
Abstract
Background
Alzheimer’s disease (AD) is a neurodegenerative disease that is clinically characterized by progressive cognitive decline. Mutations in amyloid-β precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) are the pathogenic cause of autosomal dominant AD (ADAD). However, polymorphisms also exist within these genes.
Methods
In order to distinguish polymorphisms from pathogenic mutations, the DIAN Expanded Registry has implemented an algorithm for determining ADAD pathogenicity using available information from multiple domains, including genetic, bioinformatic, clinical, imaging, and biofluid measures and in vitro analyses.
Results
We propose that PSEN1 M84V, PSEN1 A396T, PSEN2 R284G, and APP T719N are likely pathogenic mutations, whereas PSEN1 c.379_382delXXXXinsG and PSEN2 L238F have uncertain pathogenicity.
Conclusions
In defining a subset of these variants as pathogenic, individuals from these families can now be enrolled in observational and clinical trials. This study outlines a critical approach for translating genetic data into meaningful clinical outcomes.
Similar content being viewed by others


1 Background
Alzheimer’s disease (AD) is characterized clinically by progressive cognitive decline and neuropathologically by progressive neuronal loss and the accumulation of amyloid plaques and neurofibrillary tangles. Mutations in amyloid-β precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) are the pathogenic cause of autosomal dominant AD (ADAD). More than 200 pathogenic mutations have been identified in APP, PSEN1, and PSEN2 (reviewed in [1, 2]). PSEN1 and PSEN2 form the catalytic domain of the γ-secretase complex, which is involved in sequential cleavage of APP into amyloid-β (Aβ) peptides.
The Dominantly Inherited Alzheimer Network (DIAN) is an observational study designed to follow families with mutations in APP, PSEN1, and PSEN2 that cause ADAD [3]. The DIAN Expanded Registry (DIAN EXR; www.dianexr.org) is a web-based registry with global outreach to ADAD families and investigators. It functions to identify families with ADAD and to determine the causative mutations through a genetic discovery program. This program includes genetic counseling and testing for known and unknown causes of ADAD. During a period of 6 years (2011 to 2017), the DIAN Clinical/Genetics committee reviewed 150 pedigrees, 76 of which were approved for genetic counseling and testing. Forty-six probands were positive for a coding variant in APP, PSEN1, or PSEN2, including 39 individuals with known pathogenic mutations, 6 with variants of unknown pathogenic significance, and 1 variant that was determined to be an AD risk factor.
In some cases, families are identified with several generations of early-onset AD; however, at the time of enrollment, whether a pathogenic mutation is the cause of disease in these families remains unknown. We performed genetic analyses in research participants from six densely affected early-onset AD pedigrees and identified variants in APP, PSEN1, and PSEN2 with unknown pathogenicity. In all families examined, we lacked sufficient genetic material to perform segregation analyses (e.g., DNA was available from only one or two family members). To assess the pathogenicity of novel variants in APP, PSEN1, and PSEN2 when pedigree and clinical data are limited or incomplete, Guerreiro and colleagues [4] proposed a pathogenicity algorithm. In the present study, we modified and expanded this algorithm to evaluate the pathogenicity of these six variants using genetic, biochemical, biomarker, and clinical data.
2 Methods
2.1 Genetic screening
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols. All coding exons in APP, PSEN1, and/or PSEN2 were amplified and sequenced using the BigDye Terminator version 3.1 cycle sequencing kit (Life Technologies, Carlsbad, CA, USA) and analyzed on an ABI 3500 genetic analyzer (Life Technologies). Sequence analysis was performed using Sequencher software (Gene Codes, Ann Arbor, MI, USA).
2.2 Bioinformatics
To determine whether APP, PSEN1, and PSEN2 variants represented rare or common polymorphisms, we investigated two population-based exome sequencing databases: the Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) browser. Polymorphism phenotype v2 (PolyPhen-2; [5]) and Sorting Intolerant From Tolerant (SIFT) were used to predict whether the amino acid change would be disruptive to the encoded protein.
2.3 Cerebrospinal fluid
Cerebrospinal fluid (CSF) was collected by lumbar puncture under fasting conditions from the PSEN1 M84V carrier and noncarrier. CSF Aβ42, total tau, and tau phosphorylated at threonine 181 (p-tau181) were measured by immunoassay (xMAP; Luminex, Austin, TX, USA) as previously described [6].
