
Abstract
Registry data worldwide indicate an overall female predominance for pulmonary arterial hypertension (PAH) of 2–4 over men. Genetic predisposition accounts for only 1–5% of PAH cases, while autoimmune diseases and infections are closely linked to PAH. Idiopathic PAH may include patients with undiagnosed autoimmune diseases based on the relatively high presence of autoantibodies in this group. The two largest PAH registries to date report a sex ratio for autoimmune connective tissue disease-associated PAH of 9:1 female to male, highlighting the need for future studies to analyze subgroup data according to sex. Autoimmune diseases that have been associated with PAH include female-dominant systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome, and thyroiditis as well as male-dominant autoimmune diseases like myocarditis which has been linked to HIV-associated PAH. The sex-specific association of PAH to certain infections and autoimmune diseases suggests that sex hormones and inflammation may play an important role in driving the pathogenesis of disease. However, there is a paucity of data on sex differences in inflammation in PAH, and more research is needed to better understand the pathogenesis underlying PAH in men and women. This review uses data on sex differences in PAH and PAH-associated autoimmune diseases from registries to provide insight into the pathogenesis of disease.
Similar content being viewed by others

Background
Pulmonary arterial hypertension (PAH) is characterized by an elevated resting mean pulmonary arterial pressure of ≥ 25 mmHg in the presence of a normal pulmonary capillary wedge pressure of ≤ 15 mmHg [1]. Vasoconstriction of the small pulmonary arteries increases pulmonary vascular resistance leading to right heart failure and premature death [2]. Narrowing and occlusion of the vessel lumen is due largely to endothelial cell proliferation and vascular remodeling [3]. Central to the pathology of PAH is the plexiform lesion which is comprised of a proliferation of endothelial cells and vascular smooth muscle cells (VSMC), myofibroblasts, and inflammatory cells that occlude the pulmonary arteries. Pulmonary hypertension is currently divided into five diagnostic groups based on etiology. PAH is classified as group 1 and includes idiopathic, heritable (previously termed familial), drug- or toxin-induced, and PAH associated with other conditions, as well as persistent pulmonary hypertension of the newborn [4]. Conditions associated with PAH include autoimmune diseases (particularly connective tissue diseases), congenital heart disease, portopulmonary hypertension, HIV infection, and Schistosoma mansoni infection leading to schistosomiasis. Factors that may lead to vasoconstriction in PAH include genetic predisposition, sex hormones, infections, autoimmune diseases, inflammation, and/or immune complex deposition. This review compiles data on sex differences in PAH and PAH-associated autoimmune diseases from registries and how this information may provide insight into the pathogenesis of disease.
Sex differences in the epidemiology of PAH
Most cardiovascular diseases (CVDs) like atherosclerosis, myocardial infarction, myocarditis, and dilated cardiomyopathy occur predominantly in men [5,6,7]. One exception is PAH [8,9,10]. Table 1 lists the sex ratio for PAH reported from a number of registries [11,12,13,14,15,16,17,18,19,20]. These data indicate a female predominance in PAH of around 2–4 over men for all races and ethnicities and across all ages that have been studied to date except for HIV-associated and portopulmonary hypertension, which tend to occur more often in men [17, 21, 22].
Understanding sex differences in PAH subgroups may provide important information about the pathogenesis of disease. For example, the sex ratios for connective tissue disease (CTD)-associated PAH skew more strongly toward females when examined as an individual subgroup (i.e., 9:1 female to male) (Table 2). Most registries combine PAH subgroups making it difficult to ascertain percentages and sex ratios by subgroup. For this reason, future studies should report sex difference data for each subtype within PAH (Table 3). With the exception of hereditary PAH, subgroups may then additionally be combined based on sex differences (Table 3). Because most autoimmune diseases can take up to 10 years for patients to obtain a diagnosis [5, 23, 24], it is possible that some CTD-associated PAH cases are categorized as idiopathic PAH in younger patients. The pronounced gender difference that exists within PAH subgroups in epidemiologic studies indicates that more research is needed to better understand sex differences in the pathogenesis of disease.
Female-dominant autoimmune diseases associated with PAH
The reported gender difference for CTD-associated PAH ranges from 4:1 to 9:1 female to male (Table 2). The two largest registries that reported sex differences for PAH subgroups, REVEAL and the Spanish Registry of Pulmonary Arterial Hypertension, found a sex ratio for CTD-associated PAH of 9:1 female to male [18, 19, 21]. The Spanish Registry of Pulmonary Arterial Hypertension found that 61% of CTD-associated PAH patients had the autoimmune connective tissue disease systemic sclerosis (SSc), also referred to as scleroderma [19]. Other female-dominant autoimmune diseases or syndromes aside from SSc that have been associated with PAH include systemic lupus erythematosus, mixed connective tissue disease, myositis (also called dermatomyositis), rheumatoid arthritis, Raynaud’s syndrome, CREST syndrome, autoimmune hepatitis, Sjögren’s syndrome, and thyroiditis [8, 9, 25,26,27,28,29,30,31]. The sex ratio and prevalence of autoimmune diseases that may be associated with PAH are listed in Table 4 with a ratio of women to men ranging from 2:1 to 19:1 [24].
SSc has been estimated at a female dominance as high as 12:1 (Table 4), but sex ratios range from 4.6:1 to 12:1 female to male [24, 32,33,34], a range that is similar to those found in CTD-associated PAH. SSc is typically diagnosed prior to menopause in women, while men with SSc are diagnosed at an older age than women [34]. The sex ratio of women to men with SSc in their childbearing years has been estimated at 15:1 while it lowers to 2.4:1 after age 50 [35, 36], indicating the importance of analyzing data by gender and age (i.e., the cutoff for menopause is around age 50–55). SSc has been divided into two clinical forms: limited cutaneous SSc (lcSSc) which occurs more often in women and diffuse cutaneous SSc (dcSSc) which occurs more often in men (Table 3) [34, 37, 38]. Men with SSc (i.e., dcSSc) were found to have more severe disease, progress rapidly, and have a higher incidence of PAH with an increased risk of heart failure and death [37]. Testosterone is able to increase inflammation and remodeling in the myocardium (i.e., myocarditis and dilated cardiomyopathy) and vessel wall (i.e., atherosclerosis) leading to an increased rate of heart failure and death in men [6, 7, 39]. Women with the SSc (i.e., lcSSc) have more autoantibodies and CREST syndrome, develop less severe disease, and have a lower mortality [34]. The sex difference in disease presentation of SSc reflects the sex differences observed in men and women in general, with more women developing autoantibodies/autoimmune diseases and reduced mortality, while more men develop heart disease (i.e., atherosclerosis, myocarditis) and are at an increased risk of heart failure and death [6, 7, 40] (Table 5).
Elevated right heart pressures were found in 59% of patients with SSc, CREST syndrome, and systemic lupus erythematosus using exercise stress echocardiography to evaluate the change in pulmonary arterial pressure [41]. Importantly, it has been estimated that over 50% of mortality in patients with SSc is due to PAH and interstitial lung disease, with PAH alone accounting for around 30% of deaths [42, 43]. Although SSc-associated PAH was found to have a female to male ratio of 5.5:1, men had more rapid PAH progression, interstitial lung disease, dcSSc (the male-dominant clinical form of SSc), and a decreased 1-, 2-, 3-, and 5-year survival compared to women [38]. This is similar to what is known for PAH cases in general where the incidence of PAH is higher in women but 5-year survival is worse in men [19, 20, 44]. Interestingly, cardiovascular disease occurs in nearly all SSc patients [45]. In addition to PAH, SSc patients have a higher risk of developing arrhythmias, coronary artery disease (CAD), myocarditis, pericarditis, cardiomyopathy (including dilated cardiomyopathy), and heart failure [5, 37, 45,46,47,48,49,50,51]. SSc has been reported to be an independent risk factor for CAD with accelerated atherosclerosis in these patients [49]. Overall, cardiac involvement in SSc is more common in dcSSc patients, where both dcSSc and CVD occur more often in men [50]. Similarly, CAD, myocarditis, pericarditis, cardiomyopathy, and heart failure all occur more often in men [5,6,7].
