
Abstract
Background
DNA methylation in miRNA genes has been reported as a mechanism that may cause dysregulation of mature miRNAs and consequently impact the gene expression. This mechanism is largely unstudied in papillary thyroid carcinomas (PTC).
Methods
To identify differentially methylated miRNA-encoding genes, we performed global methylation analysis (Illumina 450 K), integrative analysis (TCGA database), data confirmation (pyrosequencing and RT-qPCR), and functional assays.
Results
Methylation analysis revealed 27 differentially methylated miRNA genes. The integrative analyses pointed out miR-21 and miR-146b as potentially regulated by methylation (hypomethylation and increased expression). DNA methylation and expression patterns of miR-21 and miR-146b were confirmed as altered, as well as seven of 452 mRNAs targets were down-expressed. The combined methylation and expression levels of miR-21 and miR-146b showed potential to discriminate malignant from benign lesions (91–96% sensitivity and 96–97% specificity). An increased expression of miR-146b due to methylation loss was detected in the TPC1 cell line. The miRNA mimic transfection highlighted putative target mRNAs.
Conclusions
The increased expression of miR-21 and miR-146b due to loss of DNA methylation in PTC resulted in the disruption of the transcription machinery and biological pathways. These miRNAs are potential diagnostic biomarkers, and these findings provide support for future development of targeted therapies.
Similar content being viewed by others

Background
Papillary thyroid carcinoma (PTC) is the most frequent thyroid malignant neoplasm and is responsible for the increased incidence of thyroid cancer worldwide [1, 2]. The main genetic alterations described in PTC are BRAF and RAS mutations and RET rearrangements [3]. Furthermore, TERT promoter mutations have been associated with more aggressive thyroid carcinomas [4, 5], especially in tumors harboring BRAF mutations [4].
DNA methylation and microRNA (miRNA) are events capable to regulate the expression of genes related to cancer development, as previously reported in PTC [6,7,8,9]. A recent study conducted by our group identified DNA methylation alterations related to prognosis in well differentiated thyroid lesions [10]. Using a robust methylation platform in 141 thyroid samples (non-neoplastic tissue, benign lesions, and carcinomas), we developed a prognostic classifier based on 21 CpGs. This classifier was able to distinguish well-differentiated thyroid carcinomas of patients showing worse prognosis (relapse during the follow-up) from those with good prognosis (without relapse in the follow-up) [10].
Similar to protein-encoding genes, miRNAs are transcribed and could be targets of epigenetic events that modulate their expression [11]. In the last few years, the number of miRNAs described as regulated by DNA methylation increased substantially [11,12,13]. However, the characterization of this epigenetic modification and its functional role in the control of miRNA expression are poorly explored in PTC. To our knowledge, only one study described miRNAs putatively regulated by methylation in thyroid cancer [7]. Nonetheless, neither confirmation nor functional experiments have been developed in this field. This knowledge can contribute to better understanding of PTC biology leading to the discovery of biomarkers and new therapeutic strategies.
Herein, a comprehensive DNA methylation data from PTC and matched NT (non-neoplastic tissue) samples, previously described by our group [8, 10], were re-evaluated focusing in the identification of miRNA genes potentially regulated by methylation. External molecular dataset from TCGA were assessed to perform integrative analysis using DNA methylation, miRNAs expression, and target mRNAs data. The miRNA-coding genes, MIR21 and MIR146B, were further investigated in an independent sample set, and functional assays were carried out in PTC cell lines.
Results
MicroRNA genes differentially methylated in PTC
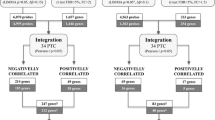
The main strategies to identify miRNAs potentially regulated by methylation are depicted in Fig. 1. DNA methylation analysis comparing paired PTC (N = 50) and NT (N = 50) samples revealed 50 CpG probes (34 miRNA genes) differentially methylated, of which 86% (42 probes mapped in 27 miRNA genes) was confirmed using the TCGA database (Table 1). A supervised hierarchical clustering analysis with all 42 probes revealed an enrichment of hypomethylation in both, our PTC cases and in the TCGA dataset (Additional file 1: Figure S1).

Flowchart showing the strategies and results obtained using bioinformatic tools to identify miRNA genes potentially regulated by methylation, their target mRNA and data confirmation studies. PTC, papillary thyroid carcinoma; NT, non-neoplastic thyroid tissue; BTL, benign thyroid lesion; P adj, adjusted P value, |Δβ|, delta beta; FC, fold-change; r−, negative correlation; *Pearson test
Impact of disrupted DNA methylation in miRNA expression
To identify alterations in the expression levels of miRNA genes in PTC, we assessed miRNA sequencing data from TCGA, revealing 58 miRNAs (27 up and 31 down expressed) (Additional file 2: Table S1). Only MIR21 (hsa-miR-21-5p) and MIR146B (hsa-miR-146b-5p and hsa-miR-146b-3p) showed differential methylation and expression (hypomethylation with increased expression). The integrative analysis revealed highly significant negative correlation between methylation and miRNAs expression (Additional file 2: Table S2).
Alterations in mRNAs targeted by miRNAs potentially regulated by methylation
The mRNA data analysis from TCGA revealed 2432 differentially expressed coding transcripts in PTC (Additional file 2: Table S3), 452 of them were considered as targets of the miRNAs affected by DNA methylation. Among these, 250, 243, and 189 mRNAs are predicted to interact with hsa-miR-146b-3p, hsa-miR-146b-5p, or hsa-miR-21-5p, respectively (681 miRNA-mRNA predicted interactions with significant negative correlation) (Additional file 2: Table S4).
