
Abstract
Background
A crucial step to understanding the mechanistic underpinnings of autism spectrum disorder (ASD), is to examine if the biological underpinnings of ASD in genetic high-risk conditions, like 22q11.2 deletion syndrome (22q11.2DS), are similar to those in idiopathic illness. This study aimed to examine if ASD symptomatology in 22q11.2DS is underpinned by the same—or distinct—neural systems that mediate these symptoms in non-deletion carriers.
Methods
We examined vertex-wise estimates of cortical volume (CV), surface area (SA), and cortical thickness across 131 individuals between 6 and 25 years of age including (1) 50 individuals with 22q11.2DS, out of which n = 25 had a diagnosis of ASD, (2) 40 non-carriers of the microdeletion with a diagnosis of ASD (i.e., idiopathic ASD), and (3) 41 typically developing (TD) controls. We employed a 2-by-2 factorial design to identify neuroanatomical variability associated with the main effects of 22q11.2DS and ASD, as well as their interaction. Further, using canonical correlation analysis (CCA), we compared neuroanatomical variability associated with the complex (i.e., multivariate) clinical phenotype of ASD between 22q11.2 deletion carriers and non-carriers.
Results
The set of brain regions associated with the main effect of 22q11.2DS was distinct from the neuroanatomical underpinnings of the main effect of ASD. Moreover, significant 22q11.2DS-by-ASD interactions were observed for CV and SA in the dorsolateral prefrontal cortex, precentral gyrus, and posterior cingulate cortex, suggesting that the neuroanatomy of ASD is significantly modulated by 22q11.2DS (p < 0.01). We further established that the multivariate patterns of neuroanatomical variability associated with differences in symptom profiles significantly differed between 22q11.2 deletion carriers and non-carriers.
Limitations
We employed a multicenter design to overcome single-site recruitment limitations; however, FreeSurfer-derived measures of surface anatomy have been shown to be highly reliable across scanner platforms and field strengths. Further, we controlled for gender to address the differing distribution between idiopathic ASD individuals and the other groups. Nonetheless, the gender distribution in our sample reflects that of the respective populations, adding to the generalizability of our results. Last, we included individuals with a relatively wide age range (i.e., 6–25 years).
Conclusions
Our findings indicate that neuroanatomical correlates of ASD symptomatology in carriers of the 22q11.2 microdeletion diverge from those in idiopathic ASD.
Similar content being viewed by others


Background
22q11.2 deletion syndrome (22q11.2DS) is a genetic condition resulting from a microdeletion at the q11.2 band of chromosome 22 [1]. The estimated prevalence of 22q11.2DS is about 1 in 4000 [2], with an equal proportion of affected males and females [3]. This makes 22q11.2DS the most common microdeletion syndrome in the general population [4, 5]. While all individuals with 22q11.2DS display a deletion within the same locus of chromosome 22, the phenotypic consequences of the deletion are both complex and variable [6]. These encompass congenital and somatic features, as well as neuropsychiatric conditions. For example, there are high rates of comorbid neuropsychiatric conditions such as the following: autism spectrum disorder (ASD, ranging from 18 to 58% until early adulthood) [7,8,9,10], attention deficit hyperactivity disorder (ADHD, 37% in childhood) [7], anxiety disorders (approximately 35% in childhood and adolescence) [7], psychotic symptoms (57% in adolescence and young adulthood) [11, 12], and psychotic spectrum disorders (around 41% in adulthood) [7, 13, 14]. These typically occur at different developmental stages, making the clinical phenotype of 22q11.2DS highly heterogeneous not only between carriers, but also within affected individuals over time.
Some evidence suggests that the complex clinical phenotypes associated with 22q11.2DS represent distinct clinical outcomes that are underpinned by separable neurobiological mechanisms. For example, it has been shown that inter-individual variability in brain structure accompanies the occurrence and severity of positive psychotic symptoms in 22q11.2DS [15]. Yet, it remains largely unknown whether the neuropsychiatric symptoms that are commonly observed in 22q11.2DS are also mediated by the same neural mechanisms that underpin these symptoms in individuals without the microdeletion. This particularly applies to ASD symptomatology, which compared to psychotic symptomatology remains currently underexplored. Despite the high prevalence, only three neuroimaging studies to date have examined the neuroanatomical underpinnings of ASD in the brains of 22q11.2DS individuals. Two of these studies report differences in right amygdala volume between 22q11.2DS individuals with and without ASD symptomatology, but there is no consensus yet with regard to the direction of the effect (i.e., enlarged or reduced in ASD) [9, 10]. A recent study by our group also reported differences in cortical volume (CV) and surface area (SA) between 22q11.2DS individuals with and without ASD symptomatology. These differences were predominantly observed in parieto-temporal regions as well as in the posterior cingulate and dorsolateral prefrontal cortices, brain regions that have previously been linked to wider autistic symptoms and traits [16]. Preliminary evidence therefore suggests that 22q11.2DS individuals with ASD symptomatology are neuroanatomically distinct from those without ASD, and may represent a distinct neurobiological subgroup. None of these studies, however, have included a comparison to individuals with idiopathic ASD and it therefore remains unknown how closely the neurobiological phenotype of ASD in 22q11.2DS resembles the ASD phenotype in those without the microdeletion. Further, if we are to better identify how microdeletions impact on the phenotype of ASD, we need to understand if abnormalities in ASD individuals with and without 22q11.2DS are shared (or not).
This study aimed to examine the neuroanatomical underpinnings of ASD symptomatology across disorders using a categorical approach that allowed us to establish the extent to which 22q11.2DS modulates the neuroanatomy of ASD. Based on prior evidence [16], it was hypothesized that ASD symptomatology in 22q11.2DS is not simply due to a higher neuroanatomical “load” or affection status (i.e., more severe behavioral impairments associated with more pronounced neuroanatomical differences), but instead significantly interacts with the microdeletion to give rise to a distinct neuroanatomical brain phenotype. Moreover, in a second analysis step, we aimed to consolidate the results of the categorical approach, where ASD is treated as a “fixed-effect” variable based on diagnostic labels, with a multivariate dimensional approach using canonical correlation analysis (CCA), where ASD is considered a complex clinical construct or phenotypic trait spanned by multiple symptom domains across disorders. This allowed us to link the complex clinical phenotype(s) of ASD with neuroanatomical variability in multiple brain regions across disorders, and to compare their multivariate association between groups.
Methods
Participants
The total sample consisted of 131 individuals between 6 and 25 years of age, including (1) 50 individuals with 22q11.2DS, where n = 25 had a diagnosis of ASD (22q11.ASD) and n = 25 individuals did not (22q11.nonASD); (2) 40 non-deletion individuals with a diagnosis of ASD (i.e., idiopathic ASD); and (3) 41 typically developing (TD) controls without the microdeletion. The 22q11.2DS group was recruited in London, UK, and in Los Angeles, USA; and the idiopathic ASD group in Frankfurt, Germany. The TD controls were pooled at equal proportions across all three sites (see Table 1 and Supplementary Methods 1). The 22q11.2 microdeletion was confirmed by in situ hybridization (FISH) or microarray. All non.22q11.2DS participants were screened for somatic features associated with the microdeletion (i.e., cleft palate abnormalities, heart surgery, characteristic facial abnormalities, and hypoparathyroidism), as well as other disorders associated with 22q11.2DS. As none of these individuals displayed somatic abnormalities associated with the 22q11.2 microdeletion, we did not perform genetic testing. ASD was assessed using the Autism Diagnostic Interview-Revised (ADI-R) [17] and the Autism Diagnostic Observation Schedule (ADOS; for information on the calculation of ADOS Calibrated Severity Scores (CSS) see Supplementary Methods 2) [18, 19]. In accordance with previously published studies examining ASD symptomatology in 22q11.2DS [9, 16], all individuals with ASD met diagnostic cutoffs in the reciprocal social interaction (cutoff = 10) and communication domain (cutoff = 8) of the ADI-R, but were allowed to fall below threshold in the repetitive behaviors domain (cutoff = 3). The 22q11.2DS sample has previously been described in Gudbrandsen et al. [16]. To capture autism along a continuum consisting of multiple symptom domains, we also administered the Social Responsiveness Scale (SRS) [20, 21] in all participants, including individuals without a diagnosis of ASD. Overall intellectual ability was assessed using the Wechsler Abbreviated Scale of Intelligence (WASI) [22]. All participants or parents for those under 18 years of age gave informed written consent in accordance with ethics approval by the respective institutional ethics boards. For further details on demographics and exclusion criteria see Supplementary Methods 1.
