
Abstract
Chemolithoautotrophic sulfur-oxidizing bacteria (SOB) are crucial key players in biotechnological processes to remove hydrogen sulfide from sour gas streams. Several different haloalkaliphilic SOB have been detected and isolated from lab- and full-scale facilities, which all performed differently considering end product yields (sulfur and sulfate) and conversion rates. Understanding and regulating bacterial community dynamics in biodesulfurization processes will enable optimization of the process operation. We developed quantitative PCR (qPCR) assays to quantify haloalkaliphilic sulfur-oxidizing gammaproteobacterial species Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus, and Thioalkalibacter halophilus that dominate bacterial communities of biodesulfurization lab- and full-scale installations at haloalkaline conditions. The specificity and PCR efficiency of novel primer sets were evaluated using pure cultures of these target species. We further validated the qPCR assays by quantification of target organisms in five globally distributed full-scale biodesulfurization installations. The qPCR assays perform a sensitive and accurate quantification of Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus, thus providing rapid and valuable insights into process performance and SOB growth dynamics in gas biodesulfurization systems.
Similar content being viewed by others


Introduction
Sulfur-oxidizing bacteria (SOB) are microorganisms that naturally occur in highly saline and alkaline environments such as soda lakes (Sorokin and Kuenen 2005; Sorokin et al. 2013). SOB are known to be present in complex and species-rich consortia of microorganisms involved in both anaerobic and aerobic processes (Janssen et al. 2009) and can grow chemolithoautotrophically using inorganic sulfur compounds as electron donor and CO2 as carbon source (Ghosh and Dam 2009). These chemolithoautotrophic SOB are specifically enriched in biotechnological processes to remove hydrogen sulfide (H2S) from industrial gas streams by producing sulfur (Van Den Bosch et al. 2007; Sorokin et al. 2011). Biodesulfurization processes of the Thiopaq® family are operated under haloalkaline conditions, i.e. high pH (≥ 8.5) and high soda concentrations (1 M Na+) (Van Den Bosch et al. 2007).
Since 2009, the number of gas biodesulfurization installations increased globally (Driessen et al. 2011). All full-scale installations operate at different process conditions i.e. pH, salinity, oxidation–reduction potential (ORP) and treat feed gas of a various compositions (i.e. presence of different organic carbon compounds or contaminants such as thiols and BTEX) (Kiragosyan et al. 2019). To investigate which factors ensure stable process operation, a number of lab- and full-scale gas biodesulfurization installations have been monitored on the microbial community composition and process conditions. In sulfide-fed lab-scale installations, Thioalkalivibrio sulfidiphilus was the dominant SOB (Sorokin et al. 2008; Kiragosyan et al. 2019). In other lab-scale installations with sulfide feed gas supplemented with thiols, Alkalilimnicola sp. and Thioalkalibacter sp. were found to dominate (Roman et al. 2016). The addition of these toxic thiols not only affected SOB community composition, but alterations of the biodesulfurization process conditions can also cause a community change. For example, changes in the bioreactor design or addition of an extra bioreactor can also result in SOB community composition shift (De Rink et al. 2019).
In most cases, these SOB community dynamics were monitored using 16S rRNA gene amplicon sequencing. This method provides only estimates of relative taxon abundances and it is time consuming and costly. Monitoring SOB dynamics with quantitative PCR (qPCR), would provide absolute numbers of key SOB species in biodesulfurization processes, and is relatively fast, less costly and results can be analyzed and interpreted with ease. The ability to monitor SOB species dynamics rapidly and inexpensively will help to monitor the key population dynamics and to optimize biodesulfurization process efficiency. qPCR is widely used for microbial quantification in many types of environmental studies (Filion 2012), but so far no SOB-specific primers are currently available to monitor dynamics of relevant SOB species. The qPCR specificity for different SOB species depends on several parameters: the primer sequences for the target, time and temperature of primer annealing, annealing, amplicon product size, concentration of Mg2+, dNTPs, fidelity of enzymes, and the purity of the DNA sample (Robertson and Walsh-Weller 1998).