MicroRNAs and target genes associated with poor prognostic features
The DNA methylation levels of MIR146B and MIR21, expression of the miRNAs (hsa-miR-146b-5p, hsa-miR-146b-3p, and hsa-miR-21), and their target transcripts (452 mRNAs from the integrative analysis) were compared with the clinical-pathological findings (TCGA dataset). Hypomethylation and increased expression of MIR146B, as well as decreased target genes expression, were significantly associated with features related to poor prognosis (advanced clinical stage, lymph node metastasis, and extrathyroidal extension) and BRAF mutation (Additional file 2: Table S5).
Data confirmation by quantitative bisulfite pyrosequencing and RT-qPCR
Hypomethylation and overexpression of MIR21 and MIR146B in PTC compared with NT and BTL were confirmed using quantitative bisulfite pyrosequencing and RT-qPCR, respectively (Fig. 2a, b). An additional analysis of PTC compared with NT samples from the same patients (18 matched samples for methylation and 17 for miRNA expression) also corroborated these findings (Additional file 1: Figure S2). A negative correlation between the methylation pattern and expression of MIR21 (r = − 0.393; P < 0.001) and MIR146B (r = − 0.649; P < 0.001) was also observed (Fig. 2c).

Quantitative bisulfite pyrosequencing and RT-qPCR confirmed the data from the large-scale analysis. MIR21 and MIR146B were significantly hypomethylated (a) and overexpressed (b) in PTC compared to non-neoplastic tissues (NT and BTL). c Significantly negative correlation between methylation and expression of MIR21 and MIR146B was observed. d Five miRNA-target transcripts showed lower expression in PTC compared to NT and BTL. No significant differences were found between NT and BTL, in exception of DOK6 and FLRT1 genes. e All target transcripts demonstrated significant negative correlation with their miRNA regulators (P < 0.001 to all genes). Scatterplot representation of MIR21 and MIR146B methylation (f) and expression (g) levels. A diagnostic classifier (dashed line) was designed to distinguish PTC from BTL using Fisher’s linear discriminant analysis. The classification performance of the methylation and miRNA-based classifier is illustrated. Seven PTC and three BTL were excluded from the methylation diagnostic classifier (low pyrosequencing quality was observed for at least one of the miRNAs). NS, not significant; *P < 0.05; ***P < 0.001 (ANOVA followed by Tukey test). PTC, papillary thyroid carcinoma (red); NT, non-neoplastic thyroid tissue (blue); BTL, benign thyroid lesions (green); r, Pearson’s correlation coefficient; P, p value obtained by Pearson’s correlation test
Seven target genes selected for RT-qPCR evaluation (DOK6, FHL1, FLRT1, MOB3B, MPPED2, MRO, and STXBP5L) showed decreased expression in PTC compared to NT and BTL (Fig. 2d). The interactions among hsa-miR-146b-5p, hsa-miR-21-5p, and the seven targets, found in the integrative analysis using the TCGA dataset, supported the findings obtained in the RT-qPCR analysis (Fig. 2e).
The pyrosequencing and RT-qPCR results were also confronted with clinical-pathological features and BRAF mutation (observed in 59% of the PTC). A significant association was observed between lower methylation and higher expression levels of MIR146B and MIR21 with BRAF mutation (Additional file 1: Figure S3).
Development of a diagnostic tool in thyroid cancer
The DNA methylation (pyrosequencing) and expression (RT-qPCR) levels of MIR21 and MIR146B were tested as a potential tool to discriminate malignant (PTC) from benign thyroid lesions (BTL). The combination of methylation values of both miRNAs allowed the discrimination of 87 out of 96 PTC and 25 out of 26 BTL (91% sensitivity and 96% specificity) (Fig. 2f). The hsa-miR-21-5p and hsa-miR-146b-5p relative expression was more efficient than methylation to classify correctly 45 of 47 PTC and 31 of 32 BTL (96% sensitivity and 97% specificity) (Fig. 2g). Curiously, the miRNA expression test correctly classified all NT samples as non-malignant (35 of 35), while the methylation test categorized 29 of 40 NT samples (73%) as malignant.
Global demethylation-induced MIR146B expression in TPC1 cell line
Firstly, we investigated the basal methylation of two PTC cell lines (TPC1 and BCPAP) to study hypomethylated CpGs. Whereas MIR146B showed high methylation levels in both cell lines (51–60% of methylated alleles), MIR21 was completely unmethylated even at basal level (5–7% of methylated alleles), rendering no change in miR expression after 5-Aza-dC treatment (Fig. 3a). Loss of global methylation (AlR1Sat and AluYB8 repetitive regions) was confirmed after treatment with 5-Aza-dC. Specific loss of methylation in CpGs mapped in the MIR146B gene and an increased expression level of the hsa-miR-146b-5p in the TPC1 cell line was also observed (Fig. 3b).

Demethylation assays in thyroid cancer cell lines (TPC1 and BCPAP). a Pyrogram showing the percentage of basal methylation of MIR21 (unmethylated in both cell lines) and MIR146B (> 50% methylation in both cell lines). b The demethylation in the mapped region of MIR146B was demonstrated after 5-Aza-dC treatment in TPC1 and BCPAP cell lines. BCPAP cell line had no changes in hsa-miR-146b-5p expression and TPC1 cell line presented seven-fold increased expression levels after the induced demethylation
No change in miR expression.
Decreased expression levels of target mRNAs after miRNA mimic transfection in the TPC1 cell line
A transfection assay using the TPC1 cell line for each miRNA mimic (hsa-miR-146b-3p, hsa-miR-146b-5p and hsa-miR-21-5p) was performed followed by global transcriptomic analysis. From the 452 mRNAs highlighted by the integrative analysis (681 miRNA-mRNA interactions), 168 also showed decreased expression levels after transfection (188 miRNA-mRNA interactions). This strategy gives additional evidence that 136, 112, and 134 mRNA targets are regulated by hsa-miR-146b-3p, hsa-miR-146b-5p, and hsa-miR-21-5p, respectively (Additional file 2: Table S6). The in silico pathway analysis (IPA and KOBAS 3.0) including the mRNA target candidates revealed the involvement of canonical pathways related to neuronal system, thyroid function (mainly for hsa-miR-146b-3p targets), and MAPK/ERK signaling (mainly for hsa-miR-146b-5p targets) (Additional file 2: Tables S7 and S8).