2.4 PiB imaging
Structural magnetic resonance imaging (MRI) was performed using 3-T scanners and following the Alzheimer’s Disease Neuroimaging Initiative (ADNI) protocol [7, 8]. T1-weighted scans were processed through FreeSurfer. Positron emission tomography with Pittsburgh compound B (PiB) was coregistered with MRI images. The standardized uptake value ratio (SUVR) was calculated for each region [9, 10]. Global burden was characterized using a summary measure [9]. These data were collected for a PSEN1 M84V carrier and noncarrier.
2.5 Biochemical analysis
2.5.1 Plasmids and mutagenesis
The full-length PSEN1 complementary DNA (cDNA) was cloned into pcDNA3.1 Myc/His vector [11]. The M84V, c.379_382delXXXXinsG, or A396T variant was introduced into the PSEN1 cDNA using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Clones were sequenced to confirm the presence of the variant and the absence of additional modifications. PSEN1 wild type (WT) and the pathogenic PSEN1 A79V mutation were included as controls.
The full-length PSEN2 cDNA was cloned into pcDNA3.1 vector [12]. The L238F or R284G variant was introduced into the PSEN2 cDNA and screened as described above. PSEN2 WT and the pathogenic PSEN2 N141I mutation were included as controls [13].
The full-length APP cDNA (isoform 695) was cloned into pcDNA3.1 [14]. The T719N variant was introduced into the APP cDNA and screened as described above. APP WT and the pathogenic APP KM670/671NL(Swe) mutation were included as controls.
2.5.2 Transient transfection
To assess novel PSEN1 and PSEN2 variants, we used neuroblastoma cells (N2A) stably expressing human APP WT (695 isoform; N2A695). To assess novel APP variants, we used N2A. N2A cells were maintained in equal amounts of DMEM and Opti-MEM, supplemented with 5% FBS, 2 mM L-glutamine, 100 μg/ml penicillin/streptomycin, and, for stable cells, 200 μg/ml G418 (Life Technologies). Upon reaching confluence, cells were transiently transfected with Lipofectamine 2000 reagent (Life Technologies). Culture media were replaced after 24 hours, and cells were incubated for another 24 hours prior to analysis. Four independent transfections were performed for each construct and used for subsequent analyses.
2.5.3 Aβ enzyme-linked immunosorbent assay
Conditioned medium was collected and centrifuged at 3000 × g at 4 °C for 10 minutes to remove cell debris. The levels of Aβ40 and Aβ42 were measured in cell culture media by sandwich enzyme-linked immunosorbent assay (ELISA) as described by the manufacturer (Life Technologies). To account for variability in transfection efficiency between experiments, ELISA values were obtained (pg/ml) and corrected for total intracellular protein (μg/ml). Statistical difference was measured using an unpaired Student’s t test.
3 Results and discussion
3.1 PSEN1 M84V
The proband was identified in a family with three generations of early-onset AD and with a mean age at onset of 59 years (Fig. 1; pedigree not shown to avoid disclosing mutation status to asymptomatic proband and other family members). Sequencing of the proband revealed a single base substitution (ATG to GTG) at codon 84 in exon 4 of PSEN1, resulting in a methionine-to-valine change (M84V). PSEN1 M84V was also identified in a family-based sequencing study of AD (National Institute of Mental Health Alzheimer’s Disease Genetics Initiative Study) [15]. Two PSEN1 M84V carriers were confirmed to have had AD at autopsy with ages at symptomatic onset of 70 and 72 years. The third carrier was cognitively normal at age 67 years [15].

Identification of APP, PSEN1, and PSEN2 variants in densely affected Alzheimer’s disease (AD) pedigrees. a–e Pedigrees. Half-shaded triangles represent individuals with a clinical diagnosis of symptomatic AD. Fully shaded triangles represent individuals with autopsy-confirmed symptomatic AD. Diagonal lines represent deceased individuals. Arrows indicate those individuals with DNA, all of whom are mutation/variant carriers. Pedigrees have been masked to maintain anonymity. The pedigree of the PSEN1 M84V family was excluded to prevent potential disclosure of mutation status in asymptomatic mutation carriers
To determine whether PSEN1 M84V represents a rare polymorphism, we examined two population-based exome sequencing databases (Table 1). PSEN1 M84V was absent from both EVS and ExAC. PSEN1 M84L was identified in one individual in the ExAC browser. PSEN1 M84V was also absent in more than 1700 AD and control samples with whole-exome sequencing.
