
Abstract
Background
Altered proteome profiles have been reported in both postmortem brain tissues and body fluids of subjects with Alzheimer disease (AD), but their broad relationships with AD pathology, amyloid pathology, and tau-related neurodegeneration have not yet been fully explored. Using a robust automated MS-based proteomic biomarker discovery workflow, we measured cerebrospinal fluid (CSF) proteomes to explore their association with well-established markers of core AD pathology.
Methods
Cross-sectional analysis was performed on CSF collected from 120 older community-dwelling adults with normal (n = 48) or impaired cognition (n = 72). LC-MS quantified hundreds of proteins in the CSF. CSF concentrations of β-amyloid 1–42 (Aβ1–42), tau, and tau phosphorylated at threonine 181 (P-tau181) were determined with immunoassays. First, we explored proteins relevant to biomarker-defined AD. Then, correlation analysis of CSF proteins with CSF markers of amyloid pathology, neuronal injury, and tau hyperphosphorylation (i.e., Aβ1–42, tau, P-tau181) was performed using Pearson’s correlation coefficient and Bonferroni correction for multiple comparisons.
Results
We quantified 790 proteins in CSF samples with MS. Four CSF proteins showed an association with CSF Aβ1–42 levels (p value ≤ 0.05 with correlation coefficient (R) ≥ 0.38). We identified 50 additional CSF proteins associated with CSF tau and 46 proteins associated with CSF P-tau181 (p value ≤ 0.05 with R ≥ 0.37). The majority of those proteins that showed such associations were brain-enriched proteins. Gene Ontology annotation revealed an enrichment for synaptic proteins and proteins originating from reelin-producing cells and the myelin sheath.
Conclusions
We used an MS-based proteomic workflow to profile the CSF proteome in relation to cerebral AD pathology. We report strong evidence of previously reported CSF proteins and several novel CSF proteins specifically associated with amyloid pathology or neuronal injury and tau hyperphosphorylation.
Similar content being viewed by others


Background
Proteome alterations have been identified in a multitude of pathologies, such as cancer, metabolic disorders, and brain diseases [1]. Several circulating protein markers of neurodegenerative diseases, such as Parkinson’s disease or Alzheimer disease (AD), have been reported [2], but the ones with consistent findings or of current clinical utility are very few [3]. AD is the most common form of dementia, and there is still an urgent need for the definition of early detection markers as well as for a better understanding of its pathogenesis. In the latter perspective, cerebrospinal fluid (CSF) represents a key biofluid to decipher altered protein levels and pathways in diseases of the central nervous system (CNS) using large-scale proteomic technologies, such as MS-based platforms.
Because of the proximity of CSF to the brain and the presence of proteins in CSF specific to the brain [4, 5], the CSF proteome can reflect the biochemical and metabolic changes in the CNS. In particular, despite the definitive confirmation of the diagnosis of AD being possible today only at brain autopsy, specific CSF peptides and proteins (i.e., β-amyloid 1–42 [Aβ1–42], total tau, and hyperphosphorylated tau [P-tau]) linked to the main hallmarks of AD pathology, such as amyloid plaques and neurofibrillary tangles, can complement clinical examination for the diagnosis of AD [6, 7].
There is now strong evidence that suggests the development of AD pathology begins years to decades prior to the onset of the first clinical signs. Thus, on one hand, elderly persons with normal cognition may already have cerebral AD pathology and be at the preclinical stage of the disease [8]; on the other hand, subjects with cognitive deficits may present with cognitive impairment suggesting AD but not primarily or only partially related to AD pathology. New research criteria consider AD as a biological continuum across the clinical spectrum from asymptomatic stage to advanced dementia and emphasize the utility of biomarkers of AD pathology for an accurate diagnosis, in particular at the preclinical and prodromal disease stages [8,9,10]. In this respect, endophenotype approaches have been proposed as innovative ways to better address AD stages using proxy measures such as the concentrations of the aforementioned CSF markers of core AD pathology [11].
Several studies have characterized the CSF proteome with MS but mainly using sample pools and/or a limited number of samples [12,13,14]. Because of technical constraints such as limited sample throughput [15], studies in larger clinical cohorts using MS-based proteomics are indeed limited [16,17,18,19,20,21]. In recent years, our group [22] and other groups [23, 24] have demonstrated that MS-based proteomics enables protein biomarker discovery in large numbers of human clinical samples, providing increased statistical power and result robustness [21, 22, 25]. Although most of these studies were performed with plasma or serum samples [26], the analysis of the CSF proteome and its alteration using MS-based proteomics in larger cohorts has been mostly unexplored.
Our aim in this study was to investigate the CSF proteome in relation to the core elements of CSF-defined AD pathology in older adults (n = 120) with normal and impaired cognition using MS-based shotgun proteomics (Fig. 1). We evaluated whether the CSF proteome could relate to AD pathology, defined as the combined presence of both amyloid pathology and tau pathology. We then explored more deeply the relationships of the quantified proteins in CSF with well-established biomarkers of amyloid pathology, neuronal injury, and tau hyperphosphorylation (i.e., Aβ1–42, tau, and tau phosphorylated at threonine 181 [P-tau181], respectively).

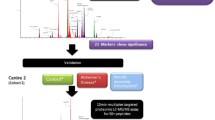
Study design and cerebrospinal fluid (CSF) proteome profiling workflow. CSF samples from 120 older individuals with or without cognitive impairment were analyzed using a highly automated shotgun MS-based proteomic workflow. The workflow consists of first removing 14 highly abundant proteins in CSF by immunoaffinity. The rest of the workflow is automated in a 96-well plate format and includes steps of (1) reduction, alkylation, and enzymatic digestion; (2) isobaric labeling and pooling; and (3) purifications. The samples are analyzed with reversed-phase LC-MS/MS, and the data are processed with standard bioinformatic tools
Methods
Study design
One hundred twenty community-dwelling participants were included in this study, of whom 48 were cognitively healthy volunteers and 72 had mild cognitive impairment (MCI) (n = 63) or mild dementia of AD type (n = 9) [27]. Diagnosis of MCI or dementia was based on neuropsychological and clinical evaluation and made by a consensus conference of psychiatrists and/or neurologists as well as neuropsychologists prior to inclusion in the study. The participants with cognitive impairment were recruited from among outpatients who were referred to the Memory Clinics, Departments of Psychiatry, and Department of Clinical Neurosciences, University Hospitals of Lausanne, Switzerland. They had no major psychiatric disorders or substance abuse or severe or unstable physical illness that might contribute to cognitive impairment, had a Clinical Dementia Rating (CDR) [28] score > 0, and met the clinical diagnostic criteria for MCI [29] or AD mild dementia according to the recommendations of the National Institute on Aging-Alzheimer’s Association [30]. In the current study, nine subjects met criteria for probable AD dementia. Because there is a clinical continuum between MCI and mild dementia, and because the participants with cognitive impairment were patients from memory clinics recruited in the same way regardless of MCI or mild dementia classification, these subjects were grouped and labeled as cognitively impaired with CDR > 0 (Table 1). The control subjects were recruited through journal announcements or word of mouth and had no history, symptoms, or signs of relevant psychiatric or neurologic disease and no cognitive impairment (CDR = 0). All participants underwent a comprehensive clinical and neuropsychological evaluation, structural brain imaging, and venous and lumbar punctures [27]. Magnetic resonance imaging (MRI) and computed tomographic scans were used to exclude cerebral pathologies possibly interfering with cognitive performance.
