f_norank, although the proportion of each phylum and genera were fluctuant (Camarinha-Silva et al. 2017; Han et al. 2017). Wang et al. reported that Firmicutes, Bacteroidetes, and Actinobacteria were the three most abundant phyla across each stage in the life of a pig, and these microbiomes were defined as core bacteria (Wang et al. 2019). At the genus level, Prevotella, Ruminococcaceae, Lactobacillus were reported as the core bacteria for fecal samples at the ages of 80, 120 and 240 days (Ke et al. 2019).
Specific fecal microbiota could potentially be linked with porcine FE. LEFse analysis found that g_1_68 in the LRFI pigs, which within the Clostridiales order, was significantly higher than in the HRFI pigs. As previously reported (McCormack et al. 2017), most of the RFI-specific OTUs were from the Clostridiales order. Clostridiales are butyric acid-producing bacteria, which can promote the functional recovery of intestinal mucosa and may inhibit the formation of inflammatory cytokines, thereby play an anti-inflammatory role in the gastrointestinal tract (Augenlicht et al. 2002; Segain et al. 2000). Moreover, the uncultured p_2534_18B5, is a member of Bacteroidales, which has been proved to have epithelial barrier function in colonic through regulation the secretion of cytokines (Kuhn et al. 2018). In addition, corresponding to the LRFI group, Chlamydia, Kitasatospora, and uncultured Erysipelotrichaceae had a higher abundance in the HRFI group, which were potential pathogenicity and may be the factors that affect porcine FE.
The involvement of different metabolic pathways in the microbiota could further justify differences in the porcine phenotype. In this study, several different KEGG pathways were observed in the LRFI and HRFI groups. “Cysteine and methionine metabolism” related genes were more abundant in the fecal microbiota of the LRFI group than those in that of the HRFI group. Several study pointed out that intestinal bacteria may affect the utilize dietary protein by producing short chain fatty acids (SCFA) and regulating the metabolism of acids (Holzer 2016). We observed that five KEGG pathways (“fructose and mannose metabolism”, “penicillin and cephalosporin biosynthesis”, “phosphotransferase system (PTS)”, “beta-Lactam resistance”, and “base excision repair”) were significantly more abundant in the microbiota of the HRFI group than those in that of the LRFI group, consistent with the results of other studies. For instance, previous studies indicated that PTS is a complex translocation system, which transports sugar to cells and phosphorylates substrates during transport by phosphotransferase (Lengeler et al. 1990). This may reduce the utilization of sugar by the host to some extent. “Penicillin and cephalosporin biosynthesis” and “beta-Lactam resistance”-related genes were more abundant in the fecal microbiota of the HRFI than those in that of the LRFI group. Penicillin and cephalosporin are synthesized by a series of enzymatic reactions that form the tripeptide δ-(l-α-aminoadipyl)-l-cysteinyl-d-valine and convert this tripeptide into the final penicillin or cephalosporin molecules (Martín et al. 1999). Interesting, we observed a significant increase Chlamydiae in the HRFI group. Demonstrably, a higher relative abundance of potentially disadvantageous bacteria, such as Chlamydiae and Bacteroidales, were found in pigs with lower FE. These results indicated that pigs that are more feed efficient are likely to have fecal microbiota with higher levels of amino acid metabolism.
Even though many studies have involved the relationship between microorganisms and FE, few FE-related microbial classifications were found, which may be due to the diet, host genetics and management strategies used in different feeding environments. In the present study, we found that the composition of fecal bacterial community was related to host (especially litter size and parity) and some environment factors. For instance, at the phylum and genus levels, the date of number, litter size and parity have a significant influence on intestinal microbial structure composition. Yang et al. reported that significant effects of sex and kinship on fecal microbial community structure (Yang et al. 2017). However, several studies have suggested no significant correlation between microbiota composition and pen or sex (Mach et al. 2015). The relationship between host or environment factors (such as pen, birth weight, parity, date of birth and litter size) and porcine fecal microbial community structure remains controversial issues. In this study, Firmicutes showed a significant negative correlation with birth weight, whereas Bacteroidetes has a significant positive correlation with birth weight. Ding et al. reported that the abundances of Firmicutes showed a significant negative correlation with pre-weaned weight gain and Bacteroidetes showed a significant positive correlation with pre-weaned weight gain in the colon (Ding et al. 2019). However, Han et al. reported that at the phylum level, the microbe of heavier weaned piglets had significantly higher levels of Firmicutes and a higher Firmicutes-to-Bacteroidetes ratio than that of the lighter piglets (Han et al. 2017). These results suggested that the host and environmental factors may play important roles in the formation of early microbial community structures in pigs.