Discussion
Aberrant microRNA expression in thyroid neoplasia was previously reported [14, 15]; however, the mechanisms underlying the regulation of these miRNAs are poorly explored. As possible impact of DNA methylation was previously noticed [7]. In this study, we investigated the methylation profiles of PTC and NT in genes encoding miRNA using a high coverage platform (Illumina 450 k). Although the use of whole genome bisulfite sequencing would encompass more miRNA gene regions, an advantage of our strategy was the inclusion of 565 PTC and 106 NT evaluated by the same platform (internal and external samples from TCGA), which strengthened our findings.
MIR21 exhibited increased expression and decreased methylation levels in PTC compared to NT and BTL. MIR21 overexpression was previously reported in thyroid cancer [16, 17]. The transcribed miRNA (hsa-miR-21) was one of the first oncomiR described and one of the most studied in several tumor types [18,19,20]. In prostate cancer cell lines, MIR21 promoter hypermethylation resulted in its repressed expression [21]. Moreover, the 5-AZA-dC treatment stimulated the expression of this miRNA in prostate and ovary cancer cell lines [21, 22]. However, these studies were restricted to cell lines, and tumor samples were not evaluated to confirm the MIR21 methylation pattern.
Similar to MIR21, MIR146B presented hypomethylation and increased expression levels in PTC. This miRNA was also previously described as overexpressed in PTC [7, 14, 15]. Contrarily, the hypermethylation and down-expression of MIR146B were reported in diffuse and anaplastic astrocytomas, gliomas, and breast cancer being the 5-AZA-dC treatment capable to induce the MIR146B expression [23, 24]. Taken together, these findings give evidences of the regulatory role of MIR146B in different tumor types, being able to act as tumor suppressor or oncomiR, depending on the context of altered regulatory pathways in each tumor type [25,26,27,28].
The identification of miRNAs target-genes is crucial to understand the regulatory mechanisms involved in thyroid cancer cells. In this context, 452 target transcripts were unveiled by the miRNA-mRNA integrative analysis (TCGA dataset). Interestingly, 168 of these 452 putative mRNA targets also exhibited decreased expression levels after the individualized mimic transfection assays using these three miRNAs (hsa-miR-146b-3p, hsa-miR-146b-5p, or hsa-miR-21-5p). Although the cell lines are very useful models, gene expression profiles are not identical with those found in primary tumor tissues. Differences based on epigenetic reprogramming have been reported as a causal effect of the in vitro conditions [29]. In addition, the induction of a single miRNA can influence the expression of many transcriptional factors, which enhances the complexity of the transcriptome regulation network and makes the outcome less predictable [30].
Seven selected transcripts were confirmed as having decreased expression levels in PTC compared to NT and BLT. Among the selected genes, MPPED2, MRO, STXBP5L, FHL1, and FLRT1 were previously reported as down-regulated in PTC [31, 32]. In prostate [33], stomach [34], and breast cancer [35], DOK6 and MOB3B were reported as putative tumor suppressor genes. In follicular tissues of the thyroid gland, these genes could have similar function of tumor suppressors.
We also investigated the association of miRNAs methylation and expression levels of the target-genes with clinical and pathological features of poor prognosis and BRAF mutation. Increased expression levels of MIR146B were related with advanced stage, tumor size, extrathyroidal extension, and BRAF mutation, as previously reported [36,37,38,39]. In our initial analysis using the TCGA cohort, MIR21 and MIR146B hypomethylation and their increased expression levels, as well as decreased levels of several target-genes, were suggested as associated with poor prognosis features. Nevertheless, a clear distinction of the molecular profiles was confirmed using pyrosequencing and RT-qPCR analysis in our BRAF-mutated cases. The association between global DNA methylation and BRAF mutation in PTC has already been extensively explored, and a global hypomethylation in tumors harboring BRAF V600E was reported [8, 40, 41]. The mechanisms possibly involved in the epigenetic control of genes by the BRAF mutation have been recently explored (42–44). In melanoma cells, the expression of 59 hypermethylated genes in the BRAF V600E knockdown were down-expressed suggesting that in mutated cells these genes were hypomethylated and over-expressed [42]. According to the authors, aberrant EZH2 (histone methyltransferase) and DNMT1 (DNA methyltransferase 1) expression were affected by the BRAF mutation [42]. A similar study performed by the same group was reported in two PTC cell lines (BCPAP and OCUT1), showing that BRAF mutation had an impact in the methylation and the expression of several genes, including HMGB2 and FDG1 [43]. A recent study demonstrated that the MAPK/ERK signaling pathway activated by the BRAF mutation was able to induce epigenetic aberrations by H3K27me3 and MYC [44]. Therefore, if BRAF mutation can trigger the epigenetic alterations in coding genes, the same effect is expected to occur in miRNAs.
MIR21 and MIR146B methylation and expression analysis revealed high sensitivity (91% and 96%, respectively) and specificity (96% and 97%, respectively) to distinguish PTC from benign lesions. Although the methylation classifier failed in distinguishing PTC from NT (73% of the NT samples were classified as malignant), the miRNA expression classifier categorized all NT as “non-malignant.” These results suggest that our methylation markers could detect cells in the preliminary steps of malignant transformation (the non-neoplastic samples were obtained from the surrounding thyroid tissue from PTC patients). Epigenetic events are known to anticipate the phenotypic manifestation of malignancy [45, 46].