MRI data acquisition
For image acquisition, we employed a contemporary MRI scanner operating at 3 T (Siemens Trio in Frankfurt and Los Angeles (UCLA), and a Signa GE Medical System in London (IoPPN)). High-resolution structural ADNI MPRAGE sequences were acquired with full head coverage. At the IoPPN, 166 contiguous slices (1.2 mm thickness, with 1.2 × 1.2 mm in-plane resolution) were acquired using a repetition time/echo time (TR/TE) of 7/2.9 ms (flip angle = 8°, FOV = 26 cm). At UCLA, 160 contiguous slices (1.2 mm thickness, with 1.2 × 1.2 mm in-plane resolution) were acquired using a TR/TE of 2300/2.9 ms (flip angle = 8°, FOV = 26 cm). In Frankfurt, 176 contiguous slices (1.0 mm thickness, with 1.0 × 1.0 mm in-plane resolution) were acquired using a TR/TE of 2300/2.2 ms (flip angle = 9°, FOV = 26 cm). Consistent image quality was ensured by a semi-automated quality control procedure at all sites, including a stringent pre-processing pipeline.
Cortical surface reconstruction using FreeSurfer
FreeSurfer v6.0.0 software (http://surfer.nmr.mgh.harvard.edu/) was used to derive models of the cortical surface for each T1-weighted image. These well validated and fully automated procedures have been extensively described elsewhere [23,24,25,26,27]. Measures of cortical thickness (CT) were computed as the closest distance from the gray-white matter boundary to the gray matter-cerebrospinal fluid boundary at each vertex on the tessellated surface [25]. Vertex-based estimates of SA were derived as outlined by Winkler et al. [28]. For each participant, we also computed mean CT across the entire brain, as well as total brain volume and total SA. To improve the ability to detect population changes, each parameter was smoothed using a 5-mm surface-based smoothing kernel. Details on excluded scans, quality assessments, and manual editing of surface models are described in the Supplementary Methods 3.
Statistical analyses
Statistical analyses were conducted using the SurfStat toolbox (http: //www.math.mcgill.ca/keith/surfstat/) for Matlab (R2017b; MathWorks). Between-group differences in age, full-scale IQ, ASD symptom severity, and total brain measures were assessed via analyses of variance (ANOVA) with group as categorical fixed-effect factor. Pair-wise differences between subgroups were examined post hoc using Scheffé test to correct for multiple comparisons in R (R3.5.2; see Supplementary Tables S1). Parameter estimates for vertex-based measures of CV, SA, and CT were estimated by regression of a general linear model (GLM) for each vertex i, with (1) group membership (i.e., having the microdeletion and/or having a diagnosis of ASD), gender, and site as categorical fixed-effect factors; (2) a 22q11.2DS-by-ASD interaction term; and (3) full-scale IQ, a linear and a quadratic age term, and the respective total brain measure (total brain volume for CV, total SA for SA, and mean CT for CT) as continuous covariates, so that
where εi is the residual error at vertex i. All included continuous covariates were mean centered across groups to improve interpretability of the coefficients. We examined between-group differences for the main effect of 22q11.2DS, estimated from the corresponding coefficient β1, as well as the main effect of ASD, estimated from the corresponding coefficient β2, normalized by the corresponding standard error respectively. We further examined the interaction effect between 22q11.2DS and ASD (coefficient β3) across parameters. Due to reasons of completeness, we also examined the main effects of ASD and 22q11.2DS in separate samples, comparing them to TD controls only (see Supplementary Figs. S1 & S2).
Corrections for multiple comparisons across the whole brain were performed using “random field theory” (RFT)-based cluster analysis for non-isotropic images using a cluster-based significance threshold of p < 0.05 (2-tailed) [29]. An RFT-based cluster correction was chosen over a permutation-based approach given its computational efficiency particularly for complex GLMs as utilized in the present study, and to make the results comparable to previous findings by our group [16, 30]. Uncorrected t maps and effect size images for the main effects are presented within the Supplementary Material (see Supplementary Figs. S3 & S4). As the 22q11.ASD and idiopathic ASD groups differed in symptom severity in the repetitive behavior domain of the ADI-R, we also performed the analysis covarying for the SRS Restricted Interests and Repetitive Behavior subscale. Further, given the large phenotypic heterogeneity typically associated with idiopathic ASD, we also tested for homogeneity of variances using the Levene’s test comparing the 22q11.ASD group to the idiopathic ASD groups (see Supplementary Fig. S5). Last, we reran the analysis with stricter matching for age and gender distribution (see Supplementary Table 2, and Supplementary Fig. S6).
Canonical correlation analysis (CCA)
In a second analysis step, we examined differences in the neural systems mediating autistic symptoms in 22q11.2DS individuals and individuals without the microdeletion (abbreviated as non22q11.2DS) within the dimensional framework of CCA. Here, ASD was not treated as a categorical fixed effect across groups, but as multivariate latent trait construct that is spanned by inter-individual differences in symptom profile. The general framework of CCA is well described elsewhere ([31]; see also Supplementary Methods 4). In the present study, we examined the relationship between neuroanatomical variability in CV, SA, and CT as predictors (Xn × p), and the five SRS subdomain scores in social awareness (SAW), social cognition (SCG), social communication (SCM), social motivation (SM), and restricted and repetitive behaviors (RRB) as clinical outcomes (Yn × q, where q = 5) (see Supplementary Fig. S7 for schematic illustration). To reduce the large number of vertex-based neuroanatomical features to a smaller subset of regions, we initially parcellated the cortex into a set of 34 cortical regions per hemisphere using the Desikan-Killiany cortical parcellation atlas [32], after correcting the data for linear and quadratic age effects, gender, site, full-scale IQ, and total brain measures across groups, which resulted in a set of n = 204 neuroanatomical features in total. As classical CCA assumes that the number of features is less than the number of samples (i.e., n ≤ max(p, q)), we employed different feature selection approaches to identify a set of neuroanatomical features that are most relevant to the prediction of the clinical SRS subdomain scores. The different feature selection approaches as well as the resulting subsets of neuroanatomical features are described in detail in the Supplementary Materials (see Supplementary Methods Section 5). We then based the analysis presented within the main manuscript on the feature selection approach that provided the largest subset of clinically relevant neuroanatomical features. This was a stepwise regression procedure with Akaike information criterion (AIC)-based model selection [33], which highlighted a set of p = 63 neuroanatomical features in total (see Supplementary Fig. S8).