Neuropsychological tests were used to assess cognitive performance in the domains of memory [31], language, and visuoconstructive functions. The Mini Mental State Examination [32] was used to assess participants’ global cognitive performance. Depression and anxiety were assessed using the Hospital Anxiety and Depression Scale [33]. The psychosocial and functional assessments included activities of daily living and instrumental activities of daily living, the Neuropsychiatric Inventory Questionnaire, and the Informant Questionnaire on Cognitive Decline in the Elderly [34], and these were completed by family members of the participants. All tests and scales are validated and widely used in the field.
CSF sample collection
Lumbar punctures were performed between 8:30 a.m. and 9:30 a.m. after overnight fasting. A standardized technique with a 22-gauge “atraumatic” spinal needle and a sitting or lying position was applied [35]. A volume of 10–12 ml of CSF was collected in polypropylene tubes. Routine cell count and protein quantification were performed. The remaining CSF was frozen in aliquots (500 μl) no later than 1 hour after collection and stored at − 80 °C without thawing until experiment and assay.
MS-based proteomics
CSF samples were prepared using a highly automated shotgun proteomic workflow as previously described [36] and isobaric tags [37] for relative quantification of proteins. Reversed-phase LC-MS/MS was performed with a hybrid linear ion trap-Orbitrap Elite and an UltiMate 3000 RSLCnano System (Thermo Scientific, Waltham, MA, USA) as recently described [38]. Protein identification was performed against the human UniProtKB/Swiss-Prot database (08/12/2014 release). All details are provided in Additional file 1: Supplementary Methods.
CSF β-amyloid 1–42, tau, tau phosphorylated at threonine 181, and APOE genotyping
The measurements were performed using commercially available enzyme-linked immunosorbent assay kits and TaqMan assays as described in Additional file 1: Supplementary Methods.
Definition of CSF biomarker profile of Alzheimer pathology
A pathological AD CSF biomarker profile was defined as CSF P-tau181/Aβ1–42 ratio > 0.0779 (i.e., “high” ratio for positive CSF profile of AD pathology), based on clinical study site data [39] and in line with previous work (i.e., 0.08) [40]. The cutoff optimized the Youden index [41] of the ROC curve for the prediction of CDR categories (CDR = 0 versus CDR > 0) as previously reported [27], where the cutoff for CSF P-tau181/Aβ1–42 ratio was further confirmed to be a highly significant predictor of cognitive decline.
Proteomic data management
Six CSF samples were removed because of aberrant values, leaving CSF proteomic data available for 114 subjects (exclusion of those 6 subjects did not induce bias on the overall population characteristics) (see Additional file 1: Table S1)). In total, 790 CSF proteins were quantified.
For exploration of CSF proteins relevant to AD pathology (see below), proteins with > 5% missingness were excluded, leaving 541 CSF proteins. The remaining missing data (5% or less per protein) were imputed by randomly drawing a value between the observed range of biomarker values. Log2 of the protein ratio fold changes were scaled to mean zero and SD of 1 prior to statistical analyses. Calculation and statistics were performed with the R version 3.3.2 statistical software (http://www.r-project.org/).
Exploratory analysis of CSF proteins relevant to Alzheimer pathology
In a first exploratory analysis, 541 CSF proteins were tested (one by one) in a logistic regression model as follows:
where positive CSF profile of AD is defined by categorizing the CSF P-tau181/Aβ1–42 ratio into two groups: P-tau181/Aβ1–42 > 0.0779 for AD CSF biomarker profile (or “high”) and P-tau181/Aβ1–42 ≤ 0.0779 for non-AD CSF biomarker profile (or “low”). p Values were corrected for multiple testing using the Benjamini-Hochberg procedure. Box plots were produced for the significant hits presenting false discovery rate (FDR) ≤ 5%.
Selection of CSF proteins relevant to Alzheimer pathology
Least absolute shrinkage and selection operator (LASSO) logistic regression [42] selected biomarkers that best predict CSF biomarker profile of AD pathology. A reference model was initially generated, testing variables that are likely to be available to clinicians and known risk factors for AD to provide a benchmark for comparison with the model that included CSF proteins. These inputs included age, gender, years of education, and presence of the apolipoprotein E (APOE) ε4 allele, such as:
In addition to all variables used to make the reference models, CSF protein measurements (i.e., 541 CSF proteins) and CSF albumin index were then included in building so-called best models:
A tenfold cross-validation process was performed for each LASSO analysis using the glmnet package [43], which allows estimating the confidence interval of the misclassification error for each value of the regularization parameter λ. The LASSO analyses were repeated 100 times (1000 times for the reference models). The model that minimized the upper limit of the cross-validated misclassification error confidence interval across the 100 runs with less than 20 features (when possible) was selected. The results were formally tested for significance against the reference model using accuracy with a McNemar test. The group differences for the CSF proteins selected in the best models were graphically illustrated in box plots and assessed using t test statistics. In addition, Kruskal-Wallis test statistics produced comparable results (see Additional file 1: Tables S2 and S3). Because the tests were applied only to the proteins selected with LASSO, p values obtained from these analyses were not corrected for multiple testing.
Statistical Pearson’s correlation and bioinformatic analysis
Correlation analysis was performed on protein fold changes of all 790 quantified proteins using Pearson’s correlation coefficient and Bonferroni correction for multiple comparisons. In addition, Spearman’s correlation analyses produced comparable results (see Additional file 1: Tables S4–S6). Several bioinformatics tools and resources were used for analysis and protein annotation (i.e., Database for Annotation, Visualization and Integrated Discovery [DAVID] 6.8 [44], UniProt tissue annotation database [45], Gene Ontology database [46], Kyoto Encyclopedia of Genes and Genomes [KEGG] database [47], tissue atlas [48], and Venny [http://bioinfogp.cnb.csic.es/tools/venny/]).