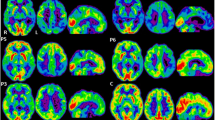
CSF Aβ, total tau, and phosphorylated tau were measured in the PSEN1 M84V carrier, who was cognitively normal at the time of lumbar puncture. CSF Aβ (361.5 pg/ml), total tau (84.93 pg/ml), and p-tau181 (22.02 pg/ml) at 11 years prior to the parental age at symptomatic onset was consistent with other presymptomatic pathogenic mutation carriers but not completely distinct from noncarriers [3, 6]. The PSEN1 M84V carrier produced a mean cortical PiB SUVR value of 1.132 at 15 years prior to the parental age at symptomatic onset and 1.209 at 11 years prior to parent age of disease onset (Fig. 2). A related noncarrier was imaged at 16 years prior to the parent age of disease onset and produced mean cortical PiB SUVR values of 1.086 and 1.039 at 13 years prior to age at disease onset in the parent (Fig. 2). Thus, fluid and imaging biomarkers were consistent with those observed in presymptomatic ADAD mutation carriers.

Pittsburgh compound B (PiB) uptake in the brain of a presymptomatic PSEN1 M84V carrier is consistent with presymptomatic autosomal dominant Alzheimer’s disease mutation carriers. 11C-PiB positron emission tomographic scans were performed longitudinally in a PSEN1 M84V noncarrier and carrier. The color scale for standardized uptake values (SUV) indicate red (high), yellow (medium), and blue (low) PiB retention. EYO Estimated years of onset
We next sought to determine whether the PSEN1 M84V variant alters Aβ isoform levels in a manner consistent with previously reported pathogenic PSEN1 mutations. We expressed vectors containing PSEN1 WT, A79V (a known pathogenic mutation), and M84V in N2A695 cells. We found that cells expressing PSEN1 M84V produced significantly more Aβ42 than cells expressing PSEN1 WT (Fig. 3a–c). We also observed a significant increase in the Aβ42/Aβ40 ratio in cells expressing PSEN1 M84V or the pathogenic PSEN1 A79V- compared with PSEN1 WT-expressing cells (Fig. 3c).

Amyloid-β 1–42 peptide (Aβ42) and Aβ40 in cells expressing APP, PSEN1, and PSEN2 variants of unknown pathogenicity. N2A695 cells were transfected with vectors expressing presenilin 1 or 2. Media was replaced 24 hours posttransfection and incubated for an additional 24 hours. Media were collected, and Aβ42 and Aβ40 were measured by enzyme-linked immunosorbent assay (ELISA) (pg/ml). Total intracellular protein was measured by bicinchoninic acid assay and used to normalize to ELISA Aβ values, resulting in a value represented as pg/μg (see the Methods section of text). a–c PSEN1 wild type (WT), pathogenic mutation A79V, and variants with unknown pathogenicity. a Aβ42. b Aβ40. c Aβ42/Aβ40 ratio. d–f PSEN2 WT, pathogenic mutation N141I, and variants with unknown pathogenicity. d Aβ42. e Aβ40. f Aβ42/Aβ40 ratio. g–i APP WT, pathogenic mutation KM670/671NL(Swe), and APP T719N. Graphs represent the mean (±SEM) of four replicate experiments. * p < 0.05. PSEN1 QR127G is the amino acid representation for PSEN1 c.379_382delXXXXinsG
Thus, we can apply a pathogenicity algorithm, modified from Guerreiro and colleagues [4], to assess the pathogenicity of PSEN1 M84V (Fig. 4). PSEN1 M84V occurs within exon 4 and the first transmembrane domain. This residue is highly conserved between PSEN1 and PSEN2 [16]. Additionally, a pathogenic mutation has been reported at this site: ΔI83/M84 [17, 18]. In a cell model, Aβ levels were consistent with pathogenic mutations (Fig. 3a–c, g). Thus, PSEN1 M84V satisfies all criteria for pathogenicity based on residue and Aβ levels [4]. Taken together, we propose that PSEN1 M84V represents a pathogenic mutation.