CCA was initially employed as a backward model that derives canonical variates as functions of the observed data based on the matrix of coefficients or weights (i.e., \( \hat{x}={\boldsymbol{W}}^Tx \)). To estimate coefficients, we applied CCA to all individuals in our sample (i.e., n = 131), resulting in a set of i = 5 canonical variate pairs, which represent the relationship between neuroanatomical variability and inter-individual differences in autism symptom profiles. The RV-coefficient [34] was firstly examined to test for the overall statistical significance of the multivariate association between X and Y. Moreover, the significance of the full canonical model was evaluated using Wilks’ lambda ( [35]) and Pillai’s Trace [36]. To identify the number of significant canonical variate pairs for the subsequent comparison between groups, a dimension reduction analysis was performed. Here, we explored the percentage of variance explained by each neuroanatomical and clinical canonical variate using Rao’s F test [37] and Bartlett’s chi-squared test [38]. To enable a better interpretation of the data, canonical variates were sorted in descending order based on their level of statistical significance, and the fraction of total clinical variance explained by each canonical variate pair (i.e., canonical variate adequacies for clinical measures). The results of the CCA were visualized based on the canonical variate scores (\( \hat{X},\hat{Y} \)), and the matrix of structural correlations or “loadings” (Λ with element λ), which represent the association between the observed neuroanatomical and clinical data with their respective canonical variates (\( \mathrm{i}.\mathrm{e}.,{\lambda}_{p,i}=\mathrm{cor}\left({x}_p,{\hat{x}}_i\right) \)), where p denotes the canonical variate number.
To examine differences in the spatially distributed patterns of neuroanatomical variability underpinning autism symptom profiles in carriers and non-carriers of the 22q11.2 microdeletion, we utilized the model coefficients and canonical variate scores resulting from the backward model, to model the association between the observed data and the derived canonical variates within groups by applying a forward model of the general form \( x=\boldsymbol{\Lambda} \hat{x}+\varepsilon \), where ε denotes the residual error [39]. If estimated latent factors are uncorrelated, which is the case in CCA, it has been shown that
which equals the correlation (i.e., loading) between X and \( \hat{X} \) if variables are standardized [40]. The forward model was applied twice, after splitting the data into carriers and non-carriers of the 22q11.2 microdeletion, which resulted in a new set of group-specific clinical and neuroanatomical loadings. The Tucker’s congruence coefficient [41] was used to compare the loading matrices between groups, where a congruence coefficient in the range of [0.85–0.94] corresponds to a fair similarity, and a value > 0.95 indicates that the structure of two factors almost equal [42]. We also tested individual loading pairs for a statistical between-group difference using Fisher-Z transformation for independent correlation coefficients (p < 0.05, one-tailed) [43]. Last, we examined the reliability of our findings across different feature selection approaches (see Supplementary Methods 5 for further details). All statistical analyses were performed using RStudio Version 1.2 (https://www.rstudio.com/products/rstudio/) using the CCA results provided by yacca: Yet Another Canonical Correlation Analysis Package toolbox (https://CRAN.R-project.org/package=yacca).
Data availability
Further details on the data and utilized software are available upon request from the corresponding author. The full set of raw data is not currently publicly available due to ethical restrictions. However, a subset of the sample can be made available upon request.
Results
Participant demographics, diagnostic group, and global brain measures
There were no significant between-group differences in participants’ age. However, groups differed significantly in gender distribution (χ2(3) = 2.26, p = 0.016), with a lower percentage of females in the idiopathic ASD group relative to the other subgroups, and in full-scale IQ (F(3) = 21.01, p < 0.001), with TD controls scoring higher than all other groups, and individuals with 22q11.2DS having a lower IQ than individuals with idiopathic ASD. Further, we found a significant effect of group for total brain volume (F(3) = 10.56, p < 0.001) and total SA (F(3) = 12.41, p < 0.001), with both 22q11.2DS groups having a significantly lower total volume and area compared to both idiopathic ASD and TD controls (p < 0.05 for all pair-wise comparisons). Last, there was a significant effect of group for mean CT (F(3) = 3.74, p < 0.05) across the cortex, with idiopathic ASD individuals having a trend towards reduced CT compared to 22q11.nonASD individuals (p = 0.088), while no other pair-wise comparison was significant (see Table 1 and Supplementary Tables S1 for all pair-wise comparisons). We thus co-varied for gender, full-scale IQ, and respective total brain measure in all subsequent analyses.
Results of the categorical fixed-effects analyses
Main effect of 22q11.2DS on CV, SA, and CT
Significant neuroanatomical differences between 22q11.2 deletion carriers (i.e., 22q11.2DS with and without ASD) and non-carriers (i.e., idiopathic ASD and TD controls) were observed in several large clusters distributed across the cortex. More specifically, CV was increased in 22q11.2DS in the bilateral superior frontal cortex, the lateral and medial orbitofrontal cortex, the pre- and postcentral gyrus, the insula, and the supramarginal gyrus, with increases being driven by a commensurate increase in SA. Increased CV in 22q11.2DS was also observed in the left middle temporal gyrus, while increased SA was further observed in the left superior temporal gyrus and the left posterior cingulate cortex (PCC). In contrast, CV was decreased in 22q11.2DS in a large cluster centered on the bilateral medial occipital and temporal lobes, as well as in the bilateral anterior cingulate cortex, and the pre- and postcentral gyrus, accompanied by commensurate decreases in SA. Further decreases in SA were observed in the bilateral dorsal anterior cingulate area and inferior temporal gyri. Last, we identified increased CT in 22q11.2DS in some scattered regions, including the bilateral lateral occipital cortex, the right postcentral gyrus, and the left supramarginal gyrus, whereas decreases in CT were observed in the bilateral superior temporal lobes, the parahippocampal gyri, and the posterior cingulate cortex (see Fig. 1, Supplementary Fig. S3, and Supplementary Table S3). Effect size images for the main effect of 22q11.2DS are shown in Supplementary Fig. S4. A similar pattern of effects was also obtained when comparing the 22q11.2DS individuals to TD controls only (see Supplementary Fig. S1), and when strictly matching for age and gender (see Supplementary Fig. S6).

Categorical results for main effect of 22q11.2DS, main effect of ASD, and 22q11.2DS-by-ASD interaction effect.a Significant differences in cortical volume (CV), b surface area (SA), and c cortical thickness (CT) for the main effect of 22q11.2DS (left panel), the main effect of ASD (middle panel), and for the 22q11.2DS-by-ASD interaction (right panel). Displayed are the random field theory (RFT)-based cluster corrected (p < 0.05, 2-tailed) difference maps following multiple comparisons, where increased parameter estimates in 22q11.2DS (or ASD) are marked in red to yellow, and decreased parameters are marked in blue to cyan
Main effect of ASD on CV, SA, and CT
For the main effect of ASD, we established that individuals with ASD symptomatology (i.e., individuals with idiopathic ASD and 22q11.ASD) were neuroanatomically distinct from those without (i.e., compared to TD controls and 22q11.nonASD), with significantly increased CV in the left insula and left superior temporal gyrus, accompanied by a more widespread increase in SA, also spanning the fusiform, parahippocampal, lingual, and supramarginal gyri. CV was further increased in the right inferior parietal cortex in ASD. In contrast, decreases in CV in ASD were observed in the left entorhinal cortex, accompanied by a commensurate decrease in SA that was more pronounced and also implicated the left fusiform gyrus. For measures of CT, individuals with ASD showed significant increases in the right isthmus cingulate cortex and the right superior temporal gyrus (see Fig. 1, Supplementary Fig. S3, and Supplementary Table S4). Effect size images for the main effect of ASD are shown in Supplementary Fig. S4. A similar pattern of effects was also obtained when comparing the idiopathic ASD individuals to TD controls only (see Supplementary Fig. S2), and when strictly matching for age and gender (see Supplementary Fig. S6).
