
Abstract
Background
There are many reasons to think that epigenetics is a key determinant of fetal growth variability across the normal population. Since IGF1 and INS genes are major determinants of intrauterine growth, we examined the methylation of selected CpGs located in the regulatory region of these two genes.
Methods
Cord blood was sampled in 159 newborns born to mothers prospectively followed during their pregnancy. A 142-item questionnaire was filled by mothers at inclusion, during the last trimester of the pregnancy and at the delivery. The methylation of selected CpGs located in the promoters of the IGF1 and INS genes was measured in cord blood mononuclear cells collected at birth using bisulfite-PCR-pyrosequencing.
Results
Methylation at IGF1 CpG-137 correlated negatively with birth length (r = 0.27, P = 3.5 × 10−4). The same effect size was found after adjustment for maternal age, parity, and smoking: a 10% increase in CpG-137 methylation was associated with a decrease of length by 0.23 SDS.
Conclusion
The current results suggest that the methylation of IGF1 CpG-137 contributes to the individual variation of fetal growth by regulating IGF1 expression in fetal tissues.
Similar content being viewed by others
Background
Since human neonates are about the same size as the birth canal, size at birth has to be a tightly regulated trait. It is controlled by a number of interacting genetic and environmental factors. Among these factors, IGF1 (insulin-like growth factor-1), IGF2 (insulin-like growth factor-2), and insulin are three key players. The expression of IGF1 and IGF2 genes in fetal tissues and placenta is essential for fetal growth, as indicated by studies of knock-out mice and genetic defects in human. Although IGF2 is more abundantly expressed in fetal serum and tissues than IGF1, IGF1 is more closely associated with fetal growth in a majority of species. Insulin secreted by the fetal ß cells is also a major growth factor during intrauterine life [1].
In animal studies, IGF1 gene ablation reduces fetal weight [2, 3]. Fetal IGF1 promotes tissue growth by stimulating anabolic events and DNA synthesis. Circulating IGF1 is most often decreased in animal models of fetal growth restriction, as well as in human fetal growth restriction. Deletions or loss-of-function mutations in the IGF1 gene cause intrauterine and postnatal growth retardation [4,5,6,7]. In mice, the deletion of the IGF2 gene reduces fetal growth [2]. The effects on fetal growth are additive to those of IGF1. IGF2 is an imprinted gene. The expression of IGF2 maternal allele is silenced during fetal life, leading to fetal growth restraint when the paternal allele is mutated or deleted. Epimutations and molecular alterations of the human chromosomal region 11p15.5, which harbors IGF2 [8], and paternally inherited nonsense mutations of the IGF2 gene [9] are associated with the Silver–Russell syndrome, a syndromic growth-retardation disorder. Heterozygous mutations in the INS gene are associated with a reduction of fetal growth [10].
While monogenic defects in the IGF1, IGF2, and INS genes demonstrate the major role of these genes for fetal growth, the genetics of the individual variability in birthweight of normal neonates is complex, as for most quantitative traits, and relies on numerous polymorphic variations of the genome. Birth size is a multifactorial phenotype resulting from many processes and gene expression patterns operating during fetal development. Genetic variation is said to account for 38–80% of birth weight variance, with a considerable variability in the estimates [11, 12]. A genome-wide association study found 60 loci to be associated with birth weight at genome-wide significance [13]. Despite their implication in monogenic disorders of fetal growth, neither IGF1 nor INS loci were among these 60 loci, but pathways involved in insulin and IGF signaling were said to be involved. As pointed out for most quantitative traits studied in humans, the genetics of birth size face the missing heritability problem. Indeed, in contrast with the estimates of birth weight heritability [12, 14], the 60 variants most recently identified could only explain 2% of the variance in birth weight [13]. When SNPs at the INS [15, 16] and IGF1 loci [17, 18] were examined specifically as candidates for their contribution to the variation of size at birth, they showed inconsistent results [15, 19].