In conclusion, the present results provided novel information of RFI-associated fecal bacterial profiles in Duroc pigs at early growth period, suggesting that the microbiota has a possible link with porcine FE. Importantly, the Clostridiales and Bacteroidales, such as g_1_68, g_norank_f_p_2534_18B5, were found to be potential early life predictive biomarkers for high FE. Predictive functional analysis also indicated that fecal microbes of the high FE pigs may have a high level of utilize dietary protein. Besides, our results indicated that the composition of fecal bacterial community was related to some host factors, especially litter size and parity. Although, as of now, more studies are required to clarify the relationship between the intestinal microbiota at a growing stage and FE at a mature stage pig, these results may provide insights into understanding the host-microbe interactions occurring in the early-life pig intestine and will be helpful for the assisted early selection of porcine FE.
Availability of data and materials
The sequencing raw data in this study was deposited in NCBI Sequence Read Archive (SRA) under accession number PRJNA593419.
Abbreviations
- RFI:
-
Residual feed intake
- FE:
-
Feed efficiency
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- RDA:
-
Redundancy analysis
- DFI:
-
Daily feed intake
- BW:
-
Body weights
- ADFI:
-
Average daily feed intake
- ADG:
-
Average daily gain
- FCR:
-
Feed conversion ratio
- BF:
-
Backfat
- PCR:
-
Polymerase chain reaction
- QIIME:
-
Quantitative Insights Into Microbial Ecology
- OTUs:
-
Operational taxonomic units
- PCA:
-
Principal component analysis
- LEfSe:
-
Linear discriminant analysis effect size
- PICRUSt:
-
Phylogenetic investigation of community by reconstruction of unobserved States
- SCFA:
-
Short chain fatty acids
References
Augenlicht LH, Mariadason JM, Wilson A, Arango D, Yang W, Heerdt BG, Velcich A (2002) Short chain fatty acids and colon cancer. J Nutr 132(12):3804S–3808S. https://doi.org/10.1093/jn/132.12.3804S
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci 107(44):18933–18938. https://doi.org/10.1073/pnas.1007028107
Buzoianu SG, Walsh MC, Rea MC, O’Sullivan O, Crispie F, Cotter PD, Ross RP, Gardiner GE, Lawlor PG (2012) The effect of feeding Bt MON810 maize to pigs for 110 days on intestinal microbiota. PLoS ONE 7(5):e33668. https://doi.org/10.1371/journal.pone.0033668
Cai W, Casey D, Dekkers J (2008) Selection response and genetic parameters for residual feed intake in Yorkshire swine. J Anim Sci 86(2):287–298. https://doi.org/10.2527/jas.2007-0396
Camarinha-Silva A, Maushammer M, Wellmann R, Vital M, Preuss S, Bennewitz J (2017) Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs. Genetics 206(3):1637–1644. https://doi.org/10.1534/genetics.117.200782/-/DC1
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335. https://doi.org/10.1038/nmeth.f.303
Chao A (1984) Nonparametric estimation of the number of classes in a population. Scan J Stat 11:265–270
Chao A, Yang MC (1993) Stopping rules and estimation for recapture debugging with unequal failure rates. Biometrika 80(1):193–201
Chatelet A, Gondret F, Merlot E, Gilbert H, Friggens NC, Le Floc’h N (2018) Impact of hygiene of housing conditions on performance and health of two pig genetic lines divergent for residual feed intake. Anim Int J Anim Biosci 12(2):350–358. https://doi.org/10.1017/S1751731117001379
Collins CL, Pluske JR, Morrison RS, McDonald TN, Smits RJ, Henman DJ, Stensland I, Dunshea FR (2017) Post-weaning and whole-of-life performance of pigs is determined by live weight at weaning and the complexity of the diet fed after weaning. Anim Nutr 3(4):372–379. https://doi.org/10.1016/j.aninu.2017.01.001
Ding X, Lan W, Liu G, Ni H, Gu J-D (2019) Exploring possible associations of the intestine bacterial microbiome with the pre-weaned weight gaining performance of piglets in intensive pig production. Sci Rep 9(1):1–10. https://doi.org/10.1038/s41598-019-52045-4