Significant interactions between 22q11.2DS and ASD
In addition to the main effects, we observed significant interactions between 22q11.2DS and ASD for measures of CV and SA. These were located in the left dorsolateral prefrontal cortex (DLPFC) for both, CV and SA, as well as in the right precentral gyrus for CV only, and in the left PCC for SA only (see Fig. 1, Supplementary Fig. S3, and Supplementary Table S5). Effect size images for the 22q11.2DS-by-ASD interaction are shown in Supplementary Fig. S4. In significant clusters, ASD was associated with increased CV and/or SA in 22q11.2DS (i.e., 22q11.ASD > 22q11.nonASD), but reduced CV and/or SA in individuals without the microdeletion (i.e., idiopathic ASD < TD controls). In the DLPFC and PCC, individuals with 22q11.nonASD were the most affected on the neuroanatomical level (i.e., had the most reduced CV and/or SA relative to all other groups), while both ASD groups were comparable in terms of their mean CV and/or SA (22q11.nonASD < 22q11.ASD = ASD ≤ TD controls). In the precentral cluster exclusively, 22q11.ASD individuals had the largest mean CV compared to all other groups, with the mean of 22q11.nonASD individuals being between idiopathic ASD and TD controls (for boxplots see Supplementary Fig. S9). As the 22q11.ASD and idiopathic ASD groups differed in symptom severity in the repetitive behavior domain of the ADI-R, we also performed the analysis covarying for the SRS Restricted Interests and Repetitive Behavior subscale. However, the patterns of significant 22q11.2DS-by-ASD interactions remained unchanged overall (see Supplementary Fig. S10). In regions with significant 22q11.2DS-by-ASD interactions, there were also no significant differences in variance between the idiopathic ASD individuals and the 22q11.ASD group (see Supplementary Fig. S5), and very little effect of age and gender (see Supplementary Fig. S6).
Results of the CCA
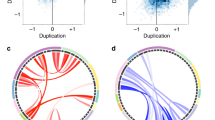
Initially, CCA was performed across all individuals within our sample (i.e., carriers and non-carriers of the 22q11.2 microdeletion). Here, we observed a significant multivariate association between the 63 regional measures of brain anatomy highlighted to be of importance by the stepwise variable selection approach, and the five symptom domains of the SRS (RVcoef = 0.082, p < 0.001; see Supplementary Fig. S11 for distribution of SRS total and subdomain scores across groups). Based on the number of clinical predictors (q = 5), the CCA yielded five canonical variate pairs with the canonical correlations of 0.822, 0.772, 0.764, 0.724, and 0.653 for each successive canonical variate pair, respectively (see Fig. 2a). Collectively, the full model including all canonical variates was statistically significant using Wilks’ λ = 0.015 (F(315,319) = 1.35, p < 0.01) and Pillai’s trace = 2.81 (F(315,335) = 1.36, p < 0.01). As Wilks’ λ indicates the variance unaccounted for by the model, the R-square type (ρ2) effect size of the model was 0.985 (i.e., 1-λ), which means that the full model explained about 98.5% of the variance shared between measures of neuroanatomy and clinical symptom profile. Moreover, the total variance in SRS scores that could be explained by neuroanatomical variation was 57.47%, which only the first two neuroanatomical canonical variates contributed to significantly (20.82% and 21.59%, respectively; see Fig. 2a). Out of all canonical variates, the 1st (Bartlett’s χ2(315) = 402.24, p < 0.001) and the 2nd (Bartlett’s χ2(248) = 294.58, p < 0.05) were also statistically significant, with the 1st clinical canonical variate explaining a total of 30.80%, and the 2nd clinical canonical variate explaining a total of 36.19% of variability within the set of clinical variables on their own (clinical canonical variate adequacy, see Fig. 2b). Thus, given the ρ2 effects for each canonical variate pair, only the first two pairs were considered noteworthy in the context of the present study. Both clinical canonical variates, and the 2nd canonical variate in particular, also provided a good discrimination between individuals with and without ASD (see Fig. 2c, d). Figure 2 e and f show the canonical loadings (λC) for each neuroanatomical predictor on the cortical surface, which highlights the set of brain regions maximally correlated with the 2nd (e) and 1st (f) neuroanatomical canonical variate. As expected, high positive loadings (i.e., > 0.25) were observed in many regions of the social brain including the right medial orbitofrontal lobe (CT, λC2 = 0.28), the right rostral middle frontal gyrus (CT, λC2 = 0.30), the left insula (CV, λC2 = 0.34), and the left transverse temporal lobe (CV, λC2 = 0.28). High negative loadings were observed in the left precuneus (CT, λC1 = − 0.27), the bilateral superior parietal lobes (CT, right: λC2 = − 0.28; CV, left: λC2 = − 0.28), and the left temporal pole (SA, λC1 = − 0.30; CV, λC1 = − 0.38).

Results of the CCA across all individuals within our sample.a Canonical correlations (subplot) and canonical variates sorted in descending order based on their canonical correlations, and based on the percentage of clinical variance explained; b clinical canonical loadings depicting correlations between each of the five clinical canonical variates (V1-V5) and the five SRS subdomain scores in social awareness (SAW), social cognition (SCG), social communication (SCM), social motivation (SM), and restricted and repetitive behaviors (RRB). Canonical variates are sorted in descending order based on the percentage of explained clinical variance, as indicated in shades of green; c and d scatter plots depicting individual observations based on their scores on the second (c) and first (d) canonical variate, which explained the largest percentage of clinical variance. Data points are colored (ASD: yes vs. no) and shaped (22q11.2DS: yes vs. no) by group membership and sized by the individual’s total SRS score; e and f display canonical loadings of each neuroanatomical feature (i.e., cortical volume (CV), surface area (SA), and cortical thickness (CT)) on the second (e) and first (f) canonical variate
After fitting the CCA in the total sample, we utilized the resulting canonical variate scores to derive group-specific factor loadings (clinical and neuroanatomical) for carriers and non-carriers of the 22q11.2 microdeletion, which were subsequently compared between groups. Overall, there was a high degree of similarity in the clinical canonical variate structure observed carriers and non-carriers, with Tucker’s congruence coefficients for the 1st and 2nd clinical covariates exceeding a value of 0.99 (see Fig. 3a, b). However, when examining the neuroanatomical underpinnings of these clinical variates between groups, we found that there was a low degree of neuroanatomical similarity overall (mean Tucker’s congruence coefficient across canonical variates = 0.336), and low levels of congruence for canonical variate 1 (Tucker’s congruence coefficient = 0.393) and variate 2 (Tucker’s congruence coefficient = 0.404). We also observed significant between-group differences in individual neuroanatomical loading pairs, which are displayed in Fig. 3c–f. More specifically, for canonical variate 2, which is the variate that explained the largest percentage of clinical variability (see Fig. 3c, d), we observed a significant difference in the loadings of the right rostral middle frontal cortex (CT; Fisher’s Z = 1.68, p < 0.05), the left precuneus (CT; Fisher’s Z = 2.01, p < 0.05), the left paracentral gyrus (SA; Fisher’s Z = 2.48, p < 0.01), the left medial orbitofrontal cortex (CT; Fisher’s Z = 1.90, p < 0.05), the left fusiform gyrus (CT; Fisher’s Z = 2.80, p < 0.01), and the right temporal pole (CT; Fisher’s Z = 1.78, p < 0.05). For canonical variate 1, the variate to explain the second most variability (Fig. 3e, f), individuals with 22q11.2DS had significantly higher neuroanatomical loadings in the left insula (CT; Fisher’s Z = 1.99, p < 0.05), the left cuneus (CT; Fisher’s Z = 1.95, p < 0.05), the right lateral orbitofrontal cortex (CT; Fisher’s Z = 1.76, p < 0.05), the left pars triangularis (SA; Fisher’s Z = 1.94, p < 0.05), and in the right rostral anterior cingulate cortex (SA; Fisher’s Z = 2.84, p < 0.01). Individuals with 22q11.2DS further had a more negative loading between the 1st canonical variate and the volume of the medial orbitofrontal cortex compared to non22q11.2DS individuals (CV; Fisher’s Z = 1.74, p < 0.05). Thus, despite the high degree of similarity in the clinical composition of autism symptoms across groups, we observed that inter-individual differences in clinical symptom profiles were underpinned by different neuroanatomical substrates in carriers and non-carriers of the 22q11.2 microdeletion.