Results
Demographic and clinical characteristics of the study population
Demographics and clinical characteristics of the patient cohort are detailed in Table 1. The cognitively impaired subjects (CDR > 0) were older and less educated and had a higher prevalence of APOE ε4 genotype than the cognitively intact group (CDR = 0). In cognitive impairment, CSF Aβ1–42 was lower, whereas CSF tau, CSF P-tau181, and CSF P-tau181/Aβ1–42 were all higher. MS-based proteomic analyses were performed in the CSF of the 120 individuals (Fig. 1). In total, we measured 790 proteins in CSF. Of those, 541 proteins presented < 5% missing values in 114 subjects (see the Methods section above).
The following classification analyses of the CSF P-tau181/Aβ1–42 ratios were aimed at separating 39 patients with high-expression AD CSF biomarker profiles (i.e., P-tau181/Aβ1–42 > 0.0779) from 75 low-expression profile subjects in the complete analysis set, regardless of the clinical diagnosis. Then, the analyses were performed on the subset of cognitively impaired patients, where 38 and 28 subjects had high and low expression of AD CSF biomarker profiles, respectively.
Identification of Alzheimer pathology with CSF proteins
First, we explored whether the CSF proteome presents specific alterations in AD, endophenotypically defined a priori as a CSF P-tau181/Aβ1–42 ratio > 0.0779 (see the Methods section above). In the whole sample, group comparisons (i.e., “high” when P-tau181/Aβ1–42 > 0.0779 and “low” when P-tau181/Aβ1–42 ≤ 0.0779) revealed 22 CSF proteins with significant differences between AD versus non-AD CSF biomarker profiles after correction for multiple testing using the Benjamini-Hochberg procedure at FDR ≤ 5% (Fig. 2a and Additional file 1: Table S7). Similarly, in the subset of cognitively impaired subjects (see the Methods section above), group comparisons provided ten CSF proteins with significant differences (Fig. 2b and Additional file 1: Table S8). All of these 10 proteins were already present among the 22 proteins (Fig. 2) previously identified in the whole sample.

Cerebrospinal fluid (CSF) proteins relevant to Alzheimer pathology. Box plots of CSF proteins according to CSF tau phosphorylated at threonine 181 (P-tau181)/β-amyloid 1–42 (Aβ1–42) ratio (i.e., “high” when P-tau181/Aβ1–42 > 0.0779 [blue dots] and “low” when P-tau181/Aβ1–42 ≤ 0.0779 [red dots]) for positive and negative CSF profiles of AD pathology, respectively, in all subjects (a) and restricted to subjects with cognitive impairment (b). In total, 541 CSF proteins were tested (one by one) in a logistic regression model. P values were corrected for multiple testing using the Benjamini-Hochberg procedure. Box plots were produced for the significant hits presenting false discovery rate ≤ 5%. Relative protein fold change ratios were used (in Log2). Human proteins in the box plots are given by their UniProtKB/Swiss-Prot entry name
As a second exploratory approach and ability assessment of the CSF proteome to identify AD, we used LASSO logistic regression to build mathematical models able to classify AD pathology, again defined a priori as a CSF P-tau181/Aβ1–42 ratio > 0.0779 (see the Methods section above). In the whole sample, the benchmark reference model for classification of CSF P-tau181/Aβ1–42 included age and presence of the APOE ε4 allele. Its prediction accuracy was 78.3% (as compared with the accuracy of a majority class prediction of 65.8%). CSF protein biomarkers were indeed able to improve the classification of AD CSF biomarker profile with respect to the reference model. The best model accuracy was 100% (McNemar p value 3.35 × 10− 7). It included 26 CSF proteins (from the 541 provided as input) in addition of age and presence of the APOE ε4 allele. Only seven selected CSF proteins displayed significant group comparison differences, i.e., 14-3-3 protein ζ/δ (1433Z) (p = 1.69 × 10− 3), SPARC-related modular calcium-binding protein 1 (SMOC1) (p = 5.26 × 10− 5), KICSTOR complex protein SZT2 (SZT2) (p = 5.47 × 10− 4), fatty acid-binding protein, heart (FABPH) (p = 8.70 × 10− 4), chitinase-3-like protein 1 (CH3L1) (p = 1.23 × 10− 3), neuromodulin (NEUM) (p = 3.40 × 10− 3), and keratin, type I cytoskeletal 10 (p = 0.025) (Additional file 1: Figure S1a). Many of these CSF proteins were correlated with each other (Additional file 1: Figure S2). Six of the seven proteins (i.e., 1433Z, SMOC1, SZT2, FABPH, CH3L1, and NEUM) were reported in the exploratory group comparisons (Fig. 2a).
In the subset of cognitively impaired subjects (see the Methods section above), the benchmark reference model to classify AD CSF biomarker profile included age, gender, years of education, and presence of APOE ε4 allele, with a prediction accuracy of 77.8% (majority class prediction of 57.6%). In cognitive impairment, inclusion of CSF protein biomarkers again improved significantly the prediction accuracy to 100% (McNemar p value of 0.0003). In total, 18 CSF proteins (from the 541 provided as input) were included in this best model in addition to gender and presence of the APOE ε4 allele. Among those proteins, four displayed significant differences between the groups: 1433Z (p = 4.04 × 10− 5), SMOC1 (p = 5.49 × 10− 5), γ-synuclein (p = 1.19× 10− 3), and macrophage colony-stimulating factor 1 receptor (p = 0.013) (Additional file 1: Figure S1b). Again, several correlations were observed between the CSF proteins retained in the model (Additional file 1: Figure S3), suggesting that models with fewer variables may still provide high classification performance. Two of the four proteins (i.e., 1433Z and SMOC1) were reported in the exploratory group comparisons (Fig. 2b). The perfect performance to classify the participants with AD pathology indicated that the reported models were very possibly overfitting the data.