SSc is a systemic autoimmune disease characterized by inflammation, remodeling, fibrosis, immune complex (IC) deposition, thrombosis, and vasculopathy that can either primarily affect the skin (i.e., scleroderma) or internal organs (i.e., SSc) like the lungs, gastrointestinal tract, and kidneys [29, 40]. SSc is part of a group of female-dominant autoimmune diseases that include myositis/dermatomyositis, rheumatoid arthritis, systemic lupus erythematosus, Raynaud’s syndrome, CREST syndrome, and Sjögren’s syndrome. All of these autoimmune diseases have been associated with PAH (reviewed in [5, 8, 9, 24,25,26]) [52]. Women with one autoimmune disease often have evidence of other autoimmune diseases with overlap occurring especially between rheumatic autoimmune diseases which include SSc, systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome, and myositis/dermatomyositis (Table 4). Up to one fifth of SSc cases have features of rheumatoid arthritis, systemic lupus erythematosus, and/or myositis [53]. Interestingly, women with Raynaud’s syndrome, rheumatoid arthritis, and thyroiditis often also have Sjögren’s [54,55,56]. Sjögren’s syndrome in particular causes inflammation, remodeling, and fibrosis of the lung [51, 57].
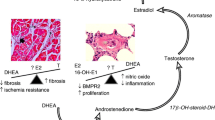
Raynaud’s syndrome frequently occurs with autoimmune diseases like mixed connective tissue disease (including SSc) (86%), systemic lupus erythematosus (31%), rheumatoid arthritis (22%), and Sjögren’s syndrome (13%) [53, 58]. The earliest clinical manifestation of microvascular dysfunction in SSc is the presence of Raynaud’s syndrome, seen in nearly 100% of SSc patients [59, 60]. Raynaud’s syndrome is associated with dysregulation of autonomic and small sensitive nerve fibers resulting in vasoconstriction of peripheral small vessels [58, 61, 62]. In patients with Raynaud’s syndrome, the presence of abnormal nail fold capillaries predicts the evolution to established SSc [63]. Anti-nuclear autoantibodies (ANA) are believed to be involved in the pathogenesis of Raynaud’s syndrome leading to IC deposition on vessels that trigger vasoconstriction [58]. Recently, ICs have been reported in the lungs of patients with PAH that were associated with perivascular macrophages and circulating monocytes [64]. Thus, autoantibodies/ICs present during autoimmune diseases like SSc, Sjögren’s syndrome, CREST syndrome, and Raynaud’s syndrome could contribute to vasoconstriction of small vessels in the lung, thereby contributing to the pathogenesis of PAH (Fig. 1).

Hypothesis for how infections and autoimmune diseases may promote PAH in women and men. Infections and autoimmune diseases like HIV and systemic sclerosis (SSc) are able to cause inflammation, immune complex (IC) deposition, and remodeling in the lung that may lead to pulmonary arterial hypertension. Estrogen increases the risk of developing autoimmune diseases like SSc following infection or other insults by promoting antibody/autoantibody and IC deposition that may contribute to the increased incidence of PAH in women, especially for autoimmune diseases that affect the lung-like SSc. PAH associated with HIV infection occurs more often in men and HIV-associated PAH patients also have myocarditis. Inflammatory mechanisms that drive myocarditis in men following infection may also drive PAH
ICs are composed of antibodies (against foreign antigen-like viruses or parasites) and/or autoantibodies (antibodies against damaged self) complexed with antigens/autoantigens and complement components (i.e., complement C3). ICs precipitate out of the blood binding to tissues when they become very large and/or sometimes when temperature changes occur, as occurs in Raynaud’s syndrome in response to cold. ICs strongly activate proinflammatory innate immune responses. Viral infections frequently lead to IC formation and may contribute to the association of viral infections with the onset/diagnosis of PAH [65]. Rheumatic autoimmune diseases like SSc, systemic lupus erythematosus, Sjögren’s syndrome, and rheumatoid arthritis are renowned for IC formation, where ICs arise from the presence of multiple autoantibodies including rheumatoid factor resulting in tissue deposition and activation of an immune response [5, 51]. Rheumatoid factor is an autoantibody formed against the Fc region of antibodies (e.g., IgM, IgG). As part of the normal immune response to infection, rheumatoid factor promotes complement fixation and clearance of ICs by macrophages, enhances antigen presentation, and amplifies the avidity of IgG, all of which protect against infections [66,67,68]. But the presence of rheumatoid factor predicts a more aggressive, destructive disease course for rheumatic autoimmune diseases where it is present in 70–90% of rheumatoid arthritis, 60–80% of Sjögren’s syndrome, 30% of systemic lupus erythematosus, 20% of myositis, and 20% of SSc patients [66, 67, 69]. Interestingly, HIV and Schistosoma mansoni infections (as well as other viral, bacterial, and parasitic infections associated with rheumatic autoimmune diseases and myocarditis) are known to form ICs with rheumatoid factor [5, 70], suggesting that CTD-, HIV-, and schistosomiasis-associated PAH may have a similar immunopathology (Fig. 1).
Autoantibodies against fibroblast, smooth muscle, and endothelial cells have been found in a significant proportion of CTD-associated PAH patients [71,72,73]. Of 380 PAH patients that underwent testing for the presence of autoantibodies, 33% of idiopathic and familial PAH (combined) tested positive for ANA in contrast to 94% of CTD-associated patients [12]. Out of 115 patients retrospectively identified with either idiopathic PAH (56%) or CTD-associated PAH (44%), 76% were women and ANA were detected more often in CTD-associated PAH cases [74]. The fact that ANA were detected in a large number of idiopathic PAH patients in both of these studies and that autoimmune diseases often take many years to diagnose suggest that some idiopathic PAH cases may develop CTD-associated PAH later in life [5, 23, 75].
Male-dominant autoimmune diseases associated with PAH
Two PAH registries reported that more men than women had HIV-associated PAH [12, 17]. In a study of HIV-associated PAH patients, all had evidence of myocarditis; 12 of 15 had myocardial inflammation confirmed by biopsy (the gold standard for diagnosis), and the other three patients had myocardial fibrosis in biopsies suggestive of healed/chronic myocarditis [76]. Ten of the patients with HIV-associated PAH and myocarditis were men and five women, a sex ratio of 2:1 male to female, which is the same as myocarditis patients in general (Table 4) [6]. HIV infection causes myocarditis, which is an autoimmune disease characterized by myocardial inflammation, remodeling, and progression to dilated cardiomyopathy [5, 77]. Myocarditis is one of the few autoimmune diseases that occurs more often in men [6]. A study of 28 asymptomatic patients with chronic HIV infection found that 82% of the patients had evidence of myocarditis and myocardial fibrosis by cardiac magnetic resonance imaging [78]. However, the study did not examine whether the HIV patients had PAH.
Myocarditis is associated with SSc and other rheumatic autoimmune diseases that are associated with PAH. Of 181 newly diagnosed cases of SSc, 7 had evidence of myocarditis [48]. Compared with SSc patients, patients with SSc and myocarditis had early disease, myositis, autoantibodies against ANA, detectable virus, pericardial effusion, and systolic and/or diastolic dysfunction [48]. Ten out of 19 female SSc patients (53%) had evidence of myocarditis and cardiac remodeling [79]. Similar to PAH, myocarditis has been reported to occur with all rheumatic autoimmune diseases (i.e., systemic lupus erythematosus, rheumatoid arthritis, SSc, myositis) except for Sjögren’s syndrome [5, 80,81,82,83,84,85]. Infections that have been associated with SSc and other rheumatic autoimmune diseases like Epstein-Barr virus, parvovirus B19, cytomegalovirus, coxsackievirus, Salmonella, and HIV are all known causes of myocarditis [5, 76, 86, 87]. Importantly, schistosomiasis, which is associated with PAH, has also been reported to cause the eosinophilic form of myocarditis in people and animal models [88,89,90,91]. The association of myocarditis with SSc-, HIV-, and schistosomiasis-associated PAH suggests the possibility that infection may increase the likelihood of PAH in men either directly or indirectly by inducing myocardial/vascular inflammation, fibrosis, and/or IC deposition. IC deposition on the exterior of the heart leads to pericarditis and occurs in animal models of myocarditis [92] and in myocarditis patients where it is termed perimyocarditis [93].
The fact that many autoimmune diseases associated with PAH like systemic lupus erythematosus, rheumatoid arthritis, and thyroiditis do not primarily cause disease in the lungs or pulmonary vasculature suggests that immune mechanisms that drive the pathogenesis of autoimmune diseases may also be important in PAH. Cytokines that are elevated in HIV infections and myocarditis include interleukin (IL)-1β, IL-6, IL-8, tumor necrosis factor (TNF), and platelet-derived growth factor (PDGF). These cytokines drive the pathogenesis of HIV/AIDs and myocarditis, as well as PAH [6, 94, 95]. Future studies should examine whether subjects with HIV- and/or Schistosoma mansoni-associated PAH also have myocarditis.