The genotype of the fetus is not the sole determinant of fetal growth. It is likely that maternal genes that regulate the environment of the womb are important determinants of birth size. Maternal environment also has a major influence, although most of the environmental factors and mechanisms of reduced fetal growth remain largely unknown. Non-inherited maternal factors found to influence birth size include parity, mother weight at birth and before pregnancy, weight gain of pregnancy, and maternal smoking, but cannot explain the vast majority of cases of intrauterine growth retardation (IUGR) [20]. Maternal nutrition, in industrialized populations seems to have only a small effect on placental and birth weights [21]. Indeed, even in affluent societies where mothers are well nourished, many children are still born with unexplained small fetal size. On the other side, extreme maternal nutrition, such as the Dutch Hunger Winter famine, is associated with a reduced birth weight [22].
As for all quantitative traits, epigenetic influences are likely to contribute to the regulation of birth size at the individual level. Few studies have yet explored the epigenetics of fetal growth in humans. To our knowledge, the only epigenome-wide association study carried out yet found that among the 485,577 CpGs analyzed in cord blood by the 450K Infinium BeadChip, 19 CpGs were significantly associated with birthweight in a large population-based cohort [23]. Another study of methylation (performed on cord blood and placenta) restricted to growth-related genes found no association between intrauterine growth retardation and IUGR and the methylation of CpGs at the IGF1, IGF2, and INS loci (note that this study investigated only 3 CpGs located within the IGF1 P1 promoter) [24].
The current study investigates the relationship between birth length and the methylation of specific CpGs of the IGF1 and INS genes. These CpGs located within promoters were selected because their level of methylation was known to control gene expression. This is the case for IGF1 CpG-137, whose methylation is associated with IGF1 gene transcription [25], circulating IGF1 [25] and childhood growth [25, 26]. This is also the case for CpG-180 in the INS promoter region, which influences INS gene transcription [27] and is associated with SNPs as rs689 [28] known to predict birth size to some degree [15]. We have not explored a potential association of birth length with CpG methylation on the non-imprinted allele of IGF2 because we did not have access to parental genotypes.
Methods
Mothers
One hundred and fifty-nine women of European ancestry aged 20–40 years, with a singleton pregnancy, were recruited at Antoine Béclère Maternity. The clinical characteristics of the 159 newborns are presented in Table 1. All were healthy neonates born after 37 weeks of amenorrhea. None had IUGR, defined as a weight or length below the 10th percentile for its gestational age [29]. Children born before 36 weeks of gestation were not included. Clinical data and biological samples were collected at inclusion (< 3 months of pregnancy) and around delivery. The main characteristics of the mothers are presented in Table 1. All mothers filled a 142-item questionnaire. All mothers were healthy and had a normal nutrition; none consumed alcohol or drugs during their pregnancy. All protocols were agreed by French ethic boards (CODECOH DC-2013-2017, CPP CO-14-001, CCTIRS no. 14-124bis, CNIL no. 914,253). Cord blood samples were taken within minutes of delivery, immediately refrigerated at 4 °C and transported to laboratory within 24 h.
Isolation of genomic DNA and bisulfite genomic conversion
Cord blood mononuclear cells (CBMC) were isolated from fresh blood using a density gradient. Five millimeters of fresh blood was mixed with 5 ml of NaCl 154 mM, and 5 ml of Lymphoprep solution (Eurobio, Paris, France) was added to the diluted blood and centrifuged for 20 min at room temperature at 800 g. After centrifugation, the interphase containing CBMC was carefully aspirated and the cells were mixed with NaCl. The cell suspension was centrifuged at 300 g, and the cell pellet washed with PBS.