Associations of CSF proteins with β-amyloid 1–42, tau, and tau phosphorylated at threonine 181
Next, we separately and more specifically studied the associations of all 790 quantified CSF proteins (no minimal missing value criteria applied) with CSF markers of core AD pathology (i.e., Aβ1–42, tau, and P-tau181). Four proteins—cannabinoid receptor 1 (CNR1, correlation coefficient [R] = 0.3929), neuroendocrine convertase 2 (NEC2, R = 0.3818), neuronal pentraxin-2 (NPTX2, R = 0.3868), and somatostatin (SMS, R = 0.4188)—showed an association with CSF Aβ1–42, which was significant (p value ≤0.05) after Bonferroni correction for multiple testing (Fig. 3a). We found 50 CSF proteins correlated with CSF tau (Fig. 3b) and 46 associated with CSF P-tau181 (Fig. 3c) in a significant manner after Bonferroni correction, of which 41 were in common (Fig. 3d). The five strongest correlations with CSF tau were CSF neurogranin (NEUG), sodium/potassium-transporting ATPase subunit α-2 (AT1A2), brain acid soluble protein 1 (BASP1), 1433Z, and NEUM. The five strongest correlations with CSF P-tau181 were CSF AT1A2, disintegrin and metalloproteinase domain-containing protein 10 (ADA10), NG,NG-dimethylarginine dimethylaminohydrolase 1 (DDAH1), NEUG, and SMOC1. In particular, CSF NEUG and NEUM [49], two synaptic proteins, were positively correlated with CSF tau (R = 0.6721 and 0.5287, respectively) and P-tau181 (R = 0.5074 and 0.4741, respectively) (Additional file 1: Figure S4). All the observed associations are summarized in the chord diagram of Additional file 1: Figure S5. With the exception of ectonucleotide pyrophosphatase/phosphodiesterase family member 2, which negatively correlated with tau, all reported correlations were positive.

Correlations of cerebrospinal fluid (CSF) proteins with β-amyloid 1–42 (Aβ1–42), tau, and tau phosphorylated at threonine 181 (P-tau181) concentrations in CSF. Correlation of CSF proteins with CSF Aβ1–42 (a), CSF tau (b), and CSF P-tau181 (c). Only significant correlations with a p value ≤ 0.05 after Bonferroni correction for multiple testing were retained and are displayed in the graphs. CSF proteins correlating with CSF Aβ1–42, tau, and P-tau181 are illustrated in a Venn diagram (d)
Annotations of CSF proteins correlating with β-amyloid 1–42, tau, and tau phosphorylated at threonine 181
Of the 59 proteins displaying correlations in those analyses (Fig. 3d), most are expressed in the brain, in particular in the fetal brain cortex and Cajal-Retzius cells (Fig. 4a). Moreover, and based on the tissue-based map of the human proteome [48], seven proteins (i.e., SLIT and NTRK-like protein 1, NEUM, NEUG, cell adhesion molecule 2, lymphocyte antigen 6H [LY6H], transgelin-3 [TAGL3], and protein lifeguard) are brain-enriched (i.e., having at least fivefold higher mRNA levels in the brain as compared with all other tissues) and a total of 22 proteins have elevated gene expression in the brain (i.e., in addition to the seven above, AT1A2, immunoglobulin superfamily containing leucine-rich repeat protein 2 [ISLR2], sodium/potassium-transporting ATPase subunit α-3 [AT1A3], BASP1, CH3L1, CNR1, ephrin type-B receptor 6 [EPHB6], NPTX2, paralemmin-1, NEC2, proline-rich transmembrane protein 2, SMOC1, VPS10 domain-containing receptor SorCS1, SMS, and V-set and transmembrane domain-containing protein 2A).

Annotations of cerebrospinal fluid (CSF) proteins correlating with β-amyloid 1–42 (Aβ1–42), tau, and/or tau phosphorylated at threonine 181 (P-tau181) concentrations in CSF. Tissue annotation using the UniProt tissue annotation database (a) and Gene Ontology (GO) (cellular component category) annotation (b) obtained with DAVID software for the 59 CSF proteins correlating with CSF Aβ1–42, tau, and/or P-tau181. Significant enrichment (Benjamini-Hochberg procedure) is indicated with an asterisk. The background used for the enrichment analysis was the 790 detected proteins in CSF. n.s. Nonsignificant
In Fig. 4b, we identified the myelin sheath as an enriched cellular component. Of the 59 CSF proteins correlating with Aβ1–42, tau, and/or P-tau181, 9 proteins pertain to the myelin sheath: TAGL3, malate dehydrogenase, cytoplasmic (MDHC), heat shock cognate 71 kDa protein (HSP7C), AT1A2, phosphoglycerate mutase 1 (PGAM1), superoxide dismutase [Cu-Zn] (SODC), AT1A3, pyruvate kinase PKM (KPYM), and L-lactate dehydrogenase B chain (LDHB). Those nine proteins were associated with tau and/or P-tau181. Pathway enrichment analysis using the KEGG database did not yield any significant results (data not shown).
Discussion
In the present study, we used MS-based shotgun proteomics to measure the CSF proteomes of 120 older adults and investigate broad CSF protein relationships with core AD pathology. Overall, human CSF proteome coverage was composed of 790 proteins. Four CSF proteins were associated with CSF Aβ1–42 levels, 50 proteins with CSF tau, and 46 proteins with CSF P-tau181 levels. The CSF proteins related to Aβ1–42 were different from those associated with tau or P-tau181.
To explore the relevance of the CSF proteome to AD pathology, we applied an approach that was unbiased by the clinical diagnosis and defined endophenotypically the disease as the presence of “core” AD pathology (i.e., the combined presence of cerebral amyloid and tau pathology). Unbiased classification based on markers of cerebral amyloid and tau pathology and neuronal injury has been proposed for use across the clinical stages [7]. We first used two exploratory approaches to evaluate and select CSF proteins that were able to stratify subjects according to levels of CSF P-tau181/Aβ1–42. Using LASSO logistic regression, we observed that CSF proteins could significantly increase the classification accuracy of non-AD versus AD CSF biomarker profiles as compared with models based only on clinical parameters and the presence of the APOE ε4 allele. Nonetheless, those statistical models relying on CSF proteins might be overfitted and should be interpreted with caution; class imbalance also affected their strict performance. Overall, with both exploratory analyses, we identified specific CSF proteome alterations that are related to AD pathology and may provide novel mechanistic insights. Assessing the whole sample and the subgroup of subjects with cognitive impairment, we could decipher the strong contribution of some CSF proteins, such as SMOC1 and 1433Z (Fig. 2 and Additional file 1: Figure S1). On the basis of this performance, we specifically investigated associations of CSF proteins with individual most validated biomarkers of amyloid pathology, neuronal injury, and tau hyperphosphorylation (i.e., Aβ1–42, tau, and P-tau181, respectively) to elaborate further on the involved mechanisms. Most of the correlations of CSF proteins were with CSF tau and P-tau181 (Fig. 3d), suggesting the CSF proteome alterations to be more representative of tau pathology than amyloid pathology. Four CSF proteins not related to tau and P-tau181 were associated with CSF Aβ1–42 levels, overall indicating distinct proteome alterations related to either amyloid pathology or tau-related neurodegeneration. The majority of these proteins were brain-enriched proteins, including synaptic proteins, and proteins involved in reelin-producing cells and the myelin sheath. Comparison of the proteins found with different levels in AD versus non-AD CSF biomarker profiles and in the models able to classify CSF-defined AD pathology with those associated with CSF Aβ1–42, tau, and P-tau181 in Venn diagrams (Additional file 1: Figures S6 and S7, respectively) revealed mixed overlaps. Interestingly, the 22 proteins with different levels in AD versus non-AD CSF biomarker profiles (Fig. 2a) were all associated with CSF tau; a large majority were associated with CSF P-tau181; but none were associated with CSF Aβ1–42 (Additional file 1: Figure S6). Nevertheless, beyond those 22 proteins, 37 proteins, still representing the majority of CSF proteins associated with CSF Aβ1–42, tau, and P-tau181, were not evidenced as having a relationship to AD, suggesting they might represent more general makers of amyloid pathology, neuronal injury, and tau hyperphosphorylation.