Comparison of factor loadings between carriers and non-carriers of the 22q11.2 microdeletion. Figures (a) and (b) display clinical canonical loadings plot depicting correlations between each of the five clinical canonical variates and each of the five SRS subdomain scores in social awareness (SAW), social cognition (SCG), social communication (SCM), social motivation (SM), and restricted and repetitive behaviors (RRB) within a all non22q11.2DS individuals (i.e., idiopathic ASD and TD controls) and within b all 22q11.2DS individuals (i.e., 22q11.ASD and 22q11.nonASD). Canonical variates are sorted in descending order based on the percentage of explained clinical variance as indicated in shades of green; b and c canonical loadings of each neuroanatomical predictor (i.e., cortical volume (CV), surface area (SA), and cortical thickness (CT)) on canonical variate 2 within c all non22q11.2DS individuals and d all 22q11.2DS individuals; e and f canonical loadings of each neuroanatomical predictor (i.e., CV, SA, and CT) on canonical variate 1 within e all non22q11.2DS individuals and f all 22q11.2DS individuals. Brain regions with a significant between-group difference in brain loadings between non22q11.2DS and 22q11.2DS individuals are indicated with an asterisk
Discussion
This study aimed to determine whether ASD symptomatology in individuals with 22q11.2DS is underpinned by similar neuroanatomical substrates that mediate ASD symptoms in non22q11.2DS. We utilized both a categorical and a dimensional approach to (1) establish the extent to which the neuroanatomy of 22q11.DS is modulated by (i.e., significantly interacts with) having a diagnosis of ASD and to (2) compare the patterns of neuroanatomical variability that mediate the complex (i.e., multi-dimensional) clinical phenotype of ASD across disorders. Within the categorical framework, we initially established that it is possible to separate the effect of 22q11.2DS from the effect of ASD on the level of neuroanatomy as characterized by regional variability in CV, SA, and CT. Notably, we also observed significant 22q11.2DS-by-ASD interactions suggesting that, 22q11.2DS individuals who also have ASD, may represent a subgroup that is neuroanatomically distinct from 22q11.nonASD individuals, and from individuals without the microdeletion. These results were confirmed by the dimensional approach, which further highlighted that, while the complex clinical phenotype of ASD might be reduced to the same underlying (i.e., latent trait) construct across disorders, the neuroanatomical substrates associated with variation in SRS scores were different between carriers and non-carriers of the 22q11.2 microdeletion.
Neuroanatomical differences associated with the 22q11.2 microdeletion are well documented in the literature and include spatially distributed differences in CV, SA, and CT in parieto-temporal and cingulate regions, as well as the bilateral insula, parahippocampal gyrus, and DLPFC [15, 16, 44]. Here, we examined the neuroanatomy of 22q11.2DS initially within a 2 × 2 factorial design that included (1) 22q11.2DS individuals with and without ASD, (2) individuals with idiopathic ASD, and (3) TD controls. While it remains a topic of debate whether ASD should be considered a categorical “fixed-effect” variable, this design allowed us to identify a set of brain regions where neuroanatomical variability in CV, SA, and CT were uniquely attributable to either the microdeletion or having ASD. By examining the main effects of groups, we were able to demonstrate that it is possible to separate the effect of 22q11.2DS from the main effect of ASD on the neuroanatomical level based on patterns of neuroanatomical differences that included extensive and spatially distributed neuroanatomical differences across all four lobes of the cortex for the effect of 22q11.2DS, and more localized atypicalities in predominantly temporal regions for the main effect of ASD. More specifically, having ASD was linked to neuroanatomical abnormalities in the superior temporal gyrus (STG), DLPFC, insula, fusiform gyrus, parahippocampal gyrus, isthmus cingulate cortex, and entorhinal cortex. Many of these brain regions have previously been reported to be integral parts of the neural systems that mediate autistic symptoms and traits. For example, the STG has been implicated in both language and social cognition in ASD [45,46,47]. Further, the parahippocampal gyrus, isthmus cingulate cortex, and entorhinal cortex, are all part of the limbic system, which has been associated with impaired socioemotional and face processing in ASD [48,49,50,51]. In addition, the DLPFC is primarily associated with executive functioning, an aspect of impairment in ASD [52], and the insula has been linked to abnormalities of emotional/affective sensory functions in ASD [53]. We also examined the effect of ASD in a subsample of non-carriers of the microdeletion, i.e., in idiopathic ASD individuals relative to TD controls, with similar results. Overall, there is a strong spatial correspondence between the set of brain regions we identified as being neuroanatomically different in individuals with ASD symptomatology, including those with 22q11.2DS, and the set of brain regions mediating autistic symptoms in idiopathic ASD.
The results reported in the present study also extend the findings of a previous neuroimaging study by our group, which was conducted in a subset of this sample, where we compared 22q11.2DS individuals with ASD symptomatology to 22q11.2DS without ASD, and to TD controls [16]. Notably, the main effect of 22q11.2DS was associated predominantly with significant reductions in SA, which have been shown to contribute more significantly to commensurate differences in CV than measures of CT [54], and is also in line with previous findings in 22q11.2DS [15, 44, 55]. However, as our comparison group for the main effect of 22q11.2DS in the present study consisted of both TD controls and individuals with idiopathic ASD, the sum of squares associated with each model term was partitioned differently across studies with regard to their main effects allocation (22q11.2DS while covarying for ASD and vice versa). We therefore also examined the main effect of 22q11.2DS compared to TD controls only, which corresponded by large with our previous findings in this sample [16]. Our findings within the 22q11.2DS sample are also in agreement with a recent large scale study conducted by the 22q11.2DS ENIGMA consortium, where neuroanatomical differences in a similar set of brain regions were reported [55]. Thus, there is strong evidence to suggest that 22q11.2DS is associated with significant structural brain abnormalities, which in turn may impact on the various clinical phenotypes associated with the syndrome. Our findings indicate that 22q11.2DS and ASD have separable neuroanatomical underpinnings, but further, it suggests that, given the increased prevalence of ASD in 22q11.2DS relative to the normative population, the effects of 22q11.2DS on brain development might impact on the risk of ASD, but in itself are not sufficient to cause the condition.