Nucleic acids were extracted from CBMC using Gentra Puregene blood kit (Qiagen, Hilden, Germany). Four hundred nanograms of genomic DNA was treated with EZ DNA Methylation-Gold Kit, according to the manufacturer’s protocol (Zymo Research Corporation, CA, USA). A bisulfite-PCR-pyrosequencing technique was used to measure the CG methylation [25]. We improved the resolution of this method from a handful of bases to up to 100 nucleotides, with the ability to quantify methylation in the same sample of blood cells with a coefficient of variation (sd/mean) as little as 1–5% [25, 28].
Pyrosequencing-based bisulfite PCR analysis
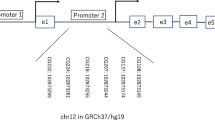
CpGs are denominated according to their position versus each promoter transcription start site (TSS). At the IGF1 locus, we studied 3 CpGs (-1044, -960, -919) located within the P1 promoter and 5 CpGs (-232, -224, -218, -207, -137) located within the P2 promoter (Additional file 1: Figure S1). We previously have shown that the methylation of the P1 promoter does not influence IGF1 gene expression [25]. At the INS locus, we studied 2 CpGs (-206 and -180) proximal to the TSS (Additional file 1: Figure S1). The bisulfite-treated genomic DNA was PCR-amplified using unbiased primers (Additional file 2: Table S1) and performed quantitative pyrosequencing using a PyroMark Q96 ID Pyrosequencing instrument (Qiagen). Pyrosequencing assay was designed using MethPrimer (htpp://urogene.org/methprimer/index1.html) [25]. Biotin-labeled single stranded amplicon was isolated according to protocol using the Qiagen Pyromark Q96 Work Station and underwent pyrosequencing with 0.5 μM of sequencing primer. The methylation percent for each CpG within the target sequence was calculated using PyroQ CpG Software (Qiagen).
Genotyping of SNPs at the IGF1 and INS loci
Genotyping of SNP rs35767 at the IGF1 gene locus was performed with TaqMan allelic discrimination using Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems, Courtaboeuf, France). SNP genotyping assay (ID: C_7999146_10) was purchased from Life technologies (Saint Aubin, France).
Genotyping of SNP rs689 at the INS gene locus was determined by the analysis of PCR products [30]. PCR amplification was in 96-well microliter plates, each 50 μl reaction contained DNA (100 ng), MgCl2 (1.5 mM), 10× reaction Buffer (Thermo Scientific), dNTPs (2.0 mM each), primers (1 μM each), and Taq Polymerase (1.25 U, Thermo Scientific, Saint Aubin, France). Amplified PCR products were digested with 1 unit of Hph1 enzyme (Thermo Scientific, Saint Aubin, France).
For genotyping quality, negative controls were included in each PCR plate. Twenty percent of samples were analyzed as duplicate for genotyping determination. The Hardy-Weinberg equilibrium (HWE) was calculated by computing the test for deviations in HWE and was shown to be present across genotypes. Allele frequencies were calculated and tested by test.
Measurement of serum IGF1 and insulin concentrations
IGF1 and insulin concentrations were measured in serum samples from the cord blood of newborns. IGF1 concentration was measured using a chemiluminescent immunometric assay after pre-treatment with acid using Immulite®2000 (Siemens Healthcare Diagnostics Products Llanberis, UK). Insulin concentration was measured using Liaison®Insulin (DiaSorin, Saluggia, Italy).
Statistical analysis
Birth weight (sds) and birth length (sds) were regressed on methylation of CpG-137, sex, folate, supplement intake before conception (yes/no), folate supplement intake during pregnancy (yes/no), parity (primiparous/non primiparous), maternal age, and smoking (yes/no) in multivariate linear regression. Results are expressed as mean ± sd. All statistical analyses were conducted using R 3.3.2.
Results
Effects of smoking and folate on birth weight
Six percent of the newborns were from smoking mothers. The mean birth length of neonates born to a smoking mother was 0.62 SDS (95% CI 0.22–1.04) lower than that of neonates born to a non-smoking mother (P = 3.0 × 10−3), a result retrieved in the multivariate analysis (P = 5.6 × 10−3). Thirty-two percent of women took a folate supplement before conception. The bivariate analysis showed a trend for increased birth weight associated with folate intake before conception (P = 0.09), an association found strengthened in the multivariate regression (P = 9.2 × 10−3). No association between birth length or birth weight was found with a supplement of folate during pregnancy.