Considering the similarities observed in the classifiers for molecular diagnosis in thyroid nodules and the high complexity of the methylation assays, miRNAs expression analysis is more easily applicable in the clinical routine. Two recent studies [47, 48] reported the clinical validation of assays based on miRNA expression (Rosetta Genomics, Philadelphia, PA, USA). In these studies, a set of 24 miRNAs were evaluated (among them, hsa-miR-146b-5p) by RT-qPCR in thyroid nodules showing indeterminate cytology. First, an analytical validation was conducted to ensure the test robustness, proving the feasibility of the assay in fine-needle aspiration smears (N = 576 nodules) [47]. Subsequently, a classifier was developed to identify nodules as benign or suspect (N = 189 cases) showing 98% of sensitivity and 78% of specificity [48]. Despite our malignant cohort was only represented by surgical specimens of PTC, we achieved a similar sensitivity and higher specificity.
Although most of studies focused in the discovery of tumor suppressor miRNAs repressed by epigenetic mechanisms [49, 50], only hypomethylated miRNA-encoding genes were detected in our study. Cell lines treated with demethylating agents, as 5-Aza-dC, is widely used to demonstrate a direct regulation by methylation [51]. Even though MIR21 and MIR146B were hypomethylated in PTC, the basal CpG methylation found in MIR146B was relatively high in two cell lines evaluated (TPC1 and BCPAP), allowing a functional assay using 5-Aza-dC. The increased expression of MIR146B after the treatment infers an association with the methylation loss in the miRNA gene promoter.
The role of hsa-mir-146b-5p in proliferation, migration, and invasiveness of thyroid cancer was previously investigated by others using mimics-miR transfection assays in thyroid cell lines [52, 53]. The functional role of hsa-mir-146b-5p in thyroid gland oncogenesis and its association with PTC aggressiveness was reported as involved with the down-regulation of PTEN and E-cadherin [53]. Likewise, it was previously described that inducing hsa-miR-21-5p in TPC1 cell line by plasmid transfection (pEZX-eGFP-miRNA-21) resulted in increased cell proliferation and invasion, and inhibited apoptosis, possibly mediated by PDCD4 repression [16].
Conclusions
The mechanism underlying the overexpression of MIR21 and MIR146B due to DNA methylation loss in PTC was explored in our study. We used, for the first time, small-scale (RT-qPCR and pyrosequencing) analysis and functional assays to corroborate the findings originated from large-scale screening in a large cohort of cases. The interconnections between these epigenetic events are potentially responsible for many deregulations in the thyroid transcriptome leading to cancer development. DNA methylation and expression levels of these miRNA-encoding genes were demonstrated as suitable PTC diagnostic markers. Moreover, the understanding of the mechanisms of upregulation of these oncomiRs in thyroid malignancies creates opportunities to develop miRNA-targeting therapies.
Methods
Sample population
Fifty matched PTC and NT samples from our previous DNA methylation profiling studies were re-evaluated as a discovery set. In addition, a confirmatory subset of 103 PTC, 40 NT, and 32 BTL (benign thyroid lesions: 14 follicular adenomas, 17 goiters, and 1 lymphocytic thyroiditis) snap-frozen tissues were obtained retrospectively from patients treated at A.C. Camargo Cancer Center, São Paulo, SP, Brazil. The Additional file 2: Table S9 summarizes the clinical features of PTC patients. Nucleic acid isolation and BRAF mutation genotyping are detailed in Additional file 3: Supplementary Methods.
Global DNA methylation analysis
DNA methylation profiling were obtained from the 50 paired PTC and NT samples using the Infinium® Human Methylation450 BeadChip Platform (Illumina, San Diego, CA, USA). The data were retrieved from previously generated studies from our group [8, 10] and are available in the Gene Expression Omnibus database (GSE86961 and GSE97466). CpG probes differentially methylated in PTC compared to NT were detected using limma package [54] with adjusted P value (Bonferroni) < 0.05 and delta-beta (Δβ) > 0.15 or < − 0.15. Quality controls and pre-processing data are detailed in Additional file 3: Supplementary Methods.
Integrative analysis using TCGA database
The available TCGA clinical and molecular data were retrieved using UCSC Xena (https://xenabrowser.net/datapages/—accessed in February 2018). DNA methylation data from 515 PTC and 56 NT (Infinium® Human Methylation450 BeadChip) from TCGA dataset were used to confirm the CpG probes differentially methylated identified in our study (t test adjusted P < 0.05, Δβ > 0.1, or < − 0.1). Two strategies using integrative analysis were developed: (i) CpG probes differentially methylated in both cohorts were compared to miRNAs expression from TCGA (miRNASeq IlluminaHiSeq, 509 PTC and 59 NT), and (ii) miRNAs differentially expressed were contrasted with target-genes expression using the TCGA data (RNASeqV2 IlluminaHiSeq) in 505 PTC versus 59 NT (t test adjusted P < 0.05; fold change FC > 2 for both miRNA and mRNA). As different prediction methods are generally uncorrelated [55], miRNA target prediction was carried out with miRWalk 2.0 tool (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/), considering predicted interactions in at least two of four selected algorithms (miRWalk, miRanda, RNAhybrid, and Targetscan). Predictions were based on interactions between miRNA seed sequences (starting from the first position) and the 3′UTR region of the target mRNA. Pearson’s correlation test was applied to investigate negatively correlated predicted interactions in PTC (P < 0.05).