Do DN, Ostersen T, Strathe AB, Mark T, Jensen J, Kadarmideen HN (2014) Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet 15(1):27. https://doi.org/10.1186/1471-2156-15-27
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Cons 61(1):1–10
Fang S, Xiong X, Su Y, Huang L, Chen C (2017) 16S rRNA gene-based association study identified microbial taxa associated with pork intramuscular fat content in feces and cecum lumen. BMC Microbiol 17(1):162. https://doi.org/10.1186/s12866-017-1055-x
Fouhse JM, Zijlstra RT, Willing BP (2016) The role of gut microbiota in the health and disease of pigs. Anim Front 6(3):30–36. https://doi.org/10.2527/af.2016-0031
Frese SA, Parker K, Calvert CC, Mills DA (2015) Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 3:28. https://doi.org/10.1186/s40168-015-0091-8
Gilbert H, Billon Y, Brossard L, Faure J, Gatellier P, Gondret F, Labussière E, Lebret B, Lefaucheur L, Le Floch N (2017) divergent selection for residual feed intake in the growing pig. Animal 11(9):1427–1439. https://doi.org/10.1017/S175173111600286X
Gilbert H, Ruesche J, Muller N, Billon Y, Begos V, Montagne L (2018) Responses to weaning in two pig lines divergently selected for residual feed intake depending on diet. J Anim Sci 97(1):43–54. https://doi.org/10.1093/jas/sky416
Han GG, Lee J-Y, Jin G-D, Park J, Choi YH, Chae BJ, Kim EB, Choi Y-J (2017) Evaluating the association between body weight and the intestinal microbiota of weaned piglets via 16S rRNA sequencing. Appl Microbiol Biotechnol 101(14):5903–5911. https://doi.org/10.1007/s00253-017-8304-7
Herd RM, Arthur PF (2009) Physiological basis for residual feed intake. J Anim Sci 87(14 Suppl):E64–E71. https://doi.org/10.2527/jas.2008-1345
Holzer P (2016) Neuropeptides, microbiota, and behavior international review of neurobiology, vol 131. Elsevier, Amsterdam, pp 67–89. https://doi.org/10.1016/bs.irn.2016.08.005
Katouli M, Foo E, Kühn I, Möllby R (1997) Evaluation of the Phene Plate generalized microplate for metabolic fingerprinting and for measuring fermentative capacity of mixed bacterial populations. J Appl Microbiol 82(4):511–518. https://doi.org/10.1046/j.1365-2672.1997.00146.x
Ke S, Fang S, He M, Huang X, Yang H, Yang B, Chen C, Huang L (2019) Age-based dynamic changes of phylogenetic composition and interaction networks of health pig gut microbiome feeding in a uniformed condition. BMC Vet Res 15(1):172. https://doi.org/10.1186/s12917-019-1918-5
Konstantinov SR, Awati AA, Williams BA, Miller BG, Jones P, Stokes CR, Akkermans AD, Smidt H, De Vos WM (2006) Post-natal development of the porcine microbiota composition and activities. Environ Microbiol 8(7):1191–1199. https://doi.org/10.1111/j.1462-2920.2006.01009.x
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17):5112–5120. https://doi.org/10.1128/AEM.01043-13
Kuhn KA, Schulz HM, Regner EH, Severs EL, Hendrickson JD, Mehta G, Whitney AK, Ir D, Ohri N, Robertson CE, Frank DN, Campbell EL, Colgan SP (2018) Bacteroidales recruit IL-6-producing intraepithelial lymphocytes in the colon to promote barrier integrity. Mucosal Immunol 11(2):357–368. https://doi.org/10.1038/mi.2017.55
Kumar H, Park W, Srikanth K, Choi B-H, Cho E-S, Lee K-T, Kim J-M, Kim K, Park J, Lim D (2019) Comparison of bacterial populations in the ceca of swine at two different stages and their functional annotations. Genes 10(5):382. https://doi.org/10.3390/genes10050382
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814. https://doi.org/10.1038/nbt.2676
Lengeler J, Titgemeyer F, Vogler A, Wohrl B (1990) Structures and homologies of carbohydrate: phosphotransferase system (PTS) proteins. Philos Trans R Soc Lond B Biol Sci 326(1236):489–504. https://doi.org/10.1098/rstb.1990.0027
Li F, Li C, Chen Y, Liu J, Zhang C, Irving B, Fitzsimmons C, Plastow G (2019) Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7(1):92. https://doi.org/10.1186/s40168-019-0699-1