The CSF proteins CNR1, NEC2, NPTX2, and SMS were associated with CSF Aβ1–42 in our study (Fig. 3a). CNR1 and the endocannabinoid system were previously identified as potential targets for treatment of neurological disorders and AD in particular [50, 51]. In line with our results, higher NPTX2, a proinflammatory protein involved in synaptic plasticity, was previously associated with higher CSF Aβ1–42 in the Alzheimer’s Disease Neuroimaging Initiative study [52]. NEC2, also known as prohormone convertase 2, is essential to the processing of pro-islet amyloid polypeptide [53]. Its role in the processing of hormones and in particular of neuropeptide precursors in the human cortex has been established, but the link with SMS deficiency in AD, for instance, was not confirmed [54]. Relevant to our observations, neuropeptide SMS is known to be decreased in the CSF of patients with AD [55] and to regulate Aβ1–42 via proteolytic degradation [56]. Together, these findings indicate amyloid-related changes in the CSF proteome that may be particularly relevant for early cerebral AD pathology as well as for disease-modifying interventions targeting amyloid and starting at preclinical disease stages.
We found that CSF Aβ1–42, tau, and P-tau181 were mainly associated with CSF proteins enriched in brain tissue (Fig. 4a), and this despite the important proportion (about 80%) of proteins in CSF originating from blood [4]. In particular, some are expressed in the fetal brain cortex. We observed positive correlations between CSF tau and/or P-tau181 with 13 CSF proteins (i.e., calmodulin, fructose-bisphosphate aldolase A [ALDOA], DDAH1, HSP7C, KPYM, LDHB, MDHC, PGAM1, phosphatidylethanolamine-binding protein 1 [PEBP1], stathmin, TAGL3, thioredoxin, and 1433Z) known also to be present in reelin-producing Cajal-Retzius cells. In early AD, a massive decline of the number of Cajal-Retzius cells was previously described [57], suggesting a link between their loss, reduction of reelin, impairment of synaptic plasticity, amyloid plaque deposition, and neurofibrillary tangle formation [58]. Interestingly, we also revealed the involvement of nine CSF proteins (i.e., AT1A2, AT1A3, HSP7C, KPYM, LDHB, MDHC, PGAM1, SODC, and TAGL3), again positively correlating with CSF tau and/or P-tau181, being specifically part of the myelin sheath. Although amyloid plaques and neurofibrillary tangles likely induce neuronal and synaptic loss, myelin alteration may also participate in the development of AD dementia. Myelin content changes in the white matter measured with MRI have been linked to CSF AD biomarkers (i.e., lower concentrations of Aβ1–42 and higher concentrations of tau and P-tau181), but mainly in association with amyloid pathology [59]. Our results, including associations of AT1A2 and KPYM with both tau and P-tau181, may suggest an underestimated connection between tau-related neurodegeneration and (de)myelination. These specific alterations provide new insights into the disease pathology and deserve further exploration.
Several single relationships between CSF proteins and Aβ1–42, tau, and/or P-tau181 levels in our study (Fig. 3) have previously been reported. A first example is the synaptic protein NEUG, which was previously proposed as a novel candidate CSF biomarker for AD and prodromal AD; high CSF NEUG was shown to predict future cognitive decline and to be more specific for AD than tau [60]. In addition, CSF NEUG was reported to be increased in AD and positively correlated with CSF tau [61] and P-tau [49]. In line with our observations, positive associations were identified with NEUM for both tau and P-tau in CSF [49]. BASP1, like NEUM, is a presynaptic membrane protein participating in axon guidance, neurodegeneration, and synaptic plasticity [62] and was found to be significantly downregulated in AD versus control brain samples [63]. Our findings of significant association of CSF BASP1 with both CSF tau and P-tau warrant further investigations. Mutations in the ADAM10 gene, which encodes the major α-secretase responsible for cleaving APP, have previously been identified in families with late-onset AD [64]. In our study, protein ADA10, which is encoded by ADAM10, was only significantly associated with CSF P-tau181. To the best of our knowledge, such an association between those CSF proteins has not been observed before [65].
Further and broader cross-validation of our findings can be made by comparing them with those of a recent study investigating CSF proteins associated with CSF AD biomarkers in 58 cognitively healthy men using an aptamer-based technology (i.e., SOMAscan; SomaLogic, Boulder, CO, USA) [66]. Of the 59 CSF proteins associated with CSF biomarkers of core AD pathology that we report, 28 were also measured with the SOMAscan in that prior study; of those, 22 proteins (i.e., 78.6% overlap) were correlated with CSF Aβ1–42, tau, and/or P-tau [66], confirming part of our observations in an independent cohort and using a different technology. Those proteins are ALDOA, dynein light chain 2, cytoplasmic, polyubiquitin B, ISLR2, EPHB6, MDHC, SH3 domain-binding glutamic acid-rich-like protein, PEBP1, NPTX2, chromogranin A, cytochrome c, SMS, 1433Z, LDHB, SMOC1, 14–3-3 protein β/α, spondin-1, FABPH, transmembrane emp24 domain-containing protein 4, PGAM1, cytokine-like protein 1, and HSP7C.