Algorithm to classify the benign or pathogenic nature of APP, PSEN1, and PSEN2 variants. This model is modified from the algorithm previously proposed by Guerreiro et al. in 2010 [4]. The modifications include the evaluation of variants in the Exome Variant Server and Exome Aggregation Consortium databases and a tiered approach to evaluating functional studies that more heavily weighs the impact of the variant on amyloid-β 1–42 peptide (Aβ42) and Aβ40 levels on pathogenicity
3.2 PSEN1 A396T
The proband was identified in a family with three generations of early-onset AD (Fig. 1a). The proband had an age at symptomatic onset of 50 years. The parent of the proband had an age at symptomatic onset of 57 years, with AD confirmed at autopsy at age 67 years. Sequencing of the proband (Fig. 1a) revealed a single base substitution (GCG to ACG) at codon 396 in exon 11 of PSEN1, resulting in an alanine-to-threonine change (A396T). This variant was reported previously in one individual with sporadic AD [19]. PSEN1 A396T was absent in the EVS and ExAC databases (Table 1). We found that cells expressing PSEN1 A396T produced significantly more Aβ42 than cells expressing PSEN1 WT (Fig. 3a–c). Thus, applying the algorithm for assessing pathogenicity (Fig. 4), we propose that the PSEN1 A396T represents a probable pathogenic mutation.
3.3 PSEN1 c.379_382delXXXXinsG
The proband had an age at symptomatic onset of 50 years, with an average age at symptomatic onset in family members aged 43 years (Fig. 1b). Sequencing of the proband (Fig. 1b) revealed a 4-bp deletion and insertion of G at codon 127 in exon 5 of PSEN1, resulting in the deletion of glutamine and arginine and insertion of glycine (c.379_382delXXXXinsG). This is a novel variant and was absent from the EVS and ExAC databases (Table 1). N2A695 expressing PSEN1 c.379_382delXXXXinsG produced Aβ42 and Aβ40 levels similar to PSEN1 WT (Fig. 3a–c). Thus, we propose that PSEN1 c.379_382delXXXXinsG is an AD risk factor or benign polymorphism.
3.4 PSEN2 R284G
The proband had an age at symptomatic onset of 58 years, with average age at symptomatic onset in the family of 56 years (Fig. 1c). Sequencing of the proband (Fig. 1c) revealed a single base pair substitution (CGG to GGG) at codon 284 in exon 8 of PSEN2, resulting in an arginine-to-glycine change (R284G). This is a novel variant and was absent from the EVS and ExAC databases (Table 1).
To assess the effects of PSEN2 variants on Aβ isoform levels, we expressed vectors containing PSEN2 WT, N141I, and R284G in N2A695 cells. Cells expressing PSEN2 N141I and R284G produced significantly more Aβ42 than cells expressing PSEN2 WT (Fig. 3d and e). The Aβ42/Aβ40 ratio was also significantly higher in cells expressing PSEN2 N141I and R284G (Fig. 3f). Thus, applying the algorithm for assessing pathogenicity (Fig. 4), we propose that the PSEN2 R284G represents a probable pathogenic mutation.
3.5 PSEN2 L238F
The proband was identified in a family with three generations of early-onset AD (Fig. 1d). The proband had an age at symptomatic onset of 49. The parent of the proband was diagnosed with AD at 57 years of age and died at age 77. Sequencing of the proband (Fig. 1d) revealed a single base pair substitution (CTT to TTT) at codon 238 in exon 7 of PSEN2, resulting in a leucine-to-phenylalanine change (L238F). PSEN2 L238F was also identified in two alleles in EVS, in two alleles in ExAC, and in one individual with sporadic AD (Table 1; [20]). CSF Aβ, total tau, and phosphorylated tau were measured in a PSEN2 L238F carrier related to the proband, who was cognitively normal at the time of lumbar puncture. CSF Aβ (506.54 pg/ml), total tau (38.75 pg/ml), and p-tau181 (21.33 pg/ml) at 17 years prior to the parental age at symptomatic onset was consistent with biomarker levels in normal controls [3, 6]. Cells expressing PSEN2 L238F produced Aβ42 and Aβ40 levels similar to PSEN2 WT (Fig. 3d–f). Thus, we propose that PSEN2 L238F represents an AD risk factor or benign polymorphism.
3.6 App T719N
The proband was identified in a family with two generations of early-onset AD (Fig. 1e). The proband had an age at symptomatic onset of 45 years. The parent of the proband was diagnosed at 45 years of age. Sequencing of the proband (Fig. 1e) revealed a single base pair substitution (ACC to AAC) at codon 719 in exon 17 of APP, resulting in a threonine-to-asparagine change (T719N). APP T719N was absent in the EVS and ExAC databases and was detected in one individual with early-onset AD (Table 1) [21]. Cells expressing APP T719N produced significantly elevated levels of Aβ42 and Aβ42/Aβ40 relative to APP WT (Fig. 3g–i ). Thus, we propose that APP T719N is a probable pathogenic mutation.