In this work we designed target-specific primers and developed specific qPCR assays to monitor absolute abundances of the three most dominant haloalkaliphilic SOB species found in operational biodesulfurization Thiopaq® installations until now: Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus. The resulting quantitative measures provide insights in species growth dynamics and interactions. Hence, developed quantitative PCR assays can be used to establish relationships between the operational conditions and the biological community in biodesulfurization processes in order to establish stable SOB communities that ensure predictable and stable process conditions.
Materials and methods
Microbial sludge sampling and sample preparation
Biomass from a lab-scale, fed-batch biodesulfurization system, which was fed with H2S and methanethiol gas, was obtained after 76 days of continuous operation. H2S gas was continuously supplied at a loading rate of 58.15 mM S day−1, whereas methanethiol loading rate was stepwise (add steps from 0 to 2 mM) increased for biomass acclimatization during the 76 days to a maximum concentration of 2 mM S day−1. On the last day of operation, microbial sludge was sampled and centrifuged for 15 min at 16,000g to obtain the cell pellet. The cell pellet was washed twice with 0.5 M Na+ buffer solution (pH 8.5) to prevent cell lysis and was subsequently stored at − 80 °C until DNA extraction.
DNA isolation and purification
Genomic DNA was extracted using the DNeasy PowerLyzer PowerSoil Kit (Qiagen, Venlo, the Netherlands) following the manufacturer’s instructions. After DNA extraction, DNA was purified with the DNA Clean and Concentrator kit (Zymo Research, Irvine, CA, USA). Extracted DNA was quantified using the QuantiFluor dsDNA system on a Quantus™ fluorometer (Promega, Leiden, the Netherlands). DNA quality was evaluated using gel electrophoresis.
Clone library construction and sequencing
To design primers targeting the most dominant SOB species present in the established mixed SOB population, full-length 16S rRNA gene sequences recovered from the community DNA. The full 16S rRNA gene was amplified from the extracted and purified DNA using universal bacterial primers 27F (5′-GTTTGATCCTGGCTCAG-3′) (Felske and Weller 2004) and 1492R (5′-CGGCTACCTTGTTACGAC-3′) (Lane 1991). The PCR program started with initial denaturation (95 °C for 2 min) followed by 30 cycles of denaturation for 30 s at 95 °C, annealing for 40 s at 52 °C, elongation for 1.30 min at 72 °C, and with a final 7 min elongation at 72 °C. The PCR products were again purified with the DNA Clean and Concentrator kit and ligated into the pGEM-T Easy Vector System, according to the manufacturer’s instructions (Promega, City, the Netherlands). The ligation mixture was used to transform Escherichia coli JM109 competent cells (Promega, City, the Netherlands). Colonies were picked and DNA was extracted and sent for Sanger sequencing to BaseClear B.V. (Leiden, the Netherlands). Obtained forward and reverse sequences were assembled into contigs, and ends were quality trimmed with the DNA Baser software (v4, Heracle Biosoft, www.DnaBaser.com, Romania). Subsequently, sequences were cleaned by cutting off primer sequences and were screened for vector contamination using VecScreen (NCBI, MD, USA).
Target species-specific primer design
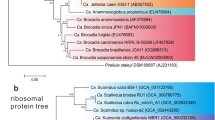
The full length 16S rRNA gene sequences were aligned and taxonomically classified using the SINA alignment tool (v1.2.11) according to the global SILVA alignment (Pruesse et al. 2007). Sequence identity was also investigated with the online nucleotide BLAST tool (Madden 2002). Aligned sequences of the species of interest (Thioalkalivibrio sulfidiphilus, Alkalilimnicola ehrlichii and Thioalkalibacter halophilus) were merged with the SILVA 16S rRNA gene database version SSU rl 28 Ref NR (Quast et al. 2013) using the ARB software package (arb-6.0.6) (Ludwig et al. 2004). Primer sets were designed based on the highest specificity (100% with 0 mismatches) for Alkalilimnicola ehrlichii and Thioalkalibacter halophilus species. For Thioalkalivibrio spp., primers were designed to target the Thioalkalivibrio denitrificans cluster (highlighted in Fig. 1). The designed primers were then validated in silico using the TestProbe and TestPrime services of Silva (Klindworth et al. 2013), Primer-BLAST (Ye et al. 2012) and in silico PCR (http://insilico.ehu.es/user_seqs/). After validation, designed primer sequences were ordered from Biolegio (Nijmegen, the Netherlands).