Altogether, our shotgun MS-based proteomic approach [22] was confirmed to provide relevant findings and to be complementary to alternative proteomic technologies. In this perspective, the identification of novel and strongly significant associations of CSF proteins with CSF biomarkers of AD core pathology in our study is of specific interest. In particular, proteins AT1A2 and KPYM implicated in energy production, as well as 1433Z, DDAH1, and SMOC1, showing some of the strongest associations with tau and/or P-tau181 in addition to NEUG and NEUM, could appear relevant. Our results in a relatively large group of subjects including both participants with cognitive impairment and healthy volunteers are therefore encouraging. Sample fractionation would have allowed deeper proteome coverage but with a throughput incompatible with the analysis of 120 clinical samples in a reasonable time frame. The proteins we have identified would deserve additional research.
Conclusions
Using an MS-based proteomic workflow, we have quantified a number of CSF proteins in 120 older adults with normal cognition and with cognitive impairment. We report strong evidence of known and new CSF proteins related to amyloid pathology, neuronal injury, and tau hyperphosphorylation. Although we confirmed several previous findings of CSF proteins related to AD pathology, our work reveals a large number of additional CSF proteome alterations involving in particular reelin-producing cells and the myelin sheath.
Abbreviations
- 1433B:
-
14-3-3 protein β/α
- 2LFG2:
-
Protein lifeguard
- AD:
-
Alzheimer disease
- ADA10:
-
Disintegrin and metalloproteinase domain-containing protein 10
- ALDOA:
-
Fructose-bisphosphate aldolase A
- APOE :
-
Apolipoprotein E
- AT1A2:
-
Sodium/potassium-transporting ATPase subunit α-2
- AT1A3:
-
Sodium/potassium-transporting ATPase subunit α-3
- AUC:
-
Area under the curve
- BASP1:
-
Brain acid soluble protein 1
- CADM2:
-
Cell adhesion molecule 2
- CALM:
-
Calmodulin
- CDR:
-
Clinical Dementia Rating
- CH3L1:
-
Chitinase-3-like protein 1
- CMGA:
-
Chromogranin-A
- CNR1:
-
Cannabinoid receptor 1
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DDAH1:
-
NG,NG-dimethylarginine dimethylaminohydrolase 1
- CSF1R:
-
Macrophage colony-stimulating factor 1 receptor
- CYC:
-
Cytochrome c
- CYTL1:
-
Cytokine-like protein 1
- DYL2:
-
Dynein light chain 2, cytoplasmic
- ENPP2:
-
Ectonucleotide pyrophosphatase/phosphodiesterase family member 2
- EPHB6:
-
Ephrin type- B receptor 6
- FABPH:
-
Fatty acid-binding protein, heart
- FDR:
-
False discovery rate
- HSP7C:
-
Heat shock cognate 71 kDa protein
- ISLR2:
-
Immunoglobulin superfamily containing leucine-rich repeat protein 2
- K1C10:
-
Keratin, type I cytoskeletal 10
- KPYM:
-
Pyruvate kinase PKM
- LASSO:
-
Least absolute shrinkage and selection operator regression
- LC:
-
Liquid chromatography
- LDHB:
-
L-lactate dehydrogenase B chain
- MCI:
-
Mild cognitive impairment
- MDHC:
-
Malate dehydrogenase, cytoplasmic
- MMSE:
-
Mini Mental State Examination
- MS:
-
Mass spectrometry
- MS/MS:
-
Tandem mass spectrometry
- MRI:
-
Magnetic resonance imaging
- NEC2:
-
Neuroendocrine convertase 2
- NEUG:
-
Neurogranin
- NEUM:
-
Neuromodulin
- NPTX2:
-
Neuronal pentraxin-2
- PALM:
-
Paralemmin-1
- PEBP1:
-
Phosphatidylethanolamine-binding protein 1
- PGAM1:
-
Phosphoglycerate mutase 1
- PRRT2:
-
Proline-rich transmembrane protein 2
- P-tau:
-
Hyperphosphorylated tau
- P-tau181:
-
Tau phosphorylated at threonine 181
- RP:
-
Reversed-phase
- SLIK1:
-
SLIT and NTRK-like protein 1
- SMOC1:
-
SPARC-related modular calcium-binding protein 1
- SMS:
-
Somatostatin
- SODC:
-
Superoxide dismutase [Cu-Zn]
- SORC1:
-
VPS10 domain-containing receptor SorCS1
- SPON1:
-
Spondin-1
- STMN1:
-
Stathmin
- SYUG:
-
γ-Synuclein
- SZT2:
-
KICSTOR complex protein SZT2
- TAGL3:
-
Lymphocyte antigen 6H (LY6H), transgelin-3
- Aβ1–-42 :
-
β-Amyloid 1–-42
- 1433Z:
-
14-3-3 protein ζ/δ
References
Aebersold R, Bader GD, Edwards AM, Van Eyk JE, Kussmann M, Qin J, Omenn GS. The biology/disease-driven Human Proteome Project (B/D-HPP): enabling protein research for the life sciences community. J Proteome Res. 2013;12:23–7.
Shi M, Caudle WM, Zhang J. Biomarker discovery in neurodegenerative diseases: a proteomic approach. Neurobiol Dis. 2009;35:157–64.
Agrawal M, Biswas A. Molecular diagnostics of neurodegenerative disorders. Front Mol Biosci. 2015;2:54.
Begcevic I, Brinc D, Drabovich AP, Batruch I, Diamandis EP. Identification of brain-enriched proteins in the cerebrospinal fluid proteome by LC-MS/MS profiling and mining of the human protein atlas. Clin Proteomics. 2016;13:11.
Fang Q, Strand A, Law W, Faca VM, Fitzgibbon MP, Hamel N, et al. Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol Cell Proteomics. 2009;8:451–66.
Galasko DR, Shaw LM. Alzheimer disease: CSF biomarkers for Alzheimer disease-approaching consensus. Nat Rev Neurol. 2017;13:131–2.
Jack CR Jr, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539–47.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9.
Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–29.
Baird AL, Westwood S, Lovestone S. Blood-based proteomic biomarkers of Alzheimer’s disease pathology. Front Neurol. 2015;6:236.
Guldbrandsen A, Vethe H, Farag Y, Oveland E, Garberg H, Berle M, et al. In-depth characterization of the cerebrospinal fluid (CSF) proteome displayed through the CSF proteome resource (CSF-PR). Mol Cell Proteomics. 2014;13:3152–63.
Schutzer SE, Liu T, Natelson BH, Angel TE, Schepmoes AA, Purvine SO, et al. Establishing the proteome of normal human cerebrospinal fluid. PLoS One. 2010;5:e10980.