Immunopathology of PAH
The primary pathophysiology of PAH involves vasoconstriction, inflammation, and vascular remodeling [2]. The histopathologic hallmark of PAH is the plexiform lesion, which consists of endothelial cell (EC) proliferation and VSMC hypertrophy in small precapillary pulmonary arterioles surrounded by inflammatory cells and fibrosis [96]. The narrowing of the lumen by proliferation of vascular cells combined with constriction of the arteriole due to remodeling and fibrosis results in elevated pulmonary arterial blood pressure. A mixed infiltrate consisting of mast cells, macrophages, dendritic cells, and T and B cells surround plexiform lesions based on histology from PAH lung biopsies [65, 96]. The presence of antibody, autoantibody, and/or IC deposition in pulmonary vessels may further occlude vessels promoting PAH (Fig. 1).
Endothelial cell proliferation
EC proliferation is a major component of the pathology of plexiform lesions. ECs require vascular endothelial growth factor (VEGF) for survival, and excess amounts of this growth factor induce EC proliferation [97]. PAH patients have elevated circulating levels of VEGF compared to controls and higher expression of the VEGF receptor in plexiform lesions [98, 99]. Evidence of EC activation includes elevated levels of sera vascular cell adhesion molecule-1 (VCAM-1) in PAH patients, which is elevated on ECs after proinflammatory cytokine stimulation where it recruits inflammatory cells [99]. Evidence of EC damage (and autoimmunity) includes increased sera levels of anti-EC autoantibodies in patients with PAH compared to controls [71]. However, the initiator of EC injury is not known and could include exposure to toxins (i.e., drugs/anorexigen and chemicals), infections, and autoantibodies, all of which promote IC formation. IC deposition on ECs may directly lead to vasoconstriction or indirectly promote PAH by damaging cells and recruiting inflammation.
Vascular smooth muscle cell hypertrophy
Another key component of the pathology of the plexiform lesion includes proliferation of VSMCs. PDGF is a potent VSCM mitogen that drives hypertrophy and proliferation of this cell population during PAH [97]. PDGF levels are significantly elevated in lung tissue from PAH patients compared to control lungs [100]. Epidermal growth factor and its receptor are important for VSMC proliferation and survival and may play a role in PAH [101, 102]. Transforming growth factor (TGF)-β1 promotes collagen deposition while Toll-like receptor (TLR)4-induced IL-1β and IL-18 enhance remodeling associated with VSMCs [103].
Fibroblast/myofibroblast activation
An important contributor to arterial constriction and elevated blood pressure in PAH patients involves vascular remodeling/fibrosis that stiffens the pulmonary arteries [2]. Pulmonary arterial adventitial fibroblasts are the major source of collagen and remodeling enzymes termed matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs) [97, 104]. Other key growth factors and cytokines that convert fibroblasts to myofibroblasts include PDGF, fibroblast growth factor (FGF), insulin-like growth factor (IGF)-1, angiotensin II, TNF, IL-4, IL-1β, IL-17, and TGF-β1 [96, 97, 105, 106]. PDGF antagonists reverse vascular remodeling in animal models of PAH [100]. TGF-β1 stimulates fibroblast proliferation and collagen synthesis and inhibits MMP-induced degradation of collagen [106]. Angiotensin II, a component of the cardiac renin-angiotensin-aldosterone system, indirectly increases TGF-β1 in response to mechanical stretch or damage [107]. TNF, IL-1β, and IL-4 also induce fibroblast proliferation resulting in collagen synthesis and remodeling [108, 109]. IL-1β and TNF have long-lasting affects by upregulating and maintaining TGF-β1 transcription for as long as 6–10 weeks [110]. In contrast, IFN-γ directly inhibits transcription of TGF-β1 and IL-4 via signal transducer and activator of transcription (STAT)1 [111]. IFN-γ prevents fibrosis by inhibiting fibroblast proliferation, collagen synthesis, and mast cell degranulation [109, 112, 113].
Inflammation
Lung biopsies from PAH patients reveal a mixed infiltrate consisting of mast cells, macrophages, dendritic cells, and T and B cells surrounding plexiform lesions [65, 96]. Mast cells and macrophages are a major source of proinflammatory and profibrotic cytokines that recruit other immune cells and promote vascular remodeling. Mast cells induce remodeling by releasing TNF, IL-1β, IL-4, IL-18, and TGF-β1 as well as collagen/matrix digesting enzymes like tryptase, chymase, and serpin A3n that stimulate perivascular fibroblast proliferation and collagen deposition [114,115,116,117,118]. Mast cell-derived tryptase induces vascular angiogenesis promoting PAH [114]. Although mast cells are typically present in small numbers surrounding vessels, they are significantly increased in patients and animal models of PAH [119, 120]. Prevention of mast cell degranulation attenuated PAH in an animal model [121], suggesting that mast cells play a role in the disease process. Mast cells act as antigen presenting cells and provide the Th2-associated cytokines IL-4 and IL-33 to drive macrophages and T cells toward an alternatively activated M2 macrophage/Th2 phenotype increasing B cells and antibody/autoantibody production [109, 117, 122].
Macrophages and dendritic cells are innate immune cells that activate the immune response and promote remodeling (reviewed in [122]). Remodeling and a B cell-induced antibody/autoantibody response require a T helper-type 2 (Th2) immune response that is initiated by IL-4 and/or IL-33 during antigen presentation. Schistosoma mansoni parasitic infections are strong inducers of M2 macrophages and Th2 responses that are associated with elevated IL-4, IL-13, and TGF-β1 [123]. In contrast, HIV infection requires an IFN-γ/Th1-type immune response for viral clearance but is known to skew the immune response to a M2/Th2 response as a strategy to evade the immune system [124]. M2 macrophages are also associated with autoimmune diseases because they scavenge damaged self-tissues following injury through the mannose receptor and scavenger receptor-A where they increase autoimmune disease by presenting self-tissue to T cells [122]. IC-mediated activation of M2 macrophages and mast cells results in the release of profibrotic mediators like IL-1β, TGF-β1, fibronectin, and MMPs.
Immunogenetics
Genetic defects/polymorphisms in the bone morphogenic protein receptor (BMPR)2/TGF-β signaling axis have been identified with a predisposition for PAH [125]. Today, over 300 different polymorphisms in BMPR2 are known to exist and are present in approximately 75% of individuals with a family history of PAH [125]. However, only 10–20% of carriers of polymorphisms in BMPR2 develop PAH [126, 127], indicating that environmental factors are critically important for disease development. Polymorphisms in BMPR2 and other members of the TGF-β superfamily such as ALK1 and SMAD9 have been associated with elevated growth factors and proinflammatory mediators that contribute to remodeling during PAH [73, 128, 129]. BMP and TGF-β have also been found to synergistically activate Foxp3+ regulatory T cells (Tregs) [128]. Tregs reduce inflammation and are critical in regulating or preventing autoimmune diseases [130]. Thus, dysfunctional BMPR2 signaling may lead to reduced Treg which could increase inflammation and the risk of developing autoimmune diseases associated with PAH [131]. Aggregations of lymphocytes, resembling lymphoid follicles, have been detected in the lung tissue of PAH patients suggesting local chronic immune stimulation [128, 132]. Thus, genetic predisposition contributes to the development of PAH and may be more closely linked to autoimmune-associated PAH than previously realized.
Sex hormone effects on inflammation and remodeling
Microarray, proteomics, and other molecular tools have revealed how sex hormones and chromosomes regulate the immune response under normal and pathologic conditions [7, 118, 133, 134]. Estrogen receptor (ER)-α, ER-β, the androgen receptor, and other steroid hormone receptors are expressed on or within endothelial cells, VSMCs, and fibroblasts in mice and humans [6, 129, 130]. Similarly, human and mouse dendritic cells, monocytes, macrophages, mast cells, and T and B cells can express ERα, ERβ, and/or the androgen receptor (reviewed in [129, 131,132,133,134,135,136,137,138]). It is important to emphasize that ERα, ERβ, and the androgen receptor are present on/in tissue and immune cells in both men and women where they function essentially as growth factors maintaining the normal physiology of cells. Cells in women should have a higher ratio of ERs to androgen receptors on/in cells and visa versa for men. In response to foreign- or self-antigens, sex hormone receptors on tissue and immune cells alter the immune response in a sex-specific manner. The effect of sex hormone receptors on individual immune cells also depends on the context of the response. For example, the dose of the hormone and whether hormone receptor signaling occurs predominantly on the surface of the cell or within the cytoplasm alters the downstream effect of sex hormones [134, 139]. In general, females have a more robust T and B cell response and correspondingly lower burden of bacterial, viral, and parasitic infections [137].