Neither smoking nor folate intake were associated with variations in CpG-137 methylation.
Epigenetic and genetic variation at the IGF1 locus
While methylation of CpG-218 located within the IGF1 P2 promoter correlated closely with methylation of CpG-137 (P = 9.4 × 10−15), methylation of CpGs -1044, -960, and -919, located within the P1 promoter (Table 2), showed no correlation with CpG-137 methylation (Additional file 3: Figure S2). We found no correlation between the rs35767 genotype and the methylation of CpG-137 of the P2 or other studied CpGs (Additional file 4: Figure S3A).
Birth length correlated negatively with methylation at CpG-137 (r = 0.2, P = 3.5 × 10−4, Fig. 1). This finding was confirmed after adjustment for several covariates: a 10% increase in methylation at CpG-137 was associated with a decrease of birth length by 0.23 SDS (95% CI 0.11–0.35; P = 1.6 × 10−4). In contrast, methylation at CpG-137 was not associated with birth weight (P = 0.36). The same result was obtained in the multivariate regression framework (P = 0.15). Cord blood IGF1 concentration showed no association with methylation at CpG-137 (r = 0.06, P = 0.61).

CpG-137 methylation correlates negatively with birth length. The correlation between CpG-137 (%) and birth length (SDS) is described by the equation: Birth length = − 0.023 × [CpG-137 methylation] + 0.68 with r = 0.2 and P = 3.5 × 10−4
Methylation at CpG-224 and CpG-218 correlated negatively with birth length (P = 0.03 and P = 0.02, respectively) (Additional file 5: Figure S4).
Epigenetic and genetic variation at the INS locus
Methylation of INS CpG-180 and CpG-206 showed no correlation with birth length or birth weight. Birth length and birth weight were not associated with rs689 alleles (Additional file 6: Figure S5).
As previously reported [28], we found that CpG-206 and CpG-180 methylation in the studied newborns was strongly influenced by rs689 alleles (Additional file 4: Figure S3B; P < 2.10−16).
Discussion
The fetal genotype explains only a limited part of intrauterine growth variability among individuals of the general population [13], which leaves an important role to maternal genotype, maternal environment, and fetal and placental epigenetics. Animal studies have demonstrated that environment can shape the epigenome, notably during the intrauterine period when it has the greatest plasticity. Epigenetic effects of the intrauterine environment can thus influence the phenotype in later life. However, it remains unclear as to how influential the fetal period is in shaping the epigenome, whether different genomic regions show varying sensitivities to this environment during this period, and the extent to which this early interaction is sensitive to genetic influences.
To our knowledge, few studies have examined the association of CpG methylation with fetal growth. The first study found an increased methylation of all CpG sites at the IGF1 P1 promoter in the placenta of fetuses with IUGR [31], associated with the previously reported decreased IGF1 gene expression [32]. Another study used the Illumina Infinium HM27 platform to profile CpG methylation in CBMC in a small number of twin pairs [33]. Array technology, however, uses CpGs that are not necessarily those having specific effects on gene expression and phenotypes. This was the case at the IGF1 locus for the array used in twins, which analyzed only 1 CpG of the P1 promoter and no CpG of the P2 promoter at the IGF1 locus, while it examined 4 CpGs at the INS locus. Findings were that none of these CpGs had their methylation level associated with twins’ birthweight. A third study of methylation restricted to growth-related genes found no association between IUGR and the methylation of CpGs at the IGF1, IGF2, and INS loci in placenta or cord blood samples [24].