DNA methylation analysis in miRNAs genes by pyrosequencing
To confirm the global DNA methylation results, 500 ng of genomic DNA samples were converted by sodium bisulfite using the EZ DNA Methylation Gold kit (Zymo, Irvine, CA, USA), according to the manufacturer’s recommendations. Only independent samples (103 PTC, 29 BTL, and 40 NT) from the previous global methylation analysis [8, 10] were included. Forward and reverse biotinylated primers (Sigma, Darmstadt, Germany) were used to amplify the region of interest (Additional file 2: Table S10 and Additional file 1: Figure S4). Two probes representative of MIR146B and MIR21 were selected, presenting the highest negative correlation with the corresponding miRNA expression. Primer design and PCR conditions are detailed in Additional file 3: Supplementary Methods. Pyrosequencing (PyroMark Q24 system, Qiagen) was performed including methylated and unmethylated DNA controls according to the manufacturer’s instructions (Zymo Research, Irvine, CA, USA).
MicroRNAs and target mRNAs expression analysis by RT-qPCR
Expression of miRNAs (hsa-miR-21-5p and hsa-miR-146b-5p) and mRNAs (MPPED2, STXBP5L, MRO, FHL1, FLRT1, DOK6, and MOB3B) were performed by RT-qPCR (detailed in Additional file 3: Supplementary Methods) using TaqMan® and SYBR® Green detection system, respectively (Applied Biosystems, CA, USA). The hsa-miR-146b-5p and hsa-miR-146b-3p showed redundant expression in the miRNA sequencing results using the TCGA dataset (PTC = 510, Pearson’s r = 0.970, Pearson’s P not computable). Based on these findings, we selected only hsa-miR-146b-5p to be evaluated by RT-qPCR (most prevalent mature form).
Seven of 452 target transcripts detected in the integrative analysis were selected according to the negative correlation coefficient values, expression levels in normal thyroid tissues (available at https://www.ncbi.nlm.nih.gov/gene), higher FC, and significant clinicopathological variables associated with poor prognosis (obtained from TCGA) (Additional file 2: Table S11). According to these parameters, all seven transcripts are targets of miR-146b-5p, and three of them (DOK6, MPPED2, and STXBP5L) are targets of miR-21-5p. The mRNA primer sequences are described in Additional file 2: Table S12.
For miRNA analysis, TaqMan® microRNA Reverse Transcription Kit (Applied Biosystems) and TaqMan® microRNA Assays (IDs: 000397 and 001097, Applied Biosystems) were used according to the manufacturer’s instructions. miRNA normalization was performed using RNU44 (ID 001094) and RNU48 (ID 001006) [36, 56]. The highly stable references, EIF2B1 and PUM1, were employed for mRNA normalization, as previously described [31]. The method proposed by Pfaffl (2001) [57] was used for normalization with a geometric mean of reference tests and efficiency equal to 100%.
Global demethylation assay in PTC cell lines
The human thyroid cancer cell lines TPC1 (BRAF wild type, received from Janete M Cerutti, Federal University of São Paulo, Brazil) and BCPAP (BRAF V600E, received from Edna T. Kimura, University of São Paulo, Brazil) were in vitro cultured in RPMI (Gibco, Grand Island, NY, USA) and DMEM/F-10 medium (Gibco), respectively, supplemented with 10% fetal bovine serum, 1% streptomycin (Gibco), and 1% penicillin (Gibco). Global demethylation assays were performed using 5-Aza-dC (Sigma, Darmstadt, Germany) at 1 μM and 3 μM determined by cell viability assays (detailed in Additional file 3: Supplementary Methods). Loss of methylation after treatment was confirmed by pyrosequencing using AluYB8 and AlR1Sat primer pairs, as described by Choi et al. (2009) [58]. Basal methylation of MIR21 and MIR146B regions and the corresponding mature miRNAs expression in the cell lines upon treatment were evaluated as described above. The samples treated with 5-Aza-dC were compared to vehicle using three replicates (independent assays) for each cell line.
MicroRNA mimics transfection in the TPC1 cell line
Due to the increased expression of MIR146B after azacitidine treatment in TPC1, this cell line was chosen to conduct the transfection experiments. The cells were seeded 24 h prior to the transfection in 6-well plates with 200,000 cells per well in the medium specific to the cell lines, without supplemented antibiotics. The transfection reagent Lipofectamine RNAiMAX (Invitrogen) was prepared in Opti-MEM medium (Invitrogen), as recommended by the manufacturer. The mirVana miRNA mimics (Invitrogen) hsa-miR-146b-3p, hsa-miR-146b-5p, and hsa-miR-21-5p were dissolved to the relevant concentrations in Opti-MEM medium. The diluted transfection reagent and mimics (final concentration of 30 nM) were then mixed and incubated at room temperature for 5 min. Afterwards, the complexes were added to each well containing cells and Opti-MEM medium. Negative control mimics (30 nM) and a mock control were included in the transfection experiments. The cells were incubated for 48 h before being harvested in trypsin followed by total RNA extraction. The mature miRNAs were reversely transcribed with targeted primers. Moreover, successful transfection was confirmed by RT-qPCR (> 100 increased expression levels for the three miRNAs).
Large-scale transcriptomic analysis after miRNA transfection
The RNA from mimics and control assays was amplified (200 ng), labeled, and hybridized using the Clariom D platform (Affymetrix, Santa Clara, CA, USA) following the manufacturer’s instructions. Two biological replicates were included for each miRNA tested individually. The arrays hybridization was performed in a GeneChip® Hybridization Oven 645 (Affymetrix) and scanned using the GeneChip Scanner 3000 (Affymetrix). The data were analyzed using the Affymetrix Transcriptome Analysis Console software (v. 3.1.0.5) and normalized by the Robust Multiarray Average module. The analysis was focused in the target mRNAs found in the integrative analysis and specifically in transcripts down-expressed (in both duplicates) after the transfection with the mimics.