ERα has been found to promote dendritic cell maturation and antigen presentation to T cells [140,141,142,143,144]. In contrast to estrogen, testosterone has a largely negative impact on dendritic cells, although the effect is complex and depends on the hormone concentration [134]. ERα, ERβ, and the androgen receptor are expressed on the membrane surface and within mast cells and macrophages [39, 40]. Testosterone increases mast cell and macrophage numbers in the heart of males with myocarditis and upregulates cardiac TLR4, IL-1β, and IL-18 [118, 145, 146]. Estrogen inhibits TNF, IL-1β, and IL-6 from macrophages by downregulating NFκB [147,148,149,150,151]. Estrogen has also been found to skew macrophages to an alternatively activated M2 phenotype by its ability to increase Th2-type cytokines like IL-4 [5, 134, 152]. Estrogen signaling through ERβ in monocytes upregulates expression of Fas ligand leading to increased monocyte apoptosis, but this does not occur in activated macrophages expressing ERα [134]. Androgen receptor expression on/in human monocytes is higher in men than in women [153,154,155]. While ERα and ERβ bind estrogen with similar affinity, the use of selective ER agonists revealed that while ERα stimulates T cell proliferation, ERβ promotes apoptosis [156, 157].
Estrogen also activates B cells in response to infection or damaged-self (i.e., autoimmunity) to increase antibody/autoantibody levels, thereby contributing to IC formation. In contrast, androgens reduce B cell synthesis of antibody/autoantibody in men [62, 135,136,137,138, 158]. Although increased antibody levels protect women from infections, they increase the risk for autoimmune diseases. In cell culture studies and animal models, estrogen has been found to activate innate immune cells like dendritic cells, stimulate T cell proliferation, skew the immune response toward Th2 and M2, and increase IL-4 and TGF-β levels [40, 138, 159,160,161,162], all of which are able to increase remodeling and fibrosis in lung cells. Far less, research has been conducted on the effect of androgens [118, 163, 164].
Menopause is defined as 1 year without a menstrual period [165,166,167] and occurs in Western cultures between age 49 and 55 [165, 166]. Decreases in estrogen (i.e., estradiol) occur in the last 6 months before menopause and thereafter [167]. In contrast, testosterone gradually decreases with age in both sexes [168]. Understanding changes in immune function following menopause are complicated by changes that occur because of aging [169, 170]. Menopause is associated with a decrease in T cell activity and an increase in IL-1, IL-6, and TNF levels [171,172,173]. That low estrogen increases proinflammatory cytokines is consistent with the known ability of high doses of estrogen via ERα to downregulate NFκB, Th1 responses, IL-1, and IL-6 in various human and murine cells [148, 150, 174,175,176]. Thus, after menopause, the protective role of estrogen is lost allowing increased proinflammatory cytokine levels while at the same time, antibody/autoantibody levels continue to rise (Fig. 1). This may promote certain autoimmune diseases like rheumatoid arthritis and Sjögren’s syndrome that peak after 50 years of age [177]. In contrast, a gradual decline in androgen levels past age 50 decreases inflammation in men without the elevated antibodies/autoantibodies found in women.
Women have higher ER expression in their arteries than men, which decreases after menopause [178]. ERβ signaling increases arterial tone and blood pressure, while ERα reduces vascular injury [179, 180]. Animal models of heart disease demonstrate that testosterone is responsible for adverse cardiac remodeling in the myocardium of males that leads to heart failure [6, 118, 181] and that estrogen signaling via ERα, in particular, prevents cardiac myocyte hypertrophy and fibrosis in females by inhibiting remodeling cytokines and collagen synthesis [180]. These findings suggest that ERβ may promote the negative effects of estrogen in pulmonary arteries in PAH, while ERα may be protective.
Sex differences in the immunopathology of PAH
PAH can occur at any age but typically between age 36 and 47 [9, 17, 18, 21, 95], suggesting that estrogen drives the pathogenesis of disease because PAH is a chronic condition that takes years to develop. SSc is also typically diagnosed prior to menopause in women, while men with SSc are diagnosed at an older age than women [34]. Recall that the sex ratio of women to men with SSc prior to menopause is estimated at 15:1 while it lowers to 2.4:1 after menopause [34,35,36], suggesting that estrogen promotes SSc. Surprisingly, circulating estradiol was found to reduce PAH in female Sprague Dawley rats that had undergone ovariectomy and estrogen replacement [182]. However, the study did not distinguish the role of ERα vs. ERβ in mediating the effect of estrogen. Estrogen via ERβ signaling has been shown to regulate arterial tone and blood pressure, while ERα protects against vascular injury, remodeling and fibrosis, and atherosclerosis [5, 6, 39, 40]. It is possible that current animal models of PAH do not fully recapitulate the clinical picture [183,184,185].
Male rats with hypoxia-induced pulmonary hypertension had more severe disease than females [186]. This finding is similar to humans, where men with PAH-associated SSc/dcSSc have more severe disease and worse survival [19, 20, 35, 37, 38, 56]. In this model, pulmonary vascular remodeling was reduced in males by 17β-estradiol and required ERα and ERβ signaling [187]. 2-Methoxyestradiol, a major non-estrogenic metabolite of estradiol, was also found to protect male rats from monocrotaline-induced PAH by decreasing pulmonary arterial hypertrophy and reducing proliferative and inflammatory responses in the lung [188]. More research is needed in animal models of PAH in order to determine the role of sex hormones in the immunopathogenesis of disease.
Importantly, recent research provides evidence that ERα via the estrogen response element is able to bind the BMPR2 promoter and modulate its activity [189]. In animal models of PAH, BMPR2 expression on lymphocytes and the pulmonary system was significantly lower in females than in males [189], suggesting that ERα may promote PAH by downregulating BMPR2. Additionally, CYP1B1, an estrogen-metabolizing enzyme, was expressed 10× less in females with BMPR2 mutations than those without [125, 190]. These data suggest that BMPR2 protects women from PAH and estrogen/ERα decreases its expression. Women with mutations/polymorphisms in BMPR2 may have a greater risk of developing PAH. Additionally, the central role for estrogen in driving a Th2 response that activates B cells and elevates autoantibody and IC levels could increase the prevalence of CTD/autoimmune disease-associated PAH in women (Fig. 1) [9, 26].
A study of 63 SSc patients that presented with isolated pulmonary hypertension found that menopause increased the risk of developing PAH, particularly in the subgroup of women with CREST (these patients also have Raynaud’s syndrome) [191]. CREST syndrome is associated with the development of SSc-associated PAH at a later age (i.e., menopause). Hormone replacement therapy improved lcSSc-associated PAH in post-menopausal women [192]. All 20 lcSSc patients that received hormone replacement did not develop PAH whereas 8/41 (19.5%) lcSSc patients that did not receive therapy developed PAH after menopause [192]. These data suggest that menopause worsens disease in women with PAH. Raynaud’s syndrome, which is associated with SSc and SSc-associated PAH, occurs more often in women than in men [55]. ANA present in Raynaud’s are believed to contribute to the pathogenesis of disease by contributing to IC deposition in vessels [58, 62]. Estrogen is thought to play a role in the pathogenesis of Raynaud’s syndrome, with the incidence of vasospastic reactions increasing during the pre-ovulatory period and with estrogen administration [193]. However, contradictory findings on the effect of sex hormones and hormone replacement therapy exist for Raynaud’s syndrome. Part of the difficulty in interpreting the effect of hormone replacement therapy on autoimmune or other chronic inflammatory diseases like PAH is that different combinations of hormones are used in therapies and they are administered to women at different ages.
There are a number of reasons why clinical and animal model data may appear to be contradictory for PAH, SSc, and Raynaud’s syndrome. High doses of estrogen have opposite effects on inflammation compared to low doses [5, 23], and so hormone replacement therapy may not exactly mimic pre-menopause conditions. Estrogen decreases inflammation by increasing regulatory Th2, Treg, IL-10, and TGF-β [62]. A recent report found that depletion of Treg in animal models of pulmonary hypertension increased disease in female rats [194], indicating the importance of Treg in preventing vascular injury in females. However, high doses of estrogen increase inflammation, as occurs during pregnancy and with hormone replacement therapy. These opposing effects of estrogen are likely due to differential signaling through ERα, ERβ, GPR30, and/or other steroid receptors. Estrogen, but not testosterone, increases antibody/autoantibody levels and thereby promotes ICs. This is true even for low doses of estrogen that occur after menopause. IC deposition activates complement, innate immune mechanisms like TLR4, decreases Treg, and increases inflammation [5]. Thus, one possible explanation for contradictory data is that estrogen in females is increasing inflammation via autoantibody/IC deposition. So even though estrogen levels drop significantly after menopause, low levels of estrogen continue to drive autoantibody diversity. Thus, estrogen may increase the incidence of PAH and SSc-associated PAH in women by increasing antibody/autoantibody production, even following menopause, leading to IC deposition, tissue damage, and inflammation (Fig. 1). In contrast, testosterone increases cell-mediated inflammation via mast cells and macrophages that release proinflammatory and profibrotic cytokines (i.e., IL-1β) that could be induced or amplified by infections (Fig. 1).