4 Conclusions
By applying genetic, bioinformatic, and functional data to an algorithm to assess pathogenicity, we propose that the PSEN1 M84V, PSEN1 A396T, PSEN2 R284G, and APP T719N are likely pathogenic mutations resulting in ADAD, whereas PSEN1 c.379_382delXXXXinsG and PSEN2 L238F are likely benign polymorphisms. This algorithm was adapted and modified from a pathogenicity algorithm originally proposed by Guerreiro and colleagues [4]. We have expanded upon this algorithm in several important ways: (1) expanding the number of controls in the association analyses from 100 to 65,000 by exploiting the EVS and ExAC databases; (2) evaluating the bioinformatic functional findings (e.g., conservation between PSEN1 and PSEN2 and the presence of other mutations at the same residue) independent of the cell-based functional findings; and (3) incorporating cell-based assays to evaluate the impact of novel variants on Aβ levels. We propose that this modified approach to assessing pathogenicity provides an important pipeline for incorporating mutation data at several levels and that this algorithm may be adapted to impute pathogenicity when extensive genetic data are missing for affected families. Designation of a variant as pathogenic will allow individuals to enroll in observational and clinical trials for AD, with clear applications in clinical and research settings.
References
Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26.
Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–4.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804.
Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31:725–31.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TL, Ghetti B, Martins RN, Masters CL, Mayeux R, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med. 2014;6:226ra230.
Jack CR Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, Whitwell JL, Ward C, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91.
Jack CR Jr, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, et al. Update on the magnetic resonance imaging core of the Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 2010;6:212–20.
Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One. 2013;8:e73377.
Su Y, Blazey TM, Owen CJ, Christensen JJ, Friedrichsen K, Joseph-Mathurin N, Wang Q, Hornbeck RC, Ances BM, Snyder AZ, et al. Quantitative amyloid imaging in autosomal dominant Alzheimer’s disease: results from the DIAN study group. PLoS One. 2016;11:e0152082.
Brunkan AL, Martinez M, Walker ES, Goate AM. Presenilin endoproteolysis is an intramolecular cleavage. Mol Cell Neurosci. 2005;29:65–73.
Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, et al. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–9.
Walker ES, Martinez M, Brunkan AL, Goate A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Aβ42/40 ratios. J Neurochem. 2005;92:294–301.
Wang J, Brunkan AL, Hecimovic S, Walker E, Goate A. Conserved “PAL” sequence in presenilins is essential for gamma-secretase activity, but not required for formation or stabilization of gamma-secretase complexes. Neurobiol Dis. 2004;15:654–66.
Hooli BV, Kovacs-Vajna ZM, Mullin K, Blumenthal MA, Mattheisen M, Zhang C, Lange C, Mohapatra G, Bertram L, Tanzi RE. Rare autosomal copy number variations in early-onset familial Alzheimer’s disease. Mol Psychiatry. 2014;19:676–81.
Hardy J, Crook R. Presenilin mutations line up along transmembrane alpha-helices. Neurosci Lett. 2001;306:203–5.
Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, Wood NW, Kumar-Singh S, Geddes J, Swash M, et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-β concentrations. Ann Neurol. 2000;48:806–8.
Steiner H, Revesz T, Neumann M, Romig H, Grim MG, Pesold B, Kretzschmar HA, Hardy J, Holton JL, Baumeister R, et al. A pathogenic presenilin-1 deletion causes abberrant Aβ42 production in the absence of congophilic amyloid plaques. J Biol Chem. 2001;276:7233–9.
Lohmann E, Guerreiro RJ, Erginel-Unaltuna N, Gurunlian N, Bilgic B, Gurvit H, Hanagasi HA, Luu N, Emre M, Singleton A. Identification of PSEN1 and PSEN2 gene mutations and variants in Turkish dementia patients. Neurobiol Aging. 2012;33(1850):e1817–27.
Sala Frigerio C, Lau P, Troakes C, Deramecourt V, Gele P, Van Loo P, Voet T, De Strooper B. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimers Dement. 2015;11:1265–76.