Phylogenetic position of Thioalkalivibrio sulfidiphilus in the cluster for which primers for Thioalkalivibrio spp. were designed (adapted from Sorokin et al. 2012). Bar indicates 2% sequence divergence
In vitro primer evaluation on target species pure cultures
To be able to test the specificity of the designed primers and to optimize the qPCR protocol, every designed primer set was tested using pure cultures. Two strains used in this study were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ): Alkalilimnicola ehrlichii strain MLHE-1 (DSM-17681) and Thioalkalibacter halophilus strain ALCO 1 (DSM-19224). Thioalkalivibrio sulfidiphilus strain HL-EbGr7 was provided from the personal collection of Prof. Dr. Gerard Muyzer (Amsterdam University, the Netherlands).
qPCR assay optimization
For the optimization of qPCR assays the workflow was as follows:
-
1.
Firstly, the annealing temperature was optimized based on the theoretical melting temperature (Tm) via gradient PCR (± 10 °C). In this step, pure cultures were used as positive controls for the selected primer sets. After each temperature gradient run, gel electrophoresis (GE) was performed to confirm the size of the PCR product(s) based on expected insert lengths of the developed primer sets.
-
2.
For each primer set, primer specificity for a target species was assessed by simultaneous testing against the non-target cultured pure strains (Thioalkalivibrio sulfidiphilus strain HL-EbGr7, Thioalkalivibrio denitrificans strain ALJD (DSM-13742), Alkalilimnicola ehrlichii strain MLHE-1 and Thioalkalibacter halophilus strain ALCO 1) and cultured clones found in our cloned biomass [Thioalkalimicrobium spp., Halomonas sp. HB. Br (GU228481)] at the selected optimal annealing temperature for each primer set. When bright bands of the positive target strain were visible on GE and no bands of non-target strain negative controls appeared, fine-tuning of the optimal annealing temperature was continued with a narrower temperature gradient ranging between ± 3 °C.
-
3.
To further assess primer specificity, melting curve analysis was performed (50–95 °C, with 0.5 °C increments) using quantitative PCR (Bio Rad CFX96 Touch™, City, the Netherlands) with SYBR® Green fluorescent dye (Bio Rad, the Netherlands). The qPCR reaction volume was 20 µl with 0.33 pmol µl−1 of forward and reverse primers. Melt curves revealed target-specific product formation at the determined optimal annealing temperatures for all three primer sets (see Additional file 1: Figures S1, S2 and S3). The optimal annealing temperatures for cultured positive control strains were then applied in the qPCR analysis of bioreactor experimental samples. For primer set Thioalkalivibrio spp. Thio-6F and Thio-8R, the optimal Tm was 55 °C, for Alkali-4AF and Alkali-6BR Tm was 53 °C, and for Tab-137-G_F and Tab-210R best Tm was at 66 °C.
-
4.
To verify that our developed primer sets specifically amplified the target species in our experimental samples, melting curves of experimental samples were analyzed for qPCR amplicon specificity, and then a random subset of (n = 4 to 6 per primer set) qPCR products were sent for Sanger sequencing to BaseClear B.V. (Leiden, The Netherlands). Sequences were assembled using the DNA Baser software (v4, Heracle Biosoft, www.DnaBaser.com, Romania) and identified with BLAST (Madden 2002). When qPCR amplicons from the experimental samples were positively identified as the target species, we continued with the preparation of standard curves using the DNA of the pure cultures.