Knowledge gaps
Understanding how sex differences in the immune response to infection and autoimmunity influence PAH subgroups may provide important information about the pathogenesis of disease (Fig. 1). Future studies should analyze data on individual PAH subgroups according to sex. Heritable cases should be analyzed separately because the pathogenesis of disease differs (i.e., genetic vs. environmental). Additionally, combining PAH groups according to sex may increase the statistical power of studies vs. analyzing data only by PAH subgroup [23, 75]. Future studies also need to consider age in the analysis of PAH. That the sex ratio of women to men with SSc is 15:1 prior to 50 years of age but lowers to 2.4:1 after 50 [35, 36] indicates the importance of analyzing data by sex and age using age 50 as a cutoff. Age 50–55 is most commonly used to study the effect of menopause when the actual menopause status of the subject is not known. Very little data exists for the effect of andropause on disease in men. Aging affects both the immune system and host tissues due to a decline in cell function with age and changing levels of sex hormones. Although substantial therapeutic advances have occurred over the past 25 years for PAH patients [195], therapies could be improved by considering sex and age differences.
Conclusions
Most PAH registries report an overall female predominance of 2–4 over men. However, CTD-associated PAH is as high as 9:1 female to male while some other PAH subtypes occur more often in men. More research examining sex differences in PAH is needed. Analysis of all PAH subtypes by sex and age, with specific efforts to identify mechanistic differences, may provide potential treatment targets.
Abbreviations
- AIHA:
-
Autoimmune hemolytic anemia
- ANA:
-
Anti-nuclear autoantibody
- BMPR:
-
Bone morphogenic protein receptor
- CAD:
-
Coronary artery disease
- CTD:
-
Connective tissue disease
- CVD:
-
Cardiovascular disease
- dcSSc:
-
Diffuse cutaneous SSc
- EC:
-
Endothelial cell
- ER:
-
Estrogen receptor
- FGF:
-
Fibroblast growth factor
- IC:
-
Immune complex
- IGF-1:
-
Insulin-like growth factor-1
- IL:
-
Interleukin
- lcSSc:
-
Limited cutaneous SSc
- NIH:
-
National Institutes of Health
- PAH:
-
Pulmonary arterial hypertension
- PDGF:
-
Platelet-derived growth factor
- SSc:
-
Systemic sclerosis
- STAT:
-
Signal transducer and activator of transcription
- TGF:
-
Transforming growth factor
- Th:
-
T helper-type cells
- TIMP:
-
Tissue inhibitors of MMP
- TNF:
-
Tumor necrosis factor
- Tregs:
-
Regulatory T cells
- VEGF:
-
Vascular endothelial growth factor
- VSMC:
-
Vascular smooth muscle cell
References
Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:S40–S7.
Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–65.
Awad KS, West JD, de Jesus PV, MacLean M. Novel signaling pathways in pulmonary arterial hypertension (2015 Grover Conference series). Pulm Circ. 2016;6:285–94.
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41.
Fairweather D, Petri MA, Coronado MJ, Cooper LT Jr. Autoimmune heart disease: role of sex hormones and autoantibodies in disease pathogenesis. Expert Rev Clin Immunol. 2012;8:269–84.
Fairweather D, Cooper LT, Blauwet LA. Sex and gender differences in myocarditis and dilated cardiomyopathy. Curr Probl Cardiol. 2013;38:7–46.
Arnold AP, Cassis LA, Eghbali M, Reue K, Sandberg K. Sex hormones and sex chromosomes cause sex differences in the development of cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2017;37:746–56.
Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307:L7–L26.
Mair KM, Johansen AKZ, Wright AF, Wallace E, MacLean MR. Pulmonary arterial hypertension: basis of sex differences in incidence and treatment response. Br J Pharmacol. 2014;171:567–79.
Wei Y-C, George NI, Chang C-W, Hicks KA, Cheung A, Cushman W. Assessing sex differences in the risk of cardiovascular disease and mortality per increment in systolic blood pressure: a systematic review and meta-analysis of follow-up studies in the United States. PLoS One. 2017;12:e0170218.
Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–23.
Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103–10.
Peacock AJ, Murphy NF, McMurray JJV, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–9.
Jing Z-C, Xu X-Q, Han Z-Y, Wu Y, Deng K-W, Wang H, et al. Registry and survival study in Chinese patients with idiopathic and familial pulmonary arterial hypertension. Chest. 2007;132:373–9.
Jansa P, Jarkovsky J, Al-Hiti H, Popelova J, Ambroz D, Zatocil T, et al. Epidemiology and long-term survival of pulmonary arterial hypertension in the Czech Republic: a retrospective analysis of a nationwide registry. BMC Pulm Med. 2014;14:45.
Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JSR, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:790–6.
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France. Am J Respir Crit Care Med. 2006;173:1023–30.
McGoon MD, Miller DP. REVEAL: a contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. 2012;21:8–18.
Escribano-Subias P, Blanco I, Lopez-Meseguer M, Lopez-Guarch CJ, Roman A, Morales P, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J. 2012;40:596–603.
Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension. Circulation. 2014;129:57–65.
Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension. Chest. 2010;137:376–87.
Krowka MJ, Miller DP, Barst RJ, Taichman D, Dweik RA, Badesch DB, et al. Portopulmonary hypertension: a report from the US-based REVEAL registry. Chest. 2012;141:906–15.
Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol. 2008;173:600–9.
Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. 2012;11:754–65.
Montani D, Günther S, Dorfmüller P, Perros F, Girerd B, Garcia G, et al. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013;8:97.
Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:456–67.
Li JH, Safford RE, Aduen JF, Heckman MG, Crook JE, Burger CD. Pulmonary hypertension and thyroid disease. Chest. 2007;132:793–7.
Gisel JJ, Daniels JT, Burger CD. Clinical utility of thyroperoxidase antibody testing in patients with pulmonary hypertension. Thyroid Sci. 2009;4:CLS1–5.
Borazan N, Furst D. Systemic sclerosis, scleroderma. In: Rose N, Mackay I, editors. The autoimmune diseases. 5th ed. London: Elsevier; 2014. p. 463–80.
Kobak S, Kalkan S, Kirilmaz B, Orman M, Ercan E. Pulmonary arterial hypertension in patients with primary Sjögren’s syndrome. Autoimmune Dis. 2014;2014:710401.
Liu Z, Yang X, Tian Z, Qian J, Wang Q, Zhao J, et al. The prognosis of pulmonary arterial hypertension associated with primary Sjögren’s syndrome: a cohort study. Lupus. 2018;1:961203318756287. [Epub ahead of print].
Roberts-Thomson PJ, et al. Scleroderma in South Australia: epidemiological observations of possible pathogenic significance. Intern Med J. 2001;31:220–9.
Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part I. Arthritis Rheum. 2008;58:15–25.
Hussein H, Lee P, Chau C, Johnson SR. The effect of male sex on survival in systemic sclerosis. J Rheumatol. 2014;41:2193–200.
Schuna A. Autoimmune rheumatic diseases in women. J Am Pharm Assoc. 2002;42:612–23.
Hln C, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum. 2008;37:223–35.
Elhai M, Avouac J, Walker UA, Matucci-Cerinic M, Riemekasten G, Airò P, et al. A gender gap in primary and secondary heart dysfunctions in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis. 2016;75:163–9.
Pasarikovski CR, Granton JT, Roos AM, Sadeghi S, Kron AT, Thenganatt J, et al. Sex disparities in systemic sclerosis-associated pulmonary arterial hypertension: a cohort study. Arthritis Res Ther. 2016;18:30.
Fairweather D. Sex differences in inflammation during atherosclerosis. Clin Med Insights Cardiol. 2014;8:49–59.
Fairweather D. Autoimmune skin diseases: role of sex hormones, vitamin D, and menopause. In: Farage M, Miller K, Woods N, Maibach H, editors. Skin, mucosa and menopause. Berlin Heidelberg: Springer; 2015. p. 359–81.
Collins N, Bastian B, Quiqueree L, Jones C, Morgan R, Reeves G. Abnormal pulmonary vascular responses in patients registered with a systemic autoimmunity database: pulmonary hypertension assessment and screening evaluation using stress echocardiography (PHASE-I). Eur J Echocardiogr. 2006;7:439–46.
Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144:1346–56.
Murdaca G, Contatore M, Gulli R, Mandich P, Puppo F. Genetic factors and systemic sclerosis. Autoimmun Rev. 2016;15:427–32.
Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141:363–73.
Barsotti S, Stagnaro C, d'Ascanio A, Della RA. One year in review 2016: systemic sclerosis. Clin Exp Rheumatol. 2004;34(Suppl 100):3–13.
Coghlan G. Does left heart disease cause most systemic sclerosis associated pulmonary hypertension? Eur Respir J. 2013;42:888–90.
Dinser R, Frerix M, Meier FMP, Klingel K, Rolf A. Endocardial and myocardial involvement in systemic sclerosis—is there a relevant inflammatory component? Joint Bone Spine. 2013;80:320–3.
Pieroni M, De Santis M, Zizzo G, Bosello S, Smaldone C, Campioni M, et al. Recognizing and treating myocarditis in recent-onset systemic sclerosis heart disease: potential utility of immunosuppressive therapy in cardiac damage progression. Semin Arthritis Rheum. 2014;43:526–35.
Ali H, Ng KR, Low AHL. A qualitative systematic review of the prevalence of coronary artery disease in systemic sclerosis. Int J Rheum Dis. 2015;18:276–86.
Bissell L-A, Md Yusof MY, Buch MH. Primary myocardial disease in scleroderma—a comprehensive review of the literature to inform the UK systemic sclerosis study group cardiac working group. Rheumatology (Oxford). 2017;56:882–95.
Ozen G, Inanc N, Unal AU, Korkmaz F, Sunbul M, Ozmen M, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res. 2016;68:1538–46.
Thakkar V, Lau EM. Connective tissue disease-related pulmonary arterial hypertension. Best Pract Res Clin Rheumatol. 2016;30:22–38.
Denton CP. Advances in pathogenesis and treatment of systemic sclerosis. Clin Med (Lond). 2015;15:s58–63.
Díaz-López C, Geli C, Corominas H, Malat N, Diaz-Torner C, Llobet JM, et al. Are there clinical or serological differences between male and female patients with primary Sjögren’s syndrome? J Rheumatol. 2004;31:1352–5.
Ramos-Casals M, Solans R, Rosas J, Camps M, Gil A, Del Pino-Montes JJ-A, et al. Primary Sjögren’s syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine. 2008;87:210–9.
Horvath IF, Szanto A, Papp G, Zeher M. Clinical course, prognosis, and cause of death in primary Sjögren’s syndrome. J Immunol Res. 2014;2014:1–8.
Kronbichler A, Mayer G, Alfredsson L, Zhang H, Gonzalez AG, Richardson M, et al. Renal involvement in autoimmune connective tissue diseases. BMC Med. 2013;11:95.
Prete M, Fatone MC, Favoino E, Perosa F. Raynaud's phenomenon: from molecular pathogenesis to therapy. Autoimmun Rev. 2014;13:655–67.
Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annu Rev Pathol. 2011;6:509–37.
Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev. 2007;6:520–3.
Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol. 2012;8:469–79.
Brandt JE, Priori R, Valesini G, Fairweather D. Sex differences in Sjögren's syndrome: a comprehensive review of immune mechanisms. Biol Sex Differ. 2015;6:19.
Koenig M, Joyal F, Fritzler MJ, Roussin A, Abrahamowicz M, Boire G, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud's phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum. 2008;58:3902–12.
Saito T, Miyagawa K, Chen SY, Tamosiuniene R, Wang L, Sharpe O, et al. Upregulation of human endogenous retrovirus-K is linked to immunity and inflammation in pulmonary arterial hypertension. Circulation. 2017;136:1920–35.
Pullamsetti SS, Savai R, Janssen W, Dahal BK, Seeger W, Grimminger F, et al. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin Microbiol Infect. 2011;17:7–14.
Newkirk MM. Rheumatoid factors: host resistance or autoimmunity? Clin Immunol. 2002;104:1–13.
Dörner T, Egerer K, Feist E, Burmester GR. Rheumatoid factor revisited. Curr Opin Rheumatol. 2004;16:246–53.
Carson DA, Chen PP, Kipps TJ, Radoux V, Jirik FR, Goldfien RD, et al. Idiotypic and genetic studies of human rheumatoid factors. Arthritis Rheum. 1987;30:1321–5.
Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61.
Lapa AT, Appenzeller S, Bértolo MB. Schistosoma mansoni infection: an immune complex disease presenting with polyarthritis. Rheumatol Int. 2013;33:1341–3.
Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60:765–72.
Tamby M, Humbert M, Guilain P, Servettaz A, Dupin N, Christner J, et al. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur Resp J. 2006;28:799–807.
Humbert M, Lau EMT, Montani D, Jais X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130:2189–208.
Pagán RJ, Lee AS, Austin CO, Burger CD. Screening for connective tissue disease in pulmonary arterial hypertension. South Med J. 2014;107:666–9.
Dube SR, Fairweather D, Pearson WS, Felitti VJ, Anda RF, Croft JB. Cumulative childhood stress and autoimmune diseases in adults. Psychosom Med. 2009;71:243–50.
Frustaci A, Petrosillo N, Vizza D, Francone M, Badagliacca R, Verardo R, et al. Myocardial and microvascular inflammation/infection in patients with HIV-associated pulmonary artery hypertension. AIDS. 2014;28:2541–9.
Ntusi NAB. HIV and myocarditis. Curr Opin HIV AIDS. 2017;12:561–5.
Luetkens JA, Doerner J, Schwarze-Zander C, Wasmuth J-C, Boesecke C, Sprinkart AM, et al. Cardiac magnetic resonance reveals signs of subclinical myocardial inflammation in asymptomatic HIV-infected patients. Circ Cardiovasc Imaging. 2016;9:e004091.
Ntusi NAB, Piechnik SK, Francis JM, Ferreira VM, Rai ABS, Matthews PM, et al. Subclinical myocardial inflammation and diffuse fibrosis are common in systemic sclerosis—a clinical study using myocardial T1-mapping and extracellular volume quantification. J Cardiovasc Magn Reson. 2014;16:21.
West SG, Killian PJ, Lawless OJ. Association of myositis and myocarditis in progressive systemic sclerosis. Arthritis Rheum. 1981;24:662–8.
Carette S, Turcotte J, Mathon G. Severe myositis and myocarditis in progressive systemic sclerosis. J Rheumatol. 1985;12:997–9.
Clemson BS, Miller WR, Luck JC, Feriss JA. Acute myocarditis in fulminant systemic sclerosis. Chest. 1992;101:872–4.
Kerr LD, Spiera H. Myocarditis as a complication in scleroderma patients with myositis. Clin Cardiol. 1993;16:895–9.
Liangos O, Neure L, Kühl U, Pauschinger M, Sieper J, Distler A, et al. The possible role of myocardial biopsy in systemic sclerosis. Rheumatology (Oxford). 2000;39:674–9.
Stack J, McLaughlin P, Sinnot C, Henry M, MacEneaney P, Eltahir A, et al. Successful control of scleroderma myocarditis using a combination of cyclophosphamide and methylprednisolone. Scand J Rheumatol. 2010;39:349–50.
Ferri C, Zakrzewska K, Longombardo G, Giuggioli D, Storino FA, Pasero G, et al. Parvovirus B19 infection of bone marrow in systemic sclerosis patients. Clin Exp Rheumatol. 1999;17:718–20.
Ohtsuka T, Yamazaki S. Increased prevalence of human parvovirus B19 DNA in systemic sclerosis skin. Br J Dermatol. 2004;150:1091–5.
Lima JP. Study of the so-called "ectopical lesions" in Manson's schistosomiasis. II. Myocardial schistosomiasis. Rev Inst Med Trop Sao Paulo. 1969;11:290–3.
Chisty MM, Nargis M, Sato H, Inaba T, Kamiya H. Occurrence of myocarditis in rodents infected with Schistosoma mansoni. Southeast Asian J Trop Med Public Health. 1999;30:556–61.
Epelboin L, Jauréguiberry S, Estève J-B, Danis M, Komajda M, Bricaire F, et al. Myocarditis during acute schistosomiasis in two travelers. Am J Trop Med Hyg. 2010;82:365–7.
Nunes MCP, Guimarães Júnior MH, Diamantino AC, Gelape CL, Ferrari TCA. Cardiac manifestations of parasitic diseases. Heart. 2017;103:651–8.
Fairweather D, Frisancho-Kiss S, Njoku DB, Nyland JF, Kaya Z, Yusung SA, et al. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1beta, and immune complex deposition in the heart. J Immunol. 2006;176:3516–24.