Zhang Y, Guo Z, Zou L, Yang Y, Zhang L, Ji N, et al. A comprehensive map and functional annotation of the normal human cerebrospinal fluid proteome. J Proteome. 2015;119:90–9.
Dayon L, Kussmann M. Proteomics of human plasma: a critical comparison of analytical workflows in terms of effort, throughput and outcome. EuPA Open Proteom. 2013;1:8–16.
Cole RN, Ruczinski I, Schulze K, Christian P, Herbrich S, Wu L, et al. The plasma proteome identifies expected and novel proteins correlated with micronutrient status in undernourished Nepalese children. J Nutr. 2013;143:1540–8.
García-Bailo B, Brenner DR, Nielsen D, Lee HJ, Domanski D, Kuzyk M, et al. Dietary patterns and ethnicity are associated with distinct plasma proteomic groups. Am J Clin Nutr. 2012;95:352–61.
Johansson Å, Enroth S, Palmblad M, Deelder AM, Bergquist J, Gyllensten U. Identification of genetic variants influencing the human plasma proteome. Proc Natl Acad Sci U S A. 2013;110:4673–8.
Lee SE, Stewart CP, Schulze KJ, Cole RN, Wu LSF, Yager JD, et al. The plasma proteome is associated with anthropometric status of undernourished Nepalese school-aged children. J Nutr. 2017;147:304–13.
Lee SE, West KP Jr, Cole RN, Schulze KJ, Christian P, Wu LSF, et al. Plasma proteome biomarkers of inflammation in school aged children in Nepal. PLoS One. 2015;10:e0144279.
Oller Moreno S, Cominetti O, Núñez Galindo A, Irincheeva I, Corthésy J, Astrup A, et al. The differential plasma proteome of obese and overweight individuals undergoing a nutritional weight loss and maintenance intervention. Proteomics Clin Appl. 2018;12:1600150.
Cominetti O, Núñez Galindo A, Corthésy J, Oller Moreno S, Irincheeva I, Valsesia A, et al. Proteomic biomarker discovery in 1000 human plasma samples with mass spectrometry. J Proteome Res. 2016;15:389–99.
Geyer PE, Wewer Albrechtsen NJ, Tyanova S, Grassl N, Iepsen EW, Lundgren J, et al. Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Mol Syst Biol. 2016;12:901.
Liu Y, Buil A, Collins BC, Gillet LCJ, Blum LC, Cheng LY, et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol Syst Biol. 2015;11:786.
Dayon L, Núñez Galindo A, Cominetti O, Corthésy J, Kussmann M. A highly automated shotgun proteomic workflow: clinical scale and robustness for biomarker discovery in blood. Methods Mol Biol. 2017;1619:433–49.
Geyer PE, Holdt LM, Teupser D, Mann M. Revisiting biomarker discovery by plasma proteomics. Mol Syst Biol. 2017;13:942.
Popp J, Oikonomidi A, Tautvydaitė D, Dayon L, Bacher M, Migliavacca E, et al. Markers of neuroinflammation associated with Alzheimer’s disease pathology in older adults. Brain Behav Immun. 2017;62:203–11.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–4.
Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, et al. Mild cognitive impairment - beyond controversies, towards a consensus: report of the international working group on mild cognitive impairment. J Intern Med. 2004;256:240–6.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9.
Buschke H, Sliwinski MJ, Kuslansky G, Lipton RB. Diagnosis of early dementia by the double memory test: encoding specificity improves diagnostic sensitivity and specificity. Neurology. 1997;48:989–97.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98.
Zigmond AS, Snaith RP. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983;67:361–70.
Jorm AF, Jacomb PA. The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): socio-demographic correlates, reliability, validity and some norms. Psychol Med. 1989;19:1015–22.
Popp J, Riad M, Freymann K, Jessen F. Diagnostic lumbar puncture performed in the outpatient setting of a memory clinic: frequency and risk factors of post-lumbar puncture headache. Nervenarzt. 2007;78:547–51.
Núñez Galindo A, Kussmann M, Dayon L. Proteomics of cerebrospinal fluid: throughput and robustness using a scalable automated analysis pipeline for biomarker discovery. Anal Chem. 2015;87:10755–61.
Dayon L, Sanchez JC. Relative protein quantification by MS/MS using the tandem mass tag technology. Methods Mol Biol. 2012;893:115–27.
Dayon L, Núñez Galindo A, Corthésy J, Cominetti O, Kussmann M. Comprehensive and scalable highly automated MS-based proteomic workflow for clinical biomarker discovery in human plasma. J Proteome Res. 2014;13:3837–45.
Tautvydaitė D, Antonietti JP, Henry H, von Gunten A, Popp J. Relations between personality changes and cerebrospinal fluid biomarkers of Alzheimer’s disease pathology. J Psychiatr Res. 2017;90:12–20.
Duits FH, Teunissen CE, Bouwman FH, Visser PJ, Mattsson N, Zetterberg H, et al. The cerebrospinal fluid “Alzheimer profile”: easily said, but what does it mean? Alzheimers Dement. 2014;10:713–23.
Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3:32–5.
Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc Ser B Stat Methodol. 2011;73:273–82.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Bateman A, Martin MJ, O’Donovan C, Magrane M, Alpi E, Antunes R, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–69.
Carbon S, Dietze H, Lewis SE, Mungall CJ, Munoz-Torres MC, Basu S, et al. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–8.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Remnestål J, Just D, Mitsios N, Fredolini C, Mulder J, Schwenk JM, et al. CSF profiling of the human brain enriched proteome reveals associations of neuromodulin and neurogranin to Alzheimer’s disease. Proteomics Clin Appl. 2016;10:1242–53.
Scotter EL, Abood ME, Glass M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br J Pharmacol. 2010;160:480–98.
Stumm C, Hiebel C, Hanstein R, Purrio M, Nagel H, Conrad A, et al. Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiol Aging. 2013;34:2574–84.
Swanson A, Willette AA. Neuronal pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer’s disease spectrum. Brain Behav Immun. 2016;58:201–8.
Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, Verchere CB. The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes. 2001;50:534–9.
Winsky-Sommerer R, Grouselle D, Rougeot C, Laurent V, David JP, Delacourte A, et al. The proprotein convertase PC2 is involved in the maturation of prosomatostatin to somatostatin-14 but not in the somatostatin deficit in Alzheimer’s disease. Neuroscience. 2003;122:437–47.