In silico canonical pathway analysis
Protein-encoding genes predicted as target of the miRNAs potentially regulated by DNA methylation, found in the integrative analysis and down-expressed after the mimic transfection, were submitted to canonical pathway evaluation using the Ingenuity Pathway Analysis (IPA v2.1, Ingenuity Systems) and KOBAS 3.0 software (http://kobas.cbi.pku.edu.cn/).
Statistical analysis
The SPSS v. 21.0 (Statistics Packet for Social Sciences, Chicago, IL, USA) and BRB Array Tools v. 4.4.0 were used for statistical analysis. Graphical representations were implemented by GraphPad Prism v.5.0 (GraphPad Software Inc., La Jolla, CA, USA). The methylation, miRNAs, and target gene expression from TCGA were compared with clinical and pathological data and BRAF mutation using t test (P < 0.001; FDR < 5%; |Δβ| > 0.10 or FC > 1.5). Pyrosequencing (percentage of methylation in each CpG) and RT-qPCR data (miRNAs and mRNAs relative expression) were evaluated by parametric tests (paired and unpaired t test, ANOVA with Turkey’s post hoc, and Pearson’s correlation test). The null hypothesis was rejected when the two-tailed P value was < 0.05. Fisher discriminant analysis was used to construct diagnostic classifier algorithms.
Abbreviations
- 5-Aza-dC:
-
5-aza-deoxycytidine
- BTL:
-
Benign thyroid lesions
- CpG:
-
5′-deoxycytosine-phosphate-deoxyguanosine-3
- IPA:
-
Ingenuity pathways analysis
- KOBAS:
-
KEEG orthology based annotation system
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- NT:
-
Non-neoplastic adjacent tissues
- PTC:
-
Papillary thyroid carcinoma
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
- TCGA:
-
The Cancer Genome Atlas
References
Ito Y, Nikiforov YE, Schlumberger M, Vigneri R. Increasing incidence of thyroid cancer: controversies explored. Nat Rev Endocrinol. 2013;9(3):178–84.
Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. 2017;317(13):1338–48.
Fagin JA, Wells SA. Biologic and clinical perspectives on thyroid cancer. N Engl J Med. 2016;375(11):1054–67.
Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun. 2018;9(1):579.
Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res. 2018;24(13):3059–68.
Rodríguez-Rodero S, Delgado-Álvarez E, Fernández AF, Fernández-Morera JL, Menéndez-Torre E, Fraga MF. Epigenetic alterations in endocrine-related cancer. Endocr Relat Cancer. 2014;21(4):R319–30.
Network CGAR. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159(3):676–90.
Beltrami CM, Dos Reis MB, Barros-Filho MC, Marchi FA, Kuasne H, Pinto CAL, et al. Integrated data analysis reveals potential drivers and pathways disrupted by DNA methylation in papillary thyroid carcinomas. Clin Epigenetics. 2017;9:45.
Buj R, Mallona I, Díez-Villanueva A, Zafon C, Mate JL, Roca M, et al. Kallikreins stepwise scoring reveals three subtypes of papillary thyroid cancer with prognostic implications. Thyroid. 2018;28(5):601–12.
Bisarro Dos Reis M, Barros-Filho MC, Marchi FA, Beltrami CM, Kuasne H, Pinto CAL, et al. Prognostic classifier based on genome-wide DNA methylation profiling in well-differentiated thyroid tumors. J Clin Endocrinol Metab. 2017;102(11):4089–99.
Wang S, Wu W, Claret FX. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics. 2017;12(3):187–97.
Barros-Silva D, Costa-Pinheiro P, Duarte H, Sousa EJ, Evangelista AF, Graça I, et al. MicroRNA-27a-5p regulation by promoter methylation and MYC signaling in prostate carcinogenesis. Cell Death Dis. 2018;9(2):167.
Loginov VI, Pronina IV, Burdennyy AM, Filippova EA, Kazubskaya TP, Kushlinsky DN, et al. Novel miRNA genes deregulated by aberrant methylation in ovarian carcinoma are involved in metastasis. Gene. 2018;662:28–36.
Wójcicka A, Kolanowska M, Jażdżewski K. Mechanisms in endocrinology: microRNA in diagnostics and therapy of thyroid cancer. Eur J Endocrinol. 2016;174(3):R89–98.
Wang T, Xu H, Qi M, Yan S, Tian X. miRNA dysregulation and the risk of metastasis and invasion in papillary thyroid cancer: a systematic review and meta-analysis. Oncotarget. 2018;9(4):5473–9.
Zhang J, Yang Y, Liu Y, Fan Y, Liu Z, Wang X, et al. MicroRNA-21 regulates biological behaviors in papillary thyroid carcinoma by targeting programmed cell death 4. J Surg Res. 2014;189(1):68–74.
Yoruker EE, Terzioglu D, Teksoz S, Uslu FE, Gezer U, Dalay N. MicroRNA expression profiles in papillary thyroid carcinoma, benign thyroid nodules and healthy controls. J Cancer. 2016;7(7):803–9.
Jiang J, Yang P, Guo Z, Yang R, Yang H, Yang F, et al. Overexpression of microRNA-21 strengthens stem cell-like characteristics in a hepatocellular carcinoma cell line. World J Surg Oncol. 2016;14(1):278.
Fang H, Xie J, Zhang M, Zhao Z, Wan Y, Yao Y. miRNA-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting PTEN. Am J Transl Res. 2017;9(3):953–61.
Koutsioumpa M, Chen HW, O'Brien N, Koinis F, Mahurkar-Joshi S, Vorvis C, et al. MKAD-21 suppresses the oncogenic activity of the miR-21/PPP2R2A/ERK molecular network in bladder cancer. Mol Cancer Ther. 2018;17(7):1430–40
Hulf T, Sibbritt T, Wiklund ED, Bert S, Strbenac D, Statham AL, et al. Discovery pipeline for epigenetically deregulated miRNAs in cancer: integration of primary miRNA transcription. BMC Genomics. 2011;12:54.
Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67(18):8699–707.
Xiang M, Birkbak NJ, Vafaizadeh V, Walker SR, Yeh JE, Liu S, et al. STAT3 induction of miR-146b forms a feedback loop to inhibit the NF-κB to IL-6 signaling axis and STAT3-driven cancer phenotypes. Sci Signal. 2014;7(310):ra11.
Wolter M, Werner T, Malzkorn B, Reifenberger G. Role of microRNAs located on chromosome arm 10q in malignant Gliomas. Brain Pathol. 2016;26(3):344–58.
Haghpanah V, Fallah P, Tavakoli R, Naderi M, Samimi H, Soleimani M, et al. Antisense-miR-21 enhances differentiation/apoptosis and reduces cancer stemness state on anaplastic thyroid cancer. Tumour Biol. 2016;37(1):1299–308.
Duan J, Zhang H, Qu Y, Deng T, Huang D, Liu R, et al. Onco-miR-130 promotes cell proliferation and migration by targeting TGFβR2 in gastric cancer. Oncotarget. 2016;7(28):44522–33.
Liu C, Zhang S, Wang Q, Zhang X. Tumor suppressor miR-1 inhibits tumor growth and metastasis by simultaneously targeting multiple genes. Oncotarget. 2017;8(26):42043–60.
Imamura T, Komatsu S, Ichikawa D, Miyamae M, Okajima W, Ohashi T, et al. Depleted tumor suppressor miR-107 in plasma relates to tumor progression and is a novel therapeutic target in pancreatic cancer. Sci Rep. 2017;7(1):5708.
Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC, et al. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015;16:11.
Nazarov PV, Reinsbach SE, Muller A, Nicot N, Philippidou D, Vallar L, et al. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013;41(5):2817–31.
Barros-Filho MC, Marchi FA, Pinto CA, Rogatto SR, Kowalski LP. High diagnostic accuracy based on CLDN10, HMGA2, and LAMB3 transcripts in papillary thyroid carcinoma. J Clin Endocrinol Metabol. 2015;100(6):E890–E9.
Lan X, Zhang H, Wang Z, Dong W, Sun W, Shao L, et al. Genome-wide analysis of long noncoding RNA expression profile in papillary thyroid carcinoma. Gene. 2015;569(1):109–17.
Kim EA, Kim YH, Kang HW, Yoon HY, Kim WT, Kim YJ, et al. Lower levels of human MOB3B are associated with prostate cancer susceptibility and aggressive Clinicopathological characteristics. J Korean Med Sci. 2015;30(7):937–42.
Leong SH, Lwin KM, Lee SS, Ng WH, Ng KM, Tan SY, et al. Chromosomal breaks at FRA18C: association with reduced. NPJ Precis Oncol. 2017;1(1):9.
Ghanem T, Bracken J, Kasem A, Jiang WG, Mokbel K. mRNA expression of DOK1-6 in human breast cancer. World J Clin Oncol. 2014;5(2):156–63.
Sheu SY, Grabellus F, Schwertheim S, Worm K, Broecker-Preuss M, Schmid KW. Differential miRNA expression profiles in variants of papillary thyroid carcinoma and encapsulated follicular thyroid tumours. Br J Cancer. 2010;102(2):376–82.
Geraldo MV, Yamashita AS, Kimura ET. MicroRNA miR-146b-5p regulates signal transduction of TGF-β by repressing SMAD4 in thyroid cancer. Oncogene. 2012;31(15):1910–22.
Chou CK, Liu RT, Kang HY. MicroRNA-146b: A Novel Biomarker and Therapeutic Target for Human Papillary Thyroid Cancer. Int J Mol Sci. 2017;18(3):e636.
Qiu Z, Li H, Wang J, Sun C. miR-146a and miR-146b in the diagnosis and prognosis of papillary thyroid carcinoma. Oncol Rep. 2017;38(5):2735–40.
White MG, Nagar S, Aschebrook-Kilfoy B, Jasmine F, Kibriya MG, Ahsan H, et al. Epigenetic Alterations and Canonical Pathway Disruption in Papillary Thyroid Cancer: A Genome-wide Methylation Analysis. Ann Surg Oncol. 2016;23(7):2302–9.
Mancikova V, Buj R, Castelblanco E, Inglada-Pérez L, Diez A, de Cubas AA, et al. DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int J Cancer. 2014;135(3):598–610.
Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012;11(2):286–95.
Hou P, Liu D, Xing M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr Relat Cancer. 2011;18(6):687–97.
Qu Y, Yang Q, Liu J, Shi B, Ji M, Li G, et al. C-Myc is required for BRAF(V600E)-induced epigenetic silencing by H3K27me3 in tumorigenesis. Theranostics. 2017;7(7):2092–107.
van Hoesel AQ, Sato Y, Elashoff DA, Turner RR, Giuliano AE, Shamonki JM, et al. Assessment of DNA methylation status in early stages of breast cancer development. Br J Cancer. 2013;108(10):2033–8.
Rodríguez-Paredes M, Bormann F, Raddatz G, Gutekunst J, Lucena-Porcel C, Köhler F, et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat Commun. 2018;9(1):577.
Benjamin H, Schnitzer-Perlman T, Shtabsky A, VandenBussche CJ, Ali SZ, Kolar Z, et al. Analytical validity of a microRNA-based assay for diagnosing indeterminate thyroid FNA smears from routinely prepared cytology slides. Cancer Cytopathol. 2016;124(10):711–21.
Lithwick-Yanai G, Dromi N, Shtabsky A, Morgenstern S, Strenov Y, Feinmesser M, et al. Multicentre validation of a microRNA-based assay for diagnosing indeterminate thyroid nodules utilising fine needle aspirate smears. J Clin Pathol. 2017;70(6):500–7.