Imazio M, Cooper LT. Management of myopericarditis. Expert Rev Cardiovasc Ther. 2013;11:193–201.
Bigna JJ, Sime PS, Koulla-Shiro S. HIV related pulmonary arterial hypertension: epidemiology in Africa, physiopathology, and role of antiretroviral treatment. AIDS Res Ther. 2015;12:36.
Martin YN, Pabelick CM. Sex differences in the pulmonary circulation: implications for pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H1253–64.
Hassoun PM, Mouthon L, Barber JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S9.
Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. 2011;147:4–12.
Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28:434–42.
Mouthon L, Guillevin L, Humbert M. Pulmonary arterial hypertension: an autoimmune disease? Eur Respir J. 2005;26:986–8.
Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21.
Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–31.
Dahal BK, Cornitescu T, Tretyn A, Pullamsetti SS, Kosanovic D, Dumitrascu R, et al. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2010;181:158–67.
Raines EW, Ferri N. Thematic review series: the immune system and atherogenesis. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res. 2005;46:1081–92.
Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009;19:247–52.
Mitra AK, Del Core MG, Agrawal DK. Cells, cytokines and cellular immunity in the pathogenesis of fibroproliferative vasculopathies. Can J Physiol Pharmacol. 2005;83:701–15.
Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–7.
Schultz JEJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–96.
Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–26.
Fairweather D, Frisancho-Kiss S, Yusung S, Barrett M, Gatewood S, Davis S, et al. IFN-γ protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines TGF-β1, IL-1β, and IL-4 in the heart. Am J Pathol. 2004;165:1883–94.
Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–13.
Eickelberg O, Pansky A, Koehler E, Bihl M, Tamm M, Hildebrand P, et al. Molecular mechanisms of TGF-β antagonism by interferon-γ and cyclosporine a in lung fibroblasts. FASEB J. 2001;15:797–806.
Oldroyd SD, Thomas GL, Gabbiani G, El Nahas AM. Interferon-γ inhibits experimental renal fibrosis. Kidney Int. 1999;56:2116–27.
Ahdieh M, Vandenbos T, Youakim A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-γ. Am J Physiol Cell Physiol. 2001;281:C2029–38.
Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, et al. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–700.
Cairns JA, Walls AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest. 1997;99:1313–21.
Garbuzenko E, Nagler A, Pickholtz D, Gillery P, Reich R, Maquart F-X, et al. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin Exp Allergy. 2002;32:237–46.
Fairweather D, Frisancho-Kiss S, Gatewood S, Njoku D, Steele R, Barrett M, et al. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following coxsackievirus B3 infection. Autoimmunity. 2004;37:131–45.
Coronado MJ, Brandt JE, Kim E, Bucek A, Bedja D, Abston ED, et al. Testosterone and interleukin-1β increase cardiac remodeling during coxsackievirus B3 myocarditis via serpin A 3n. Am J Physiol Heart Circ Physiol. 2012;302:H1726–H36.
Heath D, Yacoub M. Lung mast cells in plexogenic pulmonary arteriopathy. J Clin Pathol. 1991;44:1003–6.
Miyata M, Sakuma F, Ito M, Ohira H, Sato Y, Kasukawa R. Athymic nude rats develop severe pulmonary hypertension following monocrotaline administration. Int Arch Allergy Immunol. 2000;121:246–52.
Banasová A, Maxová H, Hampl V, Vízek M, Povýsilová V, Novotná J, et al. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration. 2008;76:102–7.
Fairweather D, Cihakova D. Alternatively activated macrophages in infection and autoimmunity. J Autoimmun. 2009;33:222–30.
Graham BB, Kumar R. Schistosomiasis and the pulmonary vasculature (2013 Grover Conference series). Pulm Circ. 2014;4:353–62.
Sandstrom TS, Ranganath N, Angel JB. Impairment of the type I interferon response by HIV-1: potential targets for HIV eradication. Cytokine Growth Factor Rev. 2017;37:1-16.
Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202.
Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis. 1984;129:194–7.
Loyd JE, Butler MG, Foroud TM, Conneally PM, Phillips JA, Newman JH. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;152:93–7.
Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165-75.
Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat. 2015;36:1113–27.
Pesenacker AM, Cook L, Levings MK. The role of FOXP3 in autoimmunity. Curr Opin Immunol. 2016;43:16–23.
Nasim MT, Ogo T, Chowdhury HM, Zhao L, Chen CN, Rhodes C, et al. BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFβ-TAK1-MAPK pathways in PAH. Hum Mol Genet. 2012;21:2548–58.
Perros F, Cohen-Kaminsky S, Gambaryan N, Girerd B, Raymond N, Klingelschmitt I, et al. Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2013;187:189–96.
Isensee J, Witt H, Pregla R, Hetzer R, Regitz-Zagrosek V, Ruiz NP. Sexually dimorphic gene expression in the heart of mice and men. J Mol Med. 2008;86:61–74.
Buskiewicz IA, Huber SA, Fairweather D. Sex hormone receptor expression in the immune system. In: Neigh G, Mitzelfelt M, editors. Sex differences in physiology. London: Elsevier; 2016. p. 45–60.
Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev. 2010;9:494–8.
Lahita R. Sex hormones and immune function. In: Legato M, editor. Principles of gender-specific medicine. 2nd ed. London: Elsevier; 2010. p. 615–26.
Klein SL. The effects of hormones on sex differences in infection: from genes to behavior. Neurosci Biobehav Rev. 2000;24:627–38.
Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–74.
Regitz-Zagrosek V, Becher E, Mahmoodzadeh S, Schubert C. Sex steroid hormones. In: Bader M, editor. Cardiovascular hormone systems: from molecular mechanisms to novel therapeutics. Wiley-Blackwell: Weinheim; 2008. p. 39–64.
Douin-Echinard V, Laffont S, Seillet C, Delpy L, Krust A, Chambon P, et al. Estrogen receptor alpha, but not beta, is required for optimal dendritic cell differentiation and [corrected] CD40-induced cytokine production. J Immunol. 2008;180:3661–9.
Carreras E, Turner S, Frank MB, Knowlton N, Osban J, Centola M, et al. Estrogen receptor signaling promotes dendritic cell differentiation by increasing expression of the transcription factor IRF4. Blood. 2010;115:238–46.
Kovats S. Estrogen receptors regulate an inflammatory pathway of dendritic cell differentiation: mechanisms and implications for immunity. Horm Behav. 2012;62:254–62.
Seillet C, Rouquie N, Foulon E, Douin-Echinard V, Krust A, Chambon P, et al. Estradiol promotes functional responses in inflammatory and steady-state dendritic cells through differential requirement for activation function-1 of estrogen receptor alpha. J Immunol. 2013;190:5459–70.
Xie H, Hua C, Sun L, Zhao X, Fan H, Dou H, et al. 17Beta-estradiol induces CD40 expression in dendritic cells via MAPK signaling pathways in a minichromosome maintenance protein 6-dependent manner. Arthritis Rheum. 2011;63:2425–35.
Frisancho-Kiss S, Davis SE, Nyland JF, Frisancho JA, Cihakova D, Barrett MA, et al. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. 2007;178:6710–4.
Frisancho-Kiss S, Coronado MJ, Frisancho JA, Lau VM, Rose NR, Klein SL, et al. Gonadectomy of male BALB/c mice increases Tim-3(+) alternatively activated M2 macrophages, Tim-3(+) T cells, Th2 cells and Treg in the heart during acute coxsackievirus-induced myocarditis. Brain Behav Immun. 2009;23:649–57.
Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol. 1997;38:46–54.
Evans MJ, Eckert A, Lai K, Adelman SJ, Harnish DC. Reciprocal antagonism between estrogen receptor and NF-kappaB activity in vivo. Circ Res. 2001;89:823–30.
Chadwick CC, Chippari S, Matelan E, Borges-Marcucci L, Eckert AM, Keith JC Jr, et al. Identification of pathway-selective estrogen receptor ligands that inhibit NF-kappaB transcriptional activity. Proc Natl Acad Sci U S A. 2005;102:2543–8.
Demyanets S, Pfaffenberger S, Kaun C, Rega G, Speidl WS, Kastl SP, et al. The estrogen metabolite 17beta-dihydroequilenin counteracts interleukin-1alpha induced expression of inflammatory mediators in human endothelial cells in vitro via NF-kappaB pathway. Thromb Haemost. 2006;95:107–16.
Huang H, He J, Yuan Y, Aoyagi E, Takenaka H, Itagaki T, et al. Opposing effects of estradiol and progesterone on the oxidative stress-induced production of chemokine and proinflammatory cytokines in murine peritoneal macrophages. J Med Investig. 2008;55:133–41.