-
5.
As a positive control, cultured pure strains were used in target species-specific qPCR assays to establish a standard curve in order to quantify each of their copy numbers in the experimental samples. The concentrations of the positive control DNA were measured using QuantiFluor dsDNA systems and a Quantus™ fluorometer (Promega, the Netherlands). Positive controls were then serially diluted in tenfold dilutions ranging between 106 and 101 copies μl−1 of the 16S rRNA gene. These serial dilutions were used to generate standard curves which allowed minimal reaction efficiencies of 90–100% and 0.997 < R2 < 0.999.
Validation of the developed qPCR assay
To validate the developed qPCR methods, we used biomass from different five full-scale gas biodesulfurization installations (Table 1). Additional research focusing on the microbial community compositions of these full-scale installations was based on 16S rRNA gene amplicon sequencing (Kiragosyan et al. 2019). The applicability of the qPCR assay was validated by comparing qPCR-based relative target abundances with relative abundances obtained from the 16S rRNA gene amplicon sequence data. To calculate relative target abundances, total bacterial 16S rRNA gene copy abundance was quantified using the universal bacterial primer set 338F/518R (Lane 1991; Muyzer et al. 1993) and an in-house protocol (Pallares-Vega et al. 2019).
Accession number
The EMBL-EBI accession numbers of the full-length 16S rRNA clonal gene sequences of Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus are LR214448–LR214450 in the project number PRJEB30777. 16S rRNA gene amplicon sequences from the full-scale installations are deposited under EMBL-EBI project accession number PRJEB27163 and PRJEB32000.
Results
Primers evaluation and qPCR assay optimization
For each of the three targets, we designed three primer sets for which we subsequently optimized the qPCR protocols. The properties of the primers that were tested in silico and in vitro are summarized in Table 2.
In silico amplification demonstrated that primer set Thio-6F/Thio8-R quantified Tv. sulfidiphilus and closely-related Tv. denitrificans. Empirical qPCR experiments and subsequent melt-curve analysis on DNA extracted from pure cultures of Tv. sulfidiphilus and Tv. denitrificans correspondingly showed amplification of both species at the same annealing temperature (Additional file 1: Figure S1). qPCR assays for Alk. ehrlichii and Th. halophilus showed species-specificity in silico and in vitro (Additional file 1: Figures S2 and S3). Yet, our temperature gradient experiment conducted for the Alk. ehrlichii-targeting primer set Alkali-4AF/Alkali-6BR suggested amplification of non-target DNA at temperatures exceeding 55 °C. Melt curve analysis of this temperature gradient demonstrated that non-target amplification reduced with decreasing temperatures. A subsequent temperature gradient including lower temperatures revealed that at 53 °C, the desired specificity for Alkalilimnicola ehrlichii was reached. Optimal reaction parameters for the developed qPCR assays were initial denaturation for 5 min at 95 °C followed by 30 amplification cycles of denaturation for 10 s at 95 °C, annealing for 30 s at 53 °C for Alkalilimnicola ehrlichii, 55 °C for Thioalkalivibrio sulfidiphilus and 66 °C for Thioalkalibacter halophilus.
Validation of the developed qPCR assay
To validate the applicability of the developed qPCR assays, we quantified absolute abundances of the target species and total bacteria in five full-scale gas biodesulfurization installations geographically distributed across Europe, Asia, and North America (Table 1). Our results demonstrated that the qPCR-based relative species abundances (i.e. ratio species-specific to total bacterial 16S rRNA amplicon count) are analogous to the relative abundances obtained by16S rRNA gene amplicon sequencing (i.e. ratio taxon-specific to total read count) of the microbial community structure in the full-scale biodesulfurization installations (Fig. 2). In both Paper mill installations, the relative abundance of Thioalkalivibrio spp. quantified by qPCR and amplicon sequence data are of the same magnitude. However, in three other full-scale installations quantified abundances by qPCR assays differed from amplicon sequencing data. Relative abundances of Thioalkalivibrio spp. detected by qPCR in Oilfield and Landfill plants were two times less than the relative abundance of Tv. sulphidophilus in amplicon sequencing data (Fig. 2). The same twofold difference was found for less abundant Th. halophilus in Oilfield—2 installation (0.22% vs. 0.09%) (Fig. 2). Collectively, these results indicate that the developed qPCR assays can be applied for detection and quantification of Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus in various gas biodesulfurization installations.