Within the factorial design, it was also possible to determine to what extent the neuroanatomy of ASD is significantly modulated by 22q11.2DS (i.e., differs from individuals with 22q11.2DS without ASD and from individuals with idiopathic ASD). For example, we observed significant 22q11.2DS-by-ASD interactions in the left DLPFC and the left PCC. In these brain regions, individuals with 22q11.2DS without ASD symptomatology were the most affected, followed by 22q11.2DS individuals with ASD, individuals with idiopathic ASD, and TD controls in terms of affection status. While these interactions are complex and difficult to interpret, it seems that ASD in 22q11.2DS is not simply due to an exacerbation of the 22q11.2DS brain phenotype per se, i.e., more severe behavioral impairments associated with more pronounced neuroanatomical atypicalities. Instead, ASD symptoms in 22q11.2DS seem to be associated with a pattern of neuroanatomical differences that cannot be explained by either the microdeletion or a diagnosis of ASD alone. We also excluded the possibility that these significant interactions are driven by differences in variance between both ASD groups, as individuals with idiopathic ASD are known to be largely heterogeneous both in terms of etiology as well as clinical phenotype in comparison to the etiologically more homogeneous phenotype associated with 22q11.2DS. This implies that individuals with 22q11.2DS and ASD may constitute a distinct neuroanatomical subgroup that is neuroanatomically different from 22q11.2DS individuals without ASD, and from individuals with idiopathic ASD. Thus, although 22q11.2DS individuals with ASD share the same clinical phenotype as idiopathic ASD individuals, the neuroanatomical underpinnings appear to differ between groups, and 22q11.2DS in itself may not be sufficient to cause ASD.
While the factorial design allowed us to disentangle the effect of ASD from the main effect of 22q11.2DS, and to explore their interaction, there has been some debate whether ASD should be considered a uniform clinical construct that is common (i.e., invariant) across idiopathic and “syndromic” forms of ASD, and can hence be encoded as a categorical main effect across groups. For example, while all individuals with ASD met cutoffs in the social and communication domains of the ADI-R, not all individuals with 22q11.2DS met cutoffs in the repetitive domain (see [16] for discussion). Although our results remain stable when covarying for repetitive symptoms, it remains unclear whether the distinct neuroanatomical phenotype of 22q11.2DS with ASD is the cause or the consequence of a potentially unique clinical profile (see [56, 57]). In a second analysis step, we also employed a dimensional approach using CCA, which allowed us to treat ASD as a continuous clinical construct spanned by multiple symptom domains rather than the binary presence or absence of a diagnosis of ASD, and to examine the multivariate association between inter-individual clinical profiles and neuroanatomical variability between 22q11.2 deletion carriers and non-carriers. Here, we based our characterization of the clinical ASD phenotype on the SRS subscales, which are particularly suited to assess autistic symptoms along a continuum, with individuals with idiopathic ASD and 22q11.2DS individuals with ASD showing a very similar profile across subscales. Our findings imply that while it is possible to reduce the complex clinical ASD phenotype to two dominant latent-trait factors with comparable clinical factor structures in both carriers and non-carriers, the underlying set of brain regions that explained maximal clinical variance differed between groups. Thus, both approaches converge in suggesting that ASD symptomatology may be mediated by different neuroanatomical substrates in individuals with and without the 22q11.2 microdeletion, even when taking inter-individual variability in clinical ASD phenotypes into account.
Limitations
Our results should be interpreted in the light of several methodological limitations of which the small sample size is the most pressing one. Although we employed a multicenter design to overcome single-site recruitment limitations, our sample size of ~ 40 individuals per group is relatively small compared to other studies, which limits the generalizability of the results and the strength of the conclusions. Even though the effects for both of our between-group comparisons are comparable to previous reports in larger samples, both in terms of size and spatial distribution, larger samples are required to replicate our findings in the future, and to provide a more robust characterization of the clinical and neuroanatomical phenotype of ASD across disorders. Moreover, the generalizability of our findings is limited by the multi-side nature of our study, which resulted in our groups being recruited at three different sites. FreeSurfer derived measures of surface anatomy have, however, been shown to be highly reliable across scanner platforms and field strengths, when MRI instrument and data processing factors are controlled for [58]. In our study, all surface reconstructions were also subjected to the same stringent type of quality assessment and pre-processing pipeline, and inter-site effects were accounted for in the statistical model. Moreover, due to the parametric nature, the dimensional approach is less biased by effects of categorical variables such as site and gender. We did also not directly test our idiopathic ASD group for copy number variations (CNVs), such as 22q11.2DS. However, we did perform extensive medical screening for somatic features associated with the 22q11.2 microdeletion across all groups (i.e., heart, palatal, and characteristic facial abnormalities), which were not observed in any of the non22q11.2DS individuals. Also, given the prevalence of the microdeletion (1 in 4000), there is a very low likelihood for the presence of 22q11.2DS in the non22q11.2DS sample. However, future studies might consider testing all individuals for CNVs.
To address the differing gender distribution between the idiopathic ASD individuals and the other groups, we controlled for gender in the categorical fixed-effects analysis. Notably, while there is a male-biased prevalence with an estimated gender distribution of 4:1 (males to females) in idiopathic ASD [59], within 22q11.2 deletion carriers the distribution of males and females with ASD is estimated to be roughly equal [60]. Thus, the gender distribution in our sample reflects the gender distribution in the respective populations, which adds to the generalizability of our results. Furthermore, we included individuals with a relatively wide age range (i.e., from 6–25 years). Even though groups were matched in terms of their respective mean, and we corrected for linear and quadratic age effects, the nature and severity of autistic symptoms and their related neuroanatomical variability might vary across the lifespan. Hence, it will be crucial in the future to examine the multivariate correlation between the clinical ASD phenotype and neuroanatomical variability in more well defined age groups, and to characterize their association across development. Last, it is important to note that our CCA analysis was restricted to examining the multivariate association between the neuroanatomical and clinical phenotype of ASD as measured by the SRS. However, although the SRS is a well validated questionnaire that is well suited as a screening tool capturing the severity of autistic symptoms along a continuum [61], it does not by any means provide a comprehensive characterization of the complex ASD phenotype. It will therefore be crucial in the future to examine the multivariate correlation between the clinical ASD phenotype and neuroanatomical variability in larger more well defined groups using additional diagnostic measures to replicate our findings, and to better characterize the complex clinical and neurobiological phenotype across disorders. Future research may also benefit from extending the multi-dimensional phenotypic representation to include common comorbidities, such as ADHD, in the clinical phenotypic characterization, which may also provide important novel insights into the underlying mechanisms that underpin the differences we observe on the phenotypic level.
Conclusions
Our findings indicate that the neuroanatomical correlates of ASD symptomatology in individuals with 22q11.2DS diverge from those in idiopathic ASD.
Availability of data and materials
Further details on the data and utilized software are available upon request from the corresponding author. The full set of raw data is not currently publicly available due to ethical restrictions. However, a subset of the sample can be made available upon request.
Abbreviations
- ASD:
-
Autism spectrum disorder
- 22q11.2DS:
-
22q11.2 deletion syndrome
- CV:
-
Cortical volume
- SA:
-
Surface area
- TD:
-
Typically developing (controls)
- CCA:
-
Canonical correlation analysis
- ADHD:
-
Attention deficit hyperactivity disorder
- 22q11.ASD:
-
Individuals with 22q11.2DS and ASD
- 22q11.nonASD:
-
Individuals with 22q11.2DS but no ASD
- Non22q11.2DS:
-
Individuals without the microdeletion (i.e., idiopathic ASD and TD controls)
- ADI-R:
-
Autism Diagnostic Interview-Revised
- ADOS:
-
Autism Diagnostic Observation Schedule
- SRS:
-
Social Responsiveness Scale
- WASI:
-
Wechsler Abbreviated Scale of Intelligence
- CT:
-
Cortical thickness
- SAW:
-
Social Awareness subscale of the SRS
- SCG:
-
Social Cognition subscale of the SRS
- SCM:
-
Social Communication subscale of the SRS
- SM:
-
Social Motivation subscale of the SRS
- RBB:
-
Restricted and Repetitive Behaviors subscale of the SRS
- PCC:
-
Posterior cingulate cortex
- DLPFC:
-
Dorsolateral prefrontal cortex
- STG:
-
Superior temporal gyrus
- CNV:
-
Copy number variation
References
Scambler PJ, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, et al. Velo-cardio-facial syndrome associated with chromosome-22 deletions encompassing the DiGeorge locus. Lancet. 1992;339(8802):1138–9.