Nilsson CL, Brinkmalm A, Minthon L, Blennow K, Ekman R. Processing of neuropeptide Y, galanin, and somatostatin in the cerebrospinal fluid of patients with Alzheimer’s disease and frontotemporal dementia. Peptides. 2001;22:2105–12.
Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang SM, et al. Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat Med. 2005;11:434–9.
Baloyannis SJ. Morphological and morphometric alterations of Cajal-Retzius cells in early cases of Alzheimer’s disease: a Golgi and electron microscope study. Int J Neurosci. 2005;115:965–80.
Kocherhans S, Madhusudan A, Doehner J, Breu KS, Nitsch RM, Fritschy JM, Knuesel I. Reduced reelin expression accelerates amyloid-β plaque formation and tau pathology in transgenic Alzheimer’s disease mice. J Neurosci. 2010;30:9228–40.
Dean DC III, Hurley SA, Kecskemeti SR, O’Grady JP, Canda C, Davenport-Sis NJ, et al. Association of amyloid pathology with myelin alteration in preclinical Alzheimer disease. JAMA Neurol. 2017;74:41–9.
Blennow K. A review of fluid biomarkers for Alzheimer’s disease: moving from CSF to blood. Neurol Ther. 2017;6:15–24.
De Vos A, Jacobs D, Struyfs H, Fransen E, Andersson K, Portelius E, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015;11:1461–9.
Forsova OS, Zakharov VV. High-order oligomers of intrinsically disordered brain proteins BASP1 and GAP-43 preserve the structural disorder. FEBS J. 2016;283:1550–69.
Musunuri S, Wetterhall M, Ingelsson M, Lannfelt L, Artemenko K, Bergquist J, et al. Quantification of the brain proteome in Alzheimer’s disease using multiplexed mass spectrometry. J Proteome Res. 2014;13:2056–68.
Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum Mol Genet. 2009;18:3987–96.
Yuan XZ, Sun S, Tan CC, Yu JT, Tan L. The role of ADAM10 in Alzheimer’s disease. J Alzheimers Dis. 2017;58:303–22.
Westwood S, Liu B, Baird AL, Anand S, Nevado-Holgado AJ, Newby D, et al. The influence of insulin resistance on cerebrospinal fluid and plasma biomarkers of Alzheimer’s pathology. Alzheimers Res Ther. 2017;9:31.
Acknowledgements
We thank Barbara Moullet and Domilė Tautvydaitė for their assistance with and contributions to data acquisition.
Funding
This study was supported by grants from the Swiss National Research Foundation (to JP) (SNF 320030_141179) and funding from the Nestlé Institute of Health Sciences.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon request.
Author information
Authors and Affiliations
Contributions
LD was responsible for study conception and design, acquisition of data, supervision of data acquisition, analysis of data, interpretation of the analysis, and writing of the manuscript. ANG was responsible for acquisition of data and critical revision of the manuscript. JW was responsible for study conception and design, the statistical analysis plan, statistical analysis, drafting of the statistical analysis section, and critical revision of the manuscript. OC was responsible for statistical analysis and critical revision of the manuscript. JC was responsible for acquisition of data and critical revision of the manuscript. AO was responsible for acquisition of data and critical revision of the manuscript. HH was responsible for supervision of data acquisition and critical revision of the manuscript. MK was responsible for supervision of data acquisition and critical revision of the manuscript. EM was responsible for the statistical analysis plan and critical revision of the manuscript. IS was responsible for interpretation of the analysis and critical revision of the manuscript. GLB was responsible for study conception and design, the statistical analysis plan, critical revision of the manuscript, and overall study supervision. JP was responsible for study conception and design, interpretation of the analysis, critical revision of the manuscript, and overall study supervision. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional ethics committee of the University Hospitals of Lausanne approved the clinical protocol (no. 171/2013), and all participants or their legally authorized representatives signed written informed consent forms.
Competing interests
LD, ANG, OC, JC, MK, EM, and IS are employees of Nestlé Institute of Health Sciences. JW is an employee and shareholder of Precision for Medicine and received consultation honoraria from Nestlé Institute of Health Sciences. AO and HH report no competing interests. GLB is an employee of Nestlé Institute of Health Sciences, an unpaid scientific advisor of the H2020 EU-funded project PROPAG-AGEING whose aim is to identify new molecular signatures for early diagnosis of neurodegenerative diseases, and receives research support related to cognitive decline from the National Institute on Aging of the National Institutes of Health. JP received consultation honoraria from Nestlé Institute of Health Sciences.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1: Supplementary Methods.
Table S1. Demographics and clinical characteristics of subjects removed from the statistical analyses. Table S2. Non-AD versus AD CSF biomarker profile group comparison after selection in all subjects of 26 proteins with LASSO. Table S3. Non-AD versus AD CSF biomarker profile group comparison after selection in subjects with cognitive impairment of 18 proteins with LASSO. Table S4. Correlation of CSF proteins with CSF Aβ1-42. Table S5. Correlation of CSF proteins with CSF tau. Table S6. Correlation of CSF proteins with CSF P-tau181. Table S7. Group comparisons of CSF protein measurements for AD versus non-AD CSF biomarker profiles in all subjects. Table S8. Group comparisons of CSF protein measurements for AD versus non-AD CSF biomarker profiles in subjects with cognitive impairment. Figure S1. Box-plots of CSF proteins (selected with LASSO analyses) for positive and negative CSF profiles of AD pathology in all subjects and subjects with cognitive impairment. Figure S2. Pairwise correlation heatmap of the 26 CSF proteins selected with LASSO for classification of non-AD versus AD CSF biomarker profiles for all subjects. Figure S3. Pairwise correlation heatmap of the 18 CSF proteins selected with LASSO for classification of non-AD versus AD CSF biomarker profiles for subjects with cognitive impairment. Figure S4. Correlations of CSF neurogranin and neuromodulin with CSF tau and P-tau181. Figure S5. Chord diagram of the relationships of 59 CSF proteins with CSF tau, P-tau181, and/or Aβ1-42. Figure S6. Venn diagrams of CSF proteins with significant group comparison differences between AD versus non-AD CSF biomarker profiles and those correlating with CSF Aβ1-42, tau, and P-tau181. Figure S7. Venn diagrams of CSF proteins selected with LASSO to classify non-AD versus AD CSF biomarker profiles and those correlating with CSF Aβ1-42, tau, and P-tau181. (DOCX 2575 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dayon, L., Núñez Galindo, A., Wojcik, J. et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alz Res Therapy 10, 66 (2018). https://doi.org/10.1186/s13195-018-0397-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-018-0397-4