Comparison of relative taxon abundances using 16S rRNA gene amplicon sequencing (NGS) and species-specific qPCR from five full-scale gas biodesulfurization installations. The results represent the average value per full-scale biodesulfurization installation for each of the target species that was positively detected. Error bars represents the standard deviation of technical duplicates. Amplicon sequence data are modified from Kiragosyan et al. 2019
Discussion
Developed qPCR assays for fast and accurate quantification of Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus were optimized to achieve maximal specificity and sensitivity to detect potentially low abundance of target species. For Alk. ehrlichii and Th. halophilus, the designed primers achieved 100% specificity by allowing 0 mismatches in the primer binding region. For Thioalkalivibrio spp., primers were designed for a subcluster of this genus (Fig. 1). Thioalkalivibrio sulfidiphilus HL-EbGR7 is genetically related to another Tv. sulfidiphilus ALJ17 and to Tv. denitrificans (Sorokin et al. 2012; Ahn et al. 2017). However, our cloning results of the biodesulfurization process sludge and finding of Sorokin et al. (2012) indicate that only Tv. sulfidiphilus was found in the lab- and full-scale installations. Hence, we can conclude that the designed primer set quantified Tv. sulfidiphilus in the samples as it is the only Thioalkalivibrio species detected and dominantly present at low salt conditions in the gas biodesulfurization lab- and full-scale installations. The targeted sub-cluster contains other two closely related species, which are Tv denitrificans and Tv. thiocyanodenitrificans (Sorokin et al. 2012). The designed primer set Thio6F/Thio8R is specific for Tv. sulfidiphilus and Tv. denitrificans, whereas Tv. thiocyanodenitrificans was not tested (see Additional file 1: Figure S1). However, Tv. denitrificans and Tv. thiocyanodenitrificans have never been detected as thiodenitrifying conditions are not provided in the full-scale biodesulfurization process. It is therefore confirmed that the primer set Thio6F/Thio8R only quantified Thioalkalivibrio sulfidiphilus in the experimental samples from biodesulfurization full- and lab-scale installations.
Optimization of the quantitative real-time PCR protocols enhanced the specificity of the designed primer sets in experimental samples. Conventionally, regular PCR temperature gradients are advisable to be performed with subsequent visualization of product bands using gel electrophoresis. However, the detection by agarose gel electrophoreses is limited for low (expected) product concentrations (Smith and Osborn 2009; Garibyan and Avashia 2013). For the Alkali-4AF/Alkali-6BR primer set targeting Alkalilimnicola ehrlichii, we reached specificity at a lower than expected annealing temperature. Similarly, Sipos et al. (2007) found that three universal bacterial primers showed better performance at lower temperatures based on temperature gradients with varying annealing temperatures (47 to 61 °C). Furthermore, Ischii and Fukui (Ishii and Fukui 2001) also found that at low temperatures, mismatch biases of primers were reduced. Apparently, decreasing annealing temperatures can yield improved target specificity of qPCR assays, which proved a beneficial property for the Alkalilimnicola ehrlichii assay. In addition, the reverse primer with relatively high GC content Alkali-6BR (56%, Table 2), was more specific for detection of Alkalilimnicola ehrlichii at lower temperatures.