Tezenas Du Montcel S, Mendizabai H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33(8):719.
Swillen A, McDonald-McGinn D. Developmental trajectories in 22q11.2 deletion syndrome. Am J Med Genet C. 2015;169(2):172–81.
Scambler PJ. The 22q11 deletion syndromes. Hum Mol Genet. 2000;9(16):2421–6.
Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79(4):348–51.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, et al. 22q11.2 deletion syndrome. Nature Reviews Disease Primers. 2015;1.
Schneider M, Debbane M, Bassett AS, Chow EWC, Fung WLA, van den Bree MBM, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiat. 2014;171(6):627–39.
Fiksinski AM, Breetvelt EJ, Duijff SN, Bassett AS, Kahn RS, Vorstman JAS. Autism spectrum and psychosis risk in the 22q11.2 deletion syndrome. Findings from a prospective longitudinal study. Schizophr Res. 2017;188:59–62.
Jalbrzikowski M, Hamzah Ahmed K, Patel A, Jonas R, Kushan L, Chow C, et al. Categorical versus dimensional approaches to autism-associated intermediate phenotypes in 22q11.2 microdeletion syndrome. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2(1):53–65.
Antshel KM, Aneja A, Strunge L, Peebles J, Fremont WP, Stallone K, et al. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion). J Autism Dev Disord. 2007;37(9):1776–86.
Gothelf D. Measuring prodromal symptoms in youth with developmental disabilities: a lesson from 22q11 deletion syndrome. J Am Acad Child Adolesc Psychiatr. 2014;53(9):945–7.
Shapiro DI, Cubells JF, Ousley OY, Rockers K, Walker EF. Prodromal symptoms in adolescents with 22q11.2 deletion syndrome and schizotypal personality disorder. Schizophr Res. 2011;129(1):20–8.
Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome (VCFS). Schizophr Res. 2000;41(1):29.
Schreiner MJ, Lazaro MT, Jalbrzikowski M, Bearden CE. Converging levels of analysis on a genomic hotspot for psychosis: Insights from 22q11.2 Deletion Syndrome. Neuropharmacology. 2013;68:157–73.
Jalbrzikowski M, Jonas R, Senturk D, Patel A, Chow C, Green MF, et al. Structural abnormalities in cortical volume, thickness, and surface area in 22q11.2 microdeletion syndrome: relationship with psychotic symptoms. NeuroImage Clinical. 2013;3:405–15.
Gudbrandsen M, Daly E, Murphy CM, Wichers RH, Stoencheva V, Perry E, et al. The neuroanatomy of autism spectrum disorder symptomatology in 22q11.2 deletion syndrome. Cereb Cortex. 2019;29(8):3655–65.
Lord C, Rutter M, Lecouteur A. Autism Diagnostic Interview-Revised - a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–85.
Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, et al. The Autism Diagnostic Observation Schedule-Generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30(3):205–23.
Gotham K, Pickles A, Lord C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J Autism Dev Disord. 2009;39(5):693–705.
Bölte S, Constantino JN, Gruber CP, Poustka F. Skala zur Erfassung sozialer Reaktivität: SRS; dimensionale Autismus-Diagnostik; deutsche Fassung der Social responsiveness scale (SRS) (Manual; von J. N. Constantino und C. P. Gruber, Ed.). Huber; 2008.
Constantino JN, Gruber CP. Social Responsiveness Scale Second Edition (SRS-2): Manual: Western Psychological Services (WPS); 2012.
Wechsler D. Wechsler abbreviated scale of intelligence (WASI) manual. San Antonio: TX: Psychological Corporation; 1999.
Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis - I. Segmentation and surface reconstruction. Neuroimage. 1999;9(2):179–94.
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97(20):11050–5.
Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis - II: inflation, flattening, and a surface-based coordinate system. Neuroimage. 1999;9(2):195–207.
Jovicich J, Czanner S, Greve D, Haley E, van der Kouwe A, Gollub R, et al. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. Neuroimage. 2006;30(2):436–43.
Segonne F, Dale AM, Busa E, Glessner M, Salat D, Hahn HK, et al. A hybrid approach to the skull stripping problem in MRI. Neuroimage. 2004;22(3):1060–75.
Winkler AM, Sabuncu MR, Yeo BTT, Fischl B, Greve DN, Kochunov P, et al. Measuring and comparing brain cortical surface area and other areal quantities. Neuroimage. 2012;61(4):1428–43.
Worsley KJ, Andermann M, Koulis T, MacDonald D, Evans AC. Detecting changes in nonisotropic images. Hum Brain Mapp. 1999;8(2-3):98–101.
Ecker C, Shahidiani A, Feng Y, Daly E, Murphy C, D'Almeida V, et al. The effect of age, diagnosis, and their interaction on vertex-based measures of cortical thickness and surface area in autism spectrum disorder. J Neural Transm. 2014;121(9):1157–70.
Wang H-T, Smallwood, J., Mourao-Miranda, J., Xia, C.H., Satterthwaite, T.D., Bassett, D.S., & Bzdok, D. Finding the needle in high-dimensional haystack: a tutorial on canonical correlation analysis. https://arxivorg/pdf/181202598pdf. 2018.
Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–80.
Harrell F. Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis. New York: Springer-Verlag New York; 2013.
Robert P, Escoufier Y. A unifying tool for linear multivariate statistical methods: the RV-coefficient. Journal of the Royal Statistical Society: Series C (Applied Statistics). 1976;25(3):257–65.
Henson RK. Demystifying parametric analyses: illustrating canonical correlation analysis as the multivariate general linear model. Multiple Linear Regression Viewpoints. 2000;26(1):11–9.
Pillai KCS. Some new test criteria in multivariate analysis. Annals of Mathematical Statistics. 1955;26(1):117–21.
Mardia KV, Kent JT, Bibby JM. Multivariate Analysis. London: Academic Press; 1979.
Bartlett MS, Fowler RH. Properties of sufficiency and statistical tests. Proceedings of the Royal Society of London Series A - Mathematical and Physical Sciences. 1937;160(901):268–82.
Parra LC, Spence CD, Gerson AD, Sajda P. Recipes for the linear analysis of EEG. Neuroimage. 2005;28(2):326–41.
Haufe S, Meinecke F, Görgen K, Dähne S, Haynes J-D, Blankertz B, et al. On the interpretation of weight vectors of linear models in multivariate neuroimaging. Neuroimage. 2014;87:96–110.
Tucker LR, Koopman RF, Linn RL. Evaluation of factor analytic research procedures by means of simulated correlation matrices. Psychometrika. 1969;34(4):421–59.
Lorenzo-Seva U, Berge J. Tucker’s congruence coefficient as a meaningful index of factor similarity. Methodology: European Journal of Research Methods for The Behavioral and Social Sciences. 2006;2:57–64.
Fisher RA. Frequency distribution of the values of the correlation coefficient in samples from an indefinitely large population. Biometrika. 1915;10(4):507–21.