Okwan-Duodu D, Umpierrez GE, Brawley OW, Diaz R. Obesity-driven inflammation and cancer risk: role of myeloid derived suppressor cells and alternately activated macrophages. Am J Cancer Res. 2013;3:21–33.
McCrohon JA, Death AK, Nakhla S, Jessup W, Handelsman DJ, Stanley KK, et al. Androgen receptor expression is greater in macrophages from male than from female donors. A sex difference with implications for atherogenesis. Circulation. 2000;101:224–6.
Sader MA, McGrath KC, Hill MD, Bradstock KF, Jimenez M, Handelsman DJ, et al. Androgen receptor gene expression in leucocytes is hormonally regulated: implications for gender differences in disease pathogenesis. Clin Endocrinol. 2005;62:56–63.
Brechenmacher SA, Bruns CJ, Van den Engel NK, Angele P, Loehe F, Jauch KW, et al. Influence of surgical trauma on the mRNA expression of sex hormone receptors in PBMCs in male and female patients. Langenbeck's Arch Surg. 2008;393:871–6.
Yakimchuk K, Jondal M, Okret S. Estrogen receptor alpha and beta in the normal immune system and in lymphoid malignancies. Mol Cell Endocrinol. 2013;375:121–9.
Aristimuno C, Teijeiro R, Valor L, Alonso B, Tejera-Alhambra M, de Andres C, et al. Sex-hormone receptors pattern on regulatory T-cells: clinical implications for multiple sclerosis. Clin Exp Med. 2012;12:247–55.
Cook IF. Sexual dimorphism of humoral immunity with human vaccines. Vaccine. 2008;26:3551–5.
Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, et al. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol. 2004;173:2227–30.
Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce TH2 and tolerogenic responses. Nat Immunol. 2010;11:647–55.
Papenfuss TL, Powell ND, McClain MA, Bedarf A, Singh A, Gienapp IE, et al. Estriol generates tolerogenic dendritic cells in vivo that protect against autoimmunity. J Immunol. 2011;186:3346–55.
Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun. 2012;38:J282–J91.
Girón-González JA, Moral FJ, Elvira J, García-Gil D, Guerrero F, Gavilán I, et al. Consistent production of a higher TH1:TH2 cytokine ratio by stimulated T cells in men compared with women. Eur J Endocrinol. 2000;143:31–6.
Giltay EJ, Fonk JC, von Blomberg BM, Drexhage HA, Schalkwijk C, Gooren LJ. In vivo effects of sex steroids on lymphocyte responsiveness and immunoglobulin levels in humans. J Clin Endocrinol Metab. 2000;85:1648–57.
Kato I, Toniolo P, Akhmedkhanov A, Koenig KL, Shore R, Zeleniuch-Jacquotte A. Prospective study of factors influencing the onset of natural menopause. J Clin Epidemiol. 1998;51:1271–6.
Jacobsen BK, Heuch I, Kvale G. Age at natural menopause and all-cause mortality: a 37-year follow-up of 19,731 Norwegian women. Am J Epidemiol. 2003;157:923–9.
Harlow SD, Gass M, Hall JE, Lobo R, Maki P, Rebar RW, et al. Executive summary of the stages of reproductive aging workshop + 10: addressing the unfinished agenda of staging reproductive aging. J Clin Endocrinol Metab. 2012;97:1159–68.
Davison SL, Bell R, Donath S, Montalto JG, Davis SR. Androgen levels in adult females: changes with age, menopause, and oophorectomy. J Clin Endocrinol Metab. 2005;90:3847–53.
Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transpl Int. 2009;22:1041–50.
Gameiro CM, Romao F, Castelo-Branco C. Menopause and aging: changes in the immune system—a review. Maturitas. 2010;67:316–20.
White HD, Crassi KM, Givan AL, Stern JE, Gonzalez JL, Memoli VA, et al. CD3+ CD8+ CTL activity within the human female reproductive tract: influence of stage of the menstrual cycle and menopause. J Immunol. 1997;158:3017–27.
Shakhar K, Shakhar G, Rosenne E, Ben-Eliyahu S. Timing within the menstrual cycle, sex, and the use of oral contraceptives determine adrenergic suppression of NK cell activity. Br J Cancer. 2000;83:1630–6.
Bove R. Autoimmune diseases and reproductive aging. Clin Immunol. 2013;149:251–64.
Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor alpha mediates estrogen's immune protection in autoimmune disease. J Immunol. 2003;171:6936–40.
Feldman I, Feldman GM, Mobarak C, Dunkelberg JC, Leslie KK. Identification of proteins within the nuclear factor-kappa B transcriptional complex including estrogen receptor-alpha. Am J Obstet Gynecol. 2007;196:394 e1–11. discussion e11-3
Paimela T, Ryhanen T, Mannermaa E, Ojala J, Kalesnykas G, Salminen A, et al. The effect of 17beta-estradiol on IL-6 secretion and NF-kappaB DNA-binding activity in human retinal pigment epithelial cells. Immunol Lett. 2007;110:139–44.
Farage M, Millwe D, Maibach H. The effects of menopause on autoimmune diseases. In: Farage M, Miller K, Woods N, Maibach H, editors. Skin, mucosa and menopause: management of clinical issues. Heidelberg: Springer; 2015. p. 299–318.
Vitale C, Mendelsohn M, Rosano G. Gender differences in the cardiovascular effect of sex hormones. Nat Rev Cardiol. 2009;6:532–42.
Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. Estrogen receptor alpha is a major mediator of 17beta-estradiol's atheroprotective effects on lesion size in Apoe-/- mice. J Clin Invest. 2001;107:333–40.
Hammes SR, Levin ER. Minireview: recent advances in extranuclear steroid receptor actions. Endocrinology. 2011;152:4489–95.
Cavasin MA, Tao Z-Y, Yu A-L, Yang X-P. Testosterone enhances early cardiac remodeling after myocardial infarction, causing rupture and degrading cardiac function. Am J Physiol Heart Circ Physiol. 2006;290:H2043–50.
Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. 2001;280:L88–97.
Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32.
Dai Z, Zhao YY. Discovery of a murine model of clinical PAH: mission impossible? Trends Cardiovasc Med. 2017;27:229–36.
Sztuka K, Jasinska-Stroschein M. Animal models of pulmonary arterial hypertension: a systematic review and meta-analysis of data from 6126 animals. Pharmacol Res. 2017;125:201–14.
Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Phys. 1981;240:H62–72.
Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, et al. 17Beta-estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–80.
Tofovic SP, Salah EM, Mady HH, Jackson EK, Melhem MF. Estradiol metabolites attenuate monocrotaline-induced pulmonary hypertension in rats. J Cardiovasc Pharmacol. 2005;46:430–7.
Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. 2012;3:6.
West J, Cogan J, Geraci M, Robinson L, Newman J, Phillips JA, et al. Gene expression in BMPR2 mutation carriers with and without evidence of pulmonary arterial hypertension suggests pathways relevant to disease penetrance. BMC Med Genet. 2008;1:45.
Scorza R, Caronni M, Bazzi S, Nador F, Beretta L, Antonioli R, et al. Post-menopause is the main risk factor for developing isolated pulmonary hypertension in systemic sclerosis. Ann N Y Acad Sci. 2002;966:238–46.
Beretta L, Caronni M, Origgi L, Ponti A, Santaniello A, Scorza R. Hormone replacement therapy may prevent the development of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Scand J Rheumatol. 2006;35:468–71.
Generini S, Seibold JR, Matucci-Cerinic M. Estrogens and neuropeptides in Raynaud's phenomenon. Rheum Dis Clin N Am. 2005;31:177–86.
Tamosiuniene R, Manouvakhova O, Mesange P, Saito T, Qian J, Sanyal M, et al. A dominant role for regulatory T cells in protecting females against pulmonary hypertension. Circ Res. 2018. [Epub ahead of print].
McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65:1976–97.
Funding
This work was supported by National Institutes of Health (R01 HL111938, R21 ES024414) and American Heart Association (16GRNT30950007) grants to DF. Funding sources were not involved in writing the manuscript or the decision to submit an article for publication.
Author information
Authors and Affiliations
Contributions
First author KAB and DF were major contributors in writing the manuscript. All other authors contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Batton, K.A., Austin, C.O., Bruno, K.A. et al. Sex differences in pulmonary arterial hypertension: role of infection and autoimmunity in the pathogenesis of disease. Biol Sex Differ 9, 15 (2018). https://doi.org/10.1186/s13293-018-0176-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-018-0176-8