Scahill RI, Ridgway GR, Bartlett JW, Barnes J, Ryan NS, Mead S, Beck J, Clarkson MJ, Crutch SJ, Schott JM, et al. Genetic influences on atrophy patterns in familial Alzheimer’s disease: a comparison of APP and PSEN1 mutations. J Alzheimers Dis. 2013;35:199–212.
Acknowledgements
We gratefully acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study. The DIAN Expanded Registry welcomes contact from any families or treating clinicians interested in research about autosomal dominant familial Alzheimer’s disease. This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders and the departments of neurology and psychiatry at Washington University School of Medicine. This article was reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN study publications. We acknowledge the altruism of the participants and their families and the contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study. We thank the DIAN Steering Committee: Ricardo Allegri, Tammie Benzinger, Sarah Berman, Virginia Buckles, Nigel Cairns, Helena Chui, Maritza Ciliberto, Anne Fagan, Howard Feldman, Bernardino Ghetti, Neill Graff-Radford, David Holtzman, Rachel Huber, Mathias Jucker, Jae-Hong Lee, Johannes Levin, Daniel Marcus, Ralph Martins, Colin Masters, Hiroshi Mori, James Noble, Nick Fox, Stephen Salloway, Peter Schofield, Michael Weiner, and Chengjie Xiong.
Funding
Funding provided by the DIAN Expanded Registry (to RJB), National Institutes of Health grant K01 AG046374 (to CMK) and DIAN-TU Pharma Consortium (https://dian.wustl.edu/our-research/the-pharma-consortium/; to RJB, CC, AMG, CMK), Fidelity Biosciences Research Initiative (RJB, CC, and CMK), and the Gerald and Henrietta Rauenhorst Foundation (RJB, CC, and CMK). Data collection and sharing for this project were supported by The Dominantly Inherited Alzheimer’s Network (DIAN; grant UF1AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), and the Institute for Neurological Research Dr. Raul Carrea (FLENI). Partial support was received through research and development grants for dementia from the Japan Agency for Medical Research and Development (AMED) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI). The funding bodies played no role in the design of the study; in the collection, analysis, and interpretation of data; or in the writing of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
SH and CMK wrote the manuscript. CMK designed cell culture studies. SH, AL, and CMK performed cell culture experiments. JBN, DL, EZ, RL, GSD, EM, JC, JCM, and RJB recruited participants. BAG, RH, and TLSB performed and analyzed brain imaging. AMF analyzed CSF biomarker data. EZ, GSD, EM, AMG, CC, and RJB reviewed pedigrees. CMK performed bioinformatic analysis. All authors contributed to critical revision of the manuscript for intellectual content, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Washington University Institutional Review Board reviewed the study protocol (IRB no. 201111194). All subjects included in this study, or their proxies, gave written informed consent.
Competing interests
AMG is a member of the scientific advisory board for Denali Therapeutics and serves on a genetic scientific advisory panel for Pfizer. RJB receives laboratory research funding from the National Institutes of Health, the Alzheimer’s Association, the BrightFocus Foundation, the Rainwater Foundation Tau Consortium, the Association for Frontotemporal Degeneration, the Cure Alzheimer’s Fund, and the tau SILK Consortium (AbbVie, Biogen, and Eli Lilly and Co.). Funding for clinical trials not related to this research include the National Institutes of Health, the Alzheimer’s Association, Eli Lilly and Co., Hoffman La-Roche, Janssen, Avid Radiopharmaceuticals, the GHR Foundation, and an anonymous foundation. RJB also receives research funding from the DIAN Pharma Consortium (AbbVie, Amgen, AstraZeneca, Biogen, Eisai, Eli Lilly and Co., Hoffman La-Roche, Janssen, Pfizer, and Sanofi). RJB has received honoraria from Janssen and Pfizer as a speaker and from Merck and Pfizer as an advisory board member. Washington University, RJB, and DMH have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on the scientific advisory board. Washington University, with RJB as coinventor, has submitted the U.S. nonprovisional patent application “Methods for measuring the metabolism of CNS derived biomolecules in vivo” and the provisional patent application “Plasma based methods for detecting CNS amyloid deposition.” The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hsu, S., Gordon, B.A., Hornbeck, R. et al. Discovery and validation of autosomal dominant Alzheimer’s disease mutations. Alz Res Therapy 10, 67 (2018). https://doi.org/10.1186/s13195-018-0392-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-018-0392-9