Schmitt JE, Vandekar S, Yi J, Calkins ME, Ruparel K, Roalf DR, et al. Aberrant cortical morphometry in the 22q11.2 deletion syndrome. Biol Psychiatry. 2015;78(2):135–43.
Bigler ED, Mortensen S, Neeley ES, Ozonoff S, Krasny L, Johnson M, et al. Superior temporal gyrus, language function, and autism. Developmental Neuropsychology. 2007;31(2):217–38.
Jou RJ, Minshew NJ, Keshavan MS, Vitale MP, Hardan AY. Enlarged right superior temporal gyrus in children and adolescents with autism. Brain research. 2010;1360:205–12.
Stigler KA, McDougle CJ. Chapter 3.1 - Structural and functional MRI studies of autism spectrum disorders. In: Buxbaum JD, Hof PR, editors. The neuroscience of autism spectrum disorders. San Diego: Academic Press; 2013. p. 251–66.
Schumann CM, Hamstra J, Goodlin-Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, et al. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. The Journal of Neuroscience. 2004;24(28):6392–401.
Palmen S, Pol HEH, Kemner C, Schnack HG, Sitskoorn MM, Appels MCM, et al. Brain anatomy in non-affected parents of autistic probands: a MRI study. Psychological Medicine. 2005;35(10):1411–20.
Nicolson R, DeVito TJ, Vidal CN, Sui Y, Hayashi KM, Drost DJ, Williamson PC, Rajakumar N, Toga AW, Thompson PM. Detection and mapping of hippocampal abnormalities in autism. Psychiatry Res. 2006;148(1):11–21.
Nair A, Carper RA, Abbott AE, Chen CP, Solders S, Nakutin S, et al. Regional specificity of aberrant thalamocortical connectivity in autism. Hum Brain Mapp. 2015;36(11):4497–511.
Demetriou EA, MM DM, Guastella AJ. Executive function in autism spectrum disorder: history, theoretical models, empirical findings, and potential as an endophenotype. Front Psychiatry. 2019;10:753.
Ha S, Sohn IJ, Kim N, Sim HJ, Cheon KA. Characteristics of brains in autism spectrum disorder: structure, function and connectivity across the lifespan. Exp Neurobiol. 2015;24(4):273–84.
Ecker C, Ginestet C, Feng Y, Johnston P, Lombardo MV, Lai MC. Brain surface anatomy in adults with autism. JAMA Psychiatry. 2013;70.
Sun D, Ching CRK, Lin A, Forsyth JK, Kushan L, Vajdi A, et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry. 2018. https://doi.org/10.1038/s41380-018-0078-5.
Bruining H, de Sonneville L, Swaab H, de Jonge M, Kas M, van Engeland H, et al. Dissecting the clinical heterogeneity of autism spectrum disorders through defined genotypes. PLoS One. 2010;5(5):7.
Bruining H, Eijkemans MJC, Kas MJH, Curran SR, Vorstman JAS, Bolton PF. Behavioral signatures related to genetic disorders in autism. Mol Autism. 2014;5.
Han X, Jovicich J, Salat D, van der Kouwe A, Quinn B, Czanner S, et al. Reliability of MRI-derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. Neuroimage. 2006;32(1):180–94.
Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013;26(2):146–53.
Vorstman JAS, Morcus MEJ, Duijff SN, Klaassen PWJ, Heineman-de Boer JA, Beemer FA, et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatr. 2006;45(9):1104–13.
Bolte S, Poustka F, Constantino JN. Assessing autistic traits: cross-cultural validation of the Social Responsiveness Scale (SRS). Autism Res. 2008;1(6):354–63.
Acknowledgements
Prof. Ecker gratefully acknowledges support by grant EC480/1-1 from the DFG under the Heisenberg Programme. Prof. Murphy acknowledges support by grants from the Mental Health Biomedical Research Centre (BRC) and the Innovative Medicines Initiative (IMI). Furthermore, we would like to thank the National Institute for Health Research, Biomedical Research Centre for Mental Health, and the Dr. Mortimer and Theresa Sackler Foundation.
The authors further gratefully acknowledge the suggestion of one of the reviewers, who drew our attention to applying the CCA forward model within groups, to derive the set of group-specific loading matrices that were subsequently examined between groups.
Funding
This work was supported by the German Research Foundation (DFG) under grant agreement EC480/2-1 awarded to Prof. Ecker, and the National Institute of Mental Health (NIMH) with grants R01MH085953, R01MH085953-S1, and R21MH116473-01A1 to Prof. Bearden.
Author information
Authors and Affiliations
Contributions
MG, AB, CM, and LK completed recruitment and data collection. ED, CB, and VS assisted in data collection, data entry, and sample preparation. AB and MG analyzed data with support from CE. CM, CEB, DGM, and MC contributed to the revisions of the manuscript. MG, AB, and CE wrote the manuscript. All authors have provided critical input into the revisions of the manuscript and approved final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants, and accompanying parents for those under 18 years of age, gave informed written consent in accordance with ethics approval by the Ethics Committee of the Faculty of Medicine of the Goethe University Frankfurt, the National Research Ethics Service (NRES) Committee South Central (study reference 12/SC/0576), or the UCLA Institutional Review Board (IRB).
Consent for publication
Not applicable
Competing interests
Prof. Murphy sat on an advisory board for Roche and received an honorarium.
No other authors reported any financial interests or conflicts of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Methods. 1.
Participant Demographics and Exclusion Criteria. 2. Calculation of ADOS Calibrated Severity Score (CSS). 3. MRI Data Quality Assessment, Exclusion of Scans, and Manual Edits. 4. Canonical Correlation Analysis (CCA). 5. Robustness of the results across feature selection algorithms. Supplementary Tables S1. Post-hoc multiple comparisons of means. Supplementary Table S2. Sample demographics after matching for age and gender. Supplementary Table S3. Clusters with significantly increased and decreased cortical volume (CV), surface area (SA), and cortical thickness (CT) for the main effect of 22q11.2DS. Supplementary Table S4. Clusters with significantly increased and decreased cortical volume (CV), surface area (SA), and cortical thickness (CT) for the main effect of ASD. Supplementary Table S5. Clusters with a significant 22q11.2DS-by-ASD interaction effect in cortical volume (CV) and surface area (SA). Supplementary Figure S1. Between-group comparison for 22q11.2DS compared to typically developing Controls. Supplementary Figure S2. Between-group comparison for idiopathic ASD compared to typically developing Controls. Supplementary Figure S3. Un-thresholded Results for the Categorical Analyses. Supplementary Figure S4. Effect Sizes for Categorial results. Supplementary Figure S5. (In)Homogeneity of Variance between idiopathic ASD and 22q11.ASD individuals. Supplementary Figure S6. Categorical analyses corrected for age and gender. Supplementary Figure S7. Schematic Overview of the Methodology behind the Canonical Correlation Analysis (CCA). Supplementary Figure S8. Reliability of the results across feature selection algorithms. Supplementary Figure S9. Boxplots for the significant 22q11.2DS-by-ASD Interaction Clusters. Supplementary Figure S10. Categorical 22q11.2DS-by-ASD Interaction Effect when covarying for repetitive symptoms. Supplementary Figure S11. Distribution of SRS subdomain and total scores across groups
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gudbrandsen, M., Bletsch, A., Mann, C. et al. Neuroanatomical underpinnings of autism symptomatology in carriers and non-carriers of the 22q11.2 microdeletion. Molecular Autism 11, 46 (2020). https://doi.org/10.1186/s13229-020-00356-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-020-00356-z