Sequencing based methods give the possibility to resolve the community composition of complex experimental samples, where further qPCR assay can be complementary applied to answer more profound questions on population dynamics of a specific target organism. In our work, we showed that with use of both techniques similar results can be achieved, which confirms the accuracy of the developed qPCR assays. Relative abundance estimates for dominant species between qPCR and 16S amplicon sequence data were comparable in both Paper mill installations, while in other full-scale installations there was a two-fold difference. Observed difference might be caused by the use of different universal primer sets for 16S rRNA gene quantification in qPCR and NGS, because no primer set is truly universal (Leray et al. 2013). Primers have different affinity for different taxonomic groups what prevents detection of certain operational taxonomic units (OTUs) in NGS (Leray and Knowlton 2017). This results in biased relative abundances especially within complex samples (Piñol et al. 2015). In addition, variation between qPCR and NGS counts can be explained by a PCR bias (in NGS) introduced by less dominant species (Stokell et al. 2015). Hence, we suggest that it is reasonable to expect that the final number of reads for the target species is higher using universal primers after a defined number of exponential amplification cycles. Detection limits of rare taxa are strongly dependent on the sequencing depths of sample (i.e. number of sequence reads per sample). In 16S rRNA gene amplicon sequencing methods, competition for primers occurs between rare and abundant taxa, where most likely abundant taxa outcompete amplification of rare taxa (Forde and O’Toole 2013). With qPCR however, species-specific absolute quantification of rare and abundant taxa can be done.
The development of three novel qPCR assays allows for accurate, sensitive, fast and cost-efficient quantification of Alkalilimnicola ehrlichii, Thioalkalivibrio spp. and Thioalkalibacter halophilus in complex samples, such as lab- and full-scale gas biodesulfurization installations. The presented qPCR assays will have ample applicability for monitoring dynamics of key SOB species in the gas biodesulfurization process, especially in the presence of common process perturbations such as thiols. Moreover, established dynamics will allow us to expand our understanding of the gas biodesulfurization process and thus, will enable us to improve process performance.
Availability of data and materials
The data supporting the conclusions of this article are included within the article. Data and materials can also be requested from the corresponding author.
References
Ahn A-C, Overmars L, Sorokin DY, Meier-Kolthoff JP, Muyzer G, Richter M, Woyke T (2017) Genomic diversity within the haloalkaliphilic genus Thioalkalivibrio. PLoS ONE 12:e0173517. https://doi.org/10.1371/journal.pone.0173517
de Rink R, Klok JBM, van Heeringen GJ, Sorokin DY, ter Heijne A, Zeijlmaker R, Mos YM, de Wilde V, Keesman KJ, Buisman CJN (2019) Increasing the selectivity for sulfur formation in biological gas desulfurization. Environ Sci Technol. https://doi.org/10.1021/acs.est.8b06749
Driessen W, Van Zessen E, Visser M (2011) Full-scale experience with biological biogas desulfurization. In: 16th European biosolids and organic resources conference
Felske A, Weller R (2004) Cloning 16S rRNA genes and untilization to type bacterial communities. In: Kowalchuk GA, De Bruijn FJ, Head IM, Akkermans ADL, Van Elsas JD (eds) Molecular microbial ecology manual, 2nd edn. Kluwer Academic Publishers, Dordrecht, pp 510–523
Filion M (ed) (2012) Quantitative real-time pcr in applied microbiology. Caiser Academic Press, Norfolk
Forde BM, O’Toole PW (2013) Next-generation sequencing technologies and their impact on microbial genomics. Brief Funct Genom 12:440–453. https://doi.org/10.1093/bfgp/els062
Garibyan L, Avashia N (2013) Research techniques made simple: polymerase chain reaction (PCR). J Invest Dermatol 133:20382. https://doi.org/10.1038/jid.2013.1.Research