Lu D, Tiezzi F, Schillebeeckx C, McNulty NP, Schwab C, Shull C, Maltecca C (2018) Host contributes to longitudinal diversity of fecal microbiota in swine selected for lean growth. Microbiome 6(1):4. https://doi.org/10.1186/s40168-017-0384-1
Mach N, Berri M, Estellé J, Levenez F, Lemonnier G, Denis C, Leplat JJ, Chevaleyre C, Billon Y, Doré J (2015) Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ Microbiol Rep 7(3):554–569. https://doi.org/10.1111/1758-2229.12285
Mann E, Schmitz-Esser S, Zebeli Q, Wagner M, Ritzmann M, Metzler-Zebeli BU (2014) Mucosa-associated bacterial microbiome of the gastrointestinal tract of weaned pigs and dynamics linked to dietary calcium-phosphorus. PLoS ONE 9(1):e86950. https://doi.org/10.1371/journal.pone.0086950
Martín JF, Casqueiro J, Kosalková K, Marcos AT (1999) Penicillin and cephalosporin biosynthesis: mechanism of carbon catabolite regulation of penicillin production. Antonie Van Leeuwenhoek 75(1–2):21–31. https://doi.org/10.1023/A:1001820109140
McCormack UM, Curião T, Buzoianu SG, Prieto ML, Ryan T, Varley P, Crispie F, Magowan E, Metzler-Zebeli BU, Berry D (2017) Exploring a possible link between the intestinal microbiota and feed efficiency in pigs. Appl Environ Microbiol 83(15):e00380-17. https://doi.org/10.1128/AEM.00380-17
Onteru SK, Gorbach DM, Young JM, Garrick DJ, Dekkers JC, Rothschild MF (2013) Whole genome association studies of residual feed intake and related traits in the pig. PLoS ONE 8(6):e61756. https://doi.org/10.1371/journal.pone.0061756
Parks DH, Tyson GW, Hugenholtz P, Beiko RG (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30(21):3123–3124. https://doi.org/10.1093/bioinformatics/btu494
Patience JF, Rossoni-Serão MC, Gutiérrez NA (2015) A review of feed efficiency in swine: biology and application. J Anim Sci Biotechnol 6(1):33. https://doi.org/10.1186/s40104-015-0031-2
Pedersen R, Andersen AD, Mølbak L, Stagsted J, Boye M (2013) Changes in the gut microbiota of cloned and non-cloned control pigs during development of obesity: gut microbiota during development of obesity in cloned pigs. BMC Microbiol 13(1):30. https://doi.org/10.1186/1471-2180-13-30
Quan J, Cai G, Ye J, Yang M, Ding R, Wang X, Zheng E, Fu D, Li S, Zhou S (2018) A global comparison of the microbiome compositions of three gut locations in commercial pigs with extreme feed conversion ratios. Sci Rep 8(1):4536. https://doi.org/10.1038/s41598-018-22692-0
Ramayo-Caldas Y, Mach N, Lepage P, Levenez F, Denis C, Lemonnier G, Leplat JJ, Billon Y, Berri M, Dore J, Rogel-Gaillard C, Estelle J (2016) Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J 10(12):2973–2977. https://doi.org/10.1038/ismej.2016.77
Reyer H, Shirali M, Ponsuksili S, Murani E, Varley PF, Jensen J, Wimmers K (2017) Exploring the genetics of feed efficiency and feeding behaviour traits in a pig line highly selected for performance characteristics. Mol Genet Genomics 292(5):1001–1011. https://doi.org/10.1007/s00438-017-1325-1
Segain J, De La Blétiere DR, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blottiere H, Galmiche J (2000) Butyrate inhibits inflammatory responses through NFκB inhibition: implications for Crohn’s disease. Gut 47(3):397–403. https://doi.org/10.1136/gut.47.3.397
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6):R60. https://doi.org/10.1186/gb-2011-12-6-r60
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27(3):379–423
Siegerstetter S-C, Schmitz-Esser S, Magowan E, Wetzels SU, Zebeli Q, Lawlor PG, O’Connell NE, Metzler-Zebeli BU (2017) Intestinal microbiota profiles associated with low and high residual feed intake in chickens across two geographical locations. PLoS ONE 12(11):e0187766. https://doi.org/10.1371/journal.pone.0187766
Simpson EH (1949) Measurement of diversity. Nature 163(4148):688