Zhang L, Yan DL, Yang F, Wang DD, Chen X, Wu JZ, et al. DNA methylation mediated silencing of microRNA-874 is a promising diagnosis and prognostic marker in breast cancer. Oncotarget. 2017;8(28):45496–505.
Toll A, Salgado R, Espinet B, Díaz-Lagares A, Hernández-Ruiz E, Andrades E, et al. MiR-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol Cancer. 2016;15(1):53.
Sato T, Issa JJ, Kropf P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb Perspect Med. 2017;7(5):a026948
Lima CR, Geraldo MV, Fuziwara CS, Kimura ET, Santos MF. MiRNA-146b-5p upregulates migration and invasion of different papillary thyroid carcinoma cells. BMC Cancer. 2016;16:108.
Ramírez-Moya J, Wert-Lamas L, Santisteban P. MicroRNA-146b promotes PI3K/AKT pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene. 2018;37(25):3369–83.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Gamazon ER, Im HK, Duan S, Lussier YA, Cox NJ, Dolan ME, et al. Exprtarget: an integrative approach to predicting human microRNA targets. PLoS One. 2010;5(10):e13534.
Dettmer M, Perren A, Moch H, Komminoth P, Nikiforov YE, Nikiforova MN. Comprehensive MicroRNA expression profiling identifies novel markers in follicular variant of papillary thyroid carcinoma. Thyroid. 2013;23(11):1383–9.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
Choi SH, Worswick S, Byun HM, Shear T, Soussa JC, Wolff EM, et al. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int J Cancer. 2009;125(3):723–9.
Acknowledgements
The authors would like to thank the A.C. Camargo Cancer Center Biobank for providing the samples. Our gratitude to Dr. Mario Hiroyuki Hirata, Dra. Gisele Medeiros Bastos, and Ms. Jéssica Bassani Borges from the Molecular Biology Laboratory at the Dante Pazzanese Institute of Cardiology to assist with the pyrosequencing.
Funding
This study was supported by grants from the São Paulo Research Foundation (FAPESP 2015/20748-5 and 2015/17707-5).
Availability of data and materials
The large-scale methylation data is available in Gene Expression Omnibus database under the GSE86961 and GSE97466 IDs. All other original data can be obtained from the authors upon request.
Author information
Authors and Affiliations
Contributions
SRR and LPK conceived and designed the experiments. IMDPO, MBR, CMB, HK, LMC, JBHM, CA, and MCBF conducted the experiments. MCBF and FAM analyzed the data. SRR contributed with reagents/materials. CALP performed the histopathological evaluation. IMDPO, MCBF, and SRR wrote and edited the manuscript. SRR and LPK supervised the study. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The institutional Ethics Committee approved the study and all patients provided a signed informed consent (protocol # 2138/15). The study was conducted in accordance with the Helsinki Declaration.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Supervised hierarchical clustering analysis heatmaps comprising 42 probes of miRNAs identified in both, internal (A) and TCGA (B) data. The clusters highlighted in red demonstrate enrichment for PTC samples and in blue for NT samples. PTC: papillary thyroid carcinoma; NT: non-neoplastic thyroid tissue. Figure S2. Matched papillary thyroid carcinomas (PTC) compared with non-neoplastic thyroid tissue (NT) samples showed hypomethylation and miRNA increased expression of MIR21 and MIR146B in PTC (***P < 0.001; paired t test). Figure S3. Methylation (A) and expression (B) analysis of MIR21 and MIR146B according to BRAF mutation status. PTC, papillary thyroid carcinoma; BRAFWT, BRAF wild type; BRAFV600E, positive for BRAF mutation. *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t test). Figure S4. Location of the probes covering the MIR21 (A) and MIR146B (B) at chromosomes 17 and 10, respectively. The probes highlighted in orange were selected for pyrosequencing confirmation. (DOCX 5782 kb)
Additional file 2:
Table S1. Identification of 58 differentially expressed microRNAs detected in the comparison between PTC and NT from the TCGA database. Table S2. Integration between DNA methylation probes and the corresponding miRNA expression. Table S3. Differentially expressed genes in PTC compared to NT from TCGA database. Table S4. MicroRNA and target-mRNA interactions obtained by the integrative analysis (TCGA database), comprising the miRNAs potentially regulated by DNA methylation. Table S5. Comparison of miRNA genes methylation, miRNA, and target gene expression with clinical features (clinical stage, extrathyroidal extension, node metastasis and BRAF mutation) using the TCGA database. Table S6. List of 452 target transcripts of miRNA potentially regulated by methylation after mimic transfection in TPC1 cell line. Table S7. Significant pathways (P value< 0.05) enriched by target genes of hsa-miR-21-5p and hsa-miR-146b (5p and 3p) identified by IPA. Table S8. Significant pathways (P value< 0.01) enriched by target genes of hsa-miR-21-5p and hsa-miR-146b (5p and 3p) identified by KOBAS 3.0 (http://kobas.cbi.pku.edu.cn). Table S9. Clinical-pathological characteristics of the patients included in the study. Table S10. Primer sequences, amplicon size, number of CpGs flanked in the amplification and PCR temperature conditions. Table S11. Selection criteria of the miRNA targets selected for RT-qPCR validation. Table S12. Characteristics of the primers used in the transcripts expression level analyses by RT-qPCR. (XLSX 295 kb)
Additional file 3:
Supplementary methods. (DOCX 29 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ortiz, I.M.D.P., Barros-Filho, M.C., dos Reis, M.B. et al. Loss of DNA methylation is related to increased expression of miR-21 and miR-146b in papillary thyroid carcinoma. Clin Epigenet 10, 144 (2018). https://doi.org/10.1186/s13148-018-0579-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0579-8