Ghosh W, Dam B (2009) Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev 33:999–1043. https://doi.org/10.1111/j.1574-6976.2009.00187.x
Ishii K, Fukui M (2001) Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl Environ Microbiol 67:3753–3755. https://doi.org/10.1128/AEM.67.8.3753-3755.2001
Janssen AJH, Lens PNL, Stams AJM, Plugge CM, Sorokin DY, Muyzer G, Dijkman H, Van Zessen E, Luimes P, Buisman CJN (2009) Application of bacteria involved in the biological sulfur cycle for paper mill effluent purification. Sci Total Environ 407:1333–1343. https://doi.org/10.1016/j.scitotenv.2008.09.054
Kiragosyan K, Klok JBM, Keesman KJ, Roman P, Janssen AJH (2019) Development and validation of a physiologically based kinetic model for starting up and operation of the biological gas desulfurization process under haloalkaline conditions. Water Res X. https://doi.org/10.1016/j.wroa.2019.100035
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:1–11. https://doi.org/10.1093/nar/gks808
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in Bacterial systematic. Wiley, New York, pp 115–175
Leray M, Knowlton N (2017) Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 5:e3006. https://doi.org/10.7717/peerj.3006
Leray M, Yang JY, Meyer CP, Mills SC, Agudelo N, Ranwez V, Boehm JT, Machida RJ (2013) A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front Zool 10:1–14. https://doi.org/10.1186/1742-9994-10-34
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar A, Buchner A, Lai T, Steppi S, Jacob G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüßbmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. https://doi.org/10.1093/nar/gkh293
Madden T (2002) The BLAST sequence analysis tool. NCBI handbook. National Center for Biotechnology Information, Bethesda, pp 1–15
Muyzer G, Waal ECDE, Uitierlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Pallares-Vega R, Blaak H, van der Plaats R, de Roda Husman AM, Leal LH, van Loosdrecht MCM, Weissbrodt DG, Schmitt H (2019) Determinants of presence and removal of antibiotic resistance genes during WWTP treatment: a cross-sectional study. Water Res. https://doi.org/10.1016/j.watres.2019.05.100
Piñol J, Mir G, Gomez-Polo P, Agustí N (2015) Universal and blocking primer mismatches limit the use of high-throughput DNA sequencing for the quantitative metabarcoding of arthropods. Mol Ecol Resour 15:819–830. https://doi.org/10.1111/1755-0998.12355
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. https://doi.org/10.1093/nar/gkm864
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:590–596. https://doi.org/10.1093/nar/gks1219
Robertson J, Walsh-Weller J (1998) An introduction to PCR primer design and optimization of amplification reactions. Forensic DNA Profiling Protoc 98:6–8. https://doi.org/10.1385/0-89603-443-7:121
Roman P, Klok JBM, Sousa JAB, Broman E, Dopson M, Van Zessen E, Bijmans MFM, Sorokin DY, Janssen AJH (2016) Selection and application of sulfide oxidizing microorganisms able to withstand thiols in gas biodesulfurization systems. Environ Sci Technol. https://doi.org/10.1021/acs.est.6b04222
Sipos R, Székely AJ, Palatinszky M, Révész S, Márialigeti K, Nikolausz M (2007) Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol 60:341–350. https://doi.org/10.1111/j.1574-6941.2007.00283.x
Smith CJ, Osborn AM (2009) Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol Ecol 67:6–20. https://doi.org/10.1111/j.1574-6941.2008.00629.x
Sorokin DY, Kuenen JG (2005) Haloalkaliphilic sulfur-oxidizing bacteria in soda lakes. FEMS Microbiol Rev 29:685–702. https://doi.org/10.1016/j.femsre.2004.10.005
Sorokin DY, Van Den Bosch PLF, Abbas B, Janssen AJH, Muyzer G (2008) Microbiological analysis of the population of extremely haloalkaliphilic sulfur-oxidizing bacteria dominating in lab-scale sulfide-removing bioreactors. Appl Microbiol Biotechnol 80:965–975. https://doi.org/10.1007/s00253-008-1598-8
Sorokin DY, Kuenen JG, Muyzer G (2011) The microbial sulfur cycle at extremely haloalkaline conditions of soda lakes. Front Microbiol. https://doi.org/10.3389/fmicb.2011.00044