Spreeuwenberg M, Verdonk J, Gaskins HR, Verstegen M (2001) Small intestine epithelial barrier function is compromised in pigs with low feed intake at weaning. J Nutr 131(5):1520–1527. https://doi.org/10.1093/jn/131.5.1520
Tan Z, Wang Y, Yang T, Ao H, Chen S, Xing K, Zhang F, Zhao X, Liu J, Wang C (2018) Differences in gut microbiota composition in finishing Landrace pigs with low and high feed conversion ratios. Antonie Van Leeuwenhoek 111(9):1673–1685. https://doi.org/10.1007/s10482-018-1057-1
Thompson CL, Wang B, Holmes AJ (2008) The immediate environment during postnatal development has long-term impact on gut community structure in pigs. ISME J 2(7):739. https://doi.org/10.1038/ismej.2008.29
Tian L, Bruggeman G, van den Berg M, Borewicz K, Scheurink AJ, Bruininx E, de Vos P, Smidt H, Schols HA, Gruppen H (2017) Effects of pectin on fermentation characteristics, carbohydrate utilization, and microbial community composition in the gastrointestinal tract of weaning pigs. Mol Nutr Food Res 61(1):1600186. https://doi.org/10.1002/mnfr.201600186
Vigors S, O’Doherty JV, Kelly AK, O’Shea CJ, Sweeney T (2016) The effect of divergence in feed efficiency on the intestinal microbiota and the intestinal immune response in both unchallenged and lipopolysaccharide challenged ileal and colonic explants. PLoS ONE 11(2):e0148145. https://doi.org/10.1371/journal.pone.0148145
Wang X, Tsai T, Deng F, Wei X, Chai J, Knapp J, Apple J, Maxwell CV, Lee JA, Li Y (2019) Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 7(1):109. https://doi.org/10.1186/s40168-019-0721-7
Xiao L, Estelle J, Kiilerich P, Ramayo-Caldas Y, Xia Z, Feng Q, Liang S, Pedersen AO, Kjeldsen NJ, Liu C, Maguin E, Dore J, Pons N, Le Chatelier E, Prifti E, Li J, Jia H, Liu X, Xu X, Ehrlich SD, Madsen L, Kristiansen K, Rogel-Gaillard C, Wang J (2016) A reference gene catalogue of the pig gut microbiome. Nature Microbiol 1:16161. https://doi.org/10.1038/nmicrobiol.2016.161
Yan W, Sun C, Yuan J, Yang N (2017) Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci Rep 7:45308. https://doi.org/10.1038/srep45308
Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, Yang M, Zhang Z, Chen C, Huang L (2017) Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front Microbiol 8:1555. https://doi.org/10.3389/fmicb.2017.01555
Yang H, Yang M, Fang S, Huang X, He M, Ke S, Gao J, Wu J, Zhou Y, Fu H (2018) Evaluating the profound effect of gut microbiome on host appetite in pigs. BMC Microbiol 18(1):215. https://doi.org/10.1186/s12866-018-1364-8
Zhao W, Wang Y, Liu S, Huang J, Zhai Z, He C, Ding J, Wang J, Wang H, Fan W (2015) The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 10(2):e0117441. https://doi.org/10.1371/journal.pone.0117441
Acknowledgements
None.
Funding
This study was supported by the National Natural Science Foundation of China (81860150), Special Project on Innovation Driven Development of Guangxi (Guike-AA17204029 and Guike-AA18118050), National Modern Agricultural Industrial Technology System (nycytxgxcxtd-15-01), Science and Technology Major Project of Guangxi (Guike-AA17292002) and Guangxi Natural Science Foundation Program (2017GXNSFBA198157 and 2018GXNSFAA294038).
Author information
Authors and Affiliations
Contributions
GL and JL conceived and designed the experiments; JM, GZ and SZ contributed analytical tools; JG and YH analyzed and WQ the data; JS and LF wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were approved by the Institutional Animal Care and Use Committee of Guangxi University (GXU2018-057).
Consent for publication
No applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Characteristics of the pigs used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Si, J., Feng, L., Gao, J. et al. Evaluating the association between feed efficiency and the fecal microbiota of early-life Duroc pigs using 16S rRNA sequencing. AMB Expr 10, 115 (2020). https://doi.org/10.1186/s13568-020-01050-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-020-01050-2