Sorokin DY, Muntyan MS, Panteleeva AN, Muyzer G (2012) Thioalkalivibrio sulfidiphilus sp. nov., a haloalkaliphilic, sulfur-oxidizing gammaproteobacterium from alkaline habitats. Int J Syst Evol Microbiol 62:1884–1889. https://doi.org/10.1099/ijs.0.034504-0
Sorokin DY, Banciu H, Robertson LA, Kuenen JG, Muntyan MS, Muyzer G (2013) Halophilic and haloalkaliphilic sulfur-oxidizing bacteria. In: Rosenberg E, DeLong EF, Stackebrandt E, Lory S, Thompson F (eds) The prokaryotes: prokaryotic physiology and biochemistry. Springer-Verlag, Berlin-Heidelberg, pp 530–555
Stokell JR, Gharaibeh RZ, Hamp TJ, Zapata MJ, Fodor AA, Steck TR (2015) Analysis of changes in diversity and abundance of the microbial community in a cystic fibrosis patient over a multiyear period. J Clin Microbiol 53:237–247. https://doi.org/10.1128/JCM.02555-14
Van Den Bosch PLF, Van Beusekom OC, Buisman CJN, Janssen AJH (2007) Sulfide oxidation at halo-alkaline conditions in a fed-batch bioreactor. Biotechnol Bioeng 97:1053–1063. https://doi.org/10.1002/bit.21326
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform 13:11. https://doi.org/10.1186/1471-2105-13-134
Acknowledgements
We would like to thank Franka Tulner (Wetsus and Van Hall Larenstein University of Applied Science, the Netherlands) for the help with Thioalkalibacter halophilus method optimization. We acknowledge Rebeca Pallarés Vega (Delft University of Technology and Wetsus, the Netherlands) and Gonçalo Macedo (Utrecht University and Wetsus, the Netherlands) for the support and guidance during qPCR methods development and data processing. We also would like to thank Prof. Dr. Gerard Muyzer (University of Amsterdam, the Netherlands) for providing Thioalkalivibrio sulfidiphilus pure strain and Dr. Caroline Plugge for fruitful discussions regarding optimization procedure.
Funding
This work has been performed within the cooperation framework of Wetsus, European Centre of Excellence for Sustainable Water Technology (wetsus.nl) and Wageningen University and Research (wur.nl). Wetsus is co-funded by the Netherlands’ Ministry of Economic Affairs and Ministry of Infrastructure and Environment, the European Union’s Regional Development Fund, the Province of Fryslan and the Northern Netherlands Provinces. Wetsus is also a coordinator of the WaterSEED project that received funding from European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement No. 665874. The research of Peer H.A. Timmers was supported by the Soehngen Institute of Anaerobic Microbiology (SIAM) Gravitation grant (024.002.002) of the Netherlands Ministry of Education, Culture and Science and the Netherlands Organisation for Scientific Research (NWO).
Author information
Authors and Affiliations
Contributions
KK conceived and designed the analyzes, performed the analysis, collected the data and wrote the paper. PV contributed with analysis of the data and wrote the paper. SG contributed with qPCR optimization. ATP guided through cloning procedure, qPCR optimization and contributed with writing. PR contributed with guidance and contributed with writing. PHAT designed the analyzes, guided through qPCR process optimization and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Figure S1.
Melting curve analysis of qPCR products of 16S rRNA gene of Thioalkalivibrio genus with use of SYBR Green. Figure S2. Melting curve analysis of qPCR products of 16S rRNA gene of Alkalilimnicola ehrlichii with use of SYBR Green. Figure S3. Melting curve analysis of qPCR products of 16S rRNA gene of Thioalkalibacter halophilus with use of SYBR Green.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kiragosyan, K., van Veelen, P., Gupta, S. et al. Development of quantitative PCR for the detection of Alkalilimnicola ehrlichii, Thioalkalivibrio sulfidiphilus and Thioalkalibacter halophilus in gas biodesulfurization processes. AMB Expr 9, 99 (2019). https://doi.org/10.1186/s13568-019-0826-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-019-0826-1