Instead, our study focused on CpGs located within the regulatory region of IGF1 and INS genes, because these two genes are known to be major contributors to fetal growth. Some of these CpGs were selected because their methylation was already known to be associated with gene expression [25, 28], the others because they were located within neighboring regulatory regions. We found that CpG-137 methylation was negatively associated with birth length in normal neonates. The methylation of CpG-137 has a strong functional role upon IGF1 gene expression in children’s PBMC and in the HEK293 cell line [25]. It was previously shown to be associated with postnatal growth variability [13]. If methylation at CpG-137 in CBMC is a proxy of the methylation in growth-promoting tissues of the fetus (which has not been shown in the current study), one can speculate that methylation at this CpG influences IGF1 gene expression and IGF1 production in fetal tissues, thus fetal growth. There is no evidence, however, that the individual variation of CpG methylation reflects that in growth-promoting tissues. While this is a major weakness of our study, this weakness is shared with a majority of studies on epigenetics in humans where blood cells are used as proxies of physiological tissues, given that clinical research does not have access to these tissues [34]. Another weakness of measuring CpG methylation in CBMC is that it is a unique cell mixture containing red blood cells in addition to other blood cells, and not a well-characterized homogeneous cell type. Our study was not able to estimate cell composition of this mixture based on methods developed for adult blood cells [35], nor to sort a specific cell category from the cord blood sample.
Several hypotheses may account for the association of methylation at CpG-137 measured at birth with fetal growth. A first possibility is that the individual variation in CpG-137 methylation is determined in the post implantatory embryo, at time of the primary shaping of the methylome. Alternatively, the variation observed in the level of methylation of CpG-137 may result from yet unknown maternal signals transmitted to the fetal tissues through the placenta at a post-embryonic stage of intrauterine life. No association of fetal growth was observed with the other CpGs of the IGF1 locus, except for CpG-218, another CpG of the P2 promoter.
Maternal smoking [36] or folate intake [37] has been shown in other studies to affect methylation of certain CpGs, but was not found to affect the methylation of CpG-137 or of other CpGs studied herein.
Since insulin is a major growth factor in fetal life and the INS VNTR has a direct effect on insulin transcription [27], it is conceivable that INS VNTR variation influences early growth. This is why CpG-180 and CpG-206 located within the INS promoter were selected for the current study, given that their level of methylation affects the expression of the insulin gene [27, 28]. As reported previously [28] the methylation of CpG-206 and CpG-180 was strongly influenced by rs689 alleles. We found that neither rs689 alleles nor CpG-206 or CpG-180 showed a relationship with birth length or birth weight, but this conclusion should await the observation of a large number of neonates. Unlike Dunger et al. [15], we observed no association (Additional file 4: Figure S3) between birth size and insulin VNTR classes or rs689 (in complete linkage disequilibrium with VNTR classes). This may be due to the fact that we were not able to distinguish “non-changers” [15] among the studied neonates: Non-changers is the term used to describe infants that do not show a catch-up growth after being born small for gestational age.
Conclusions
In conclusion, fetal growth in normal neonates is associated with the methylation of CpG-137 located in the IGF1 P2 promoter. This observation supports a significant role of IGF1 epigenetics in the regulation of fetal growth that does not seem to be dependent on cis-genetic variation at the IGF1 locus. Although small, this epigenetic effect is of an order of magnitude comparable with that of many genomic variants associated with human quantitative traits.
Change history
04 June 2018
After publication of the original article [1], it came to the publishers’ attention that the below author’s corrections provided at the proofing stage had been misinterpreted.
Abbreviations
- CBMC:
-
Cord blood mononuclear cells
- IGF1:
-
Insulin-like growth factor 1
- IUGR:
-
Intrauterine growth retardation
References
Gatford KL, Simmons RA. Prenatal programming of insulin secretion in intrauterine growth restriction. Clin Obstet Gynecol. 2013;56(3):520–8.
Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82.
Fowden AL. The insulin-like growth factors and feto-placental growth. Placenta. 2003;24(8–9):803–12.
Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W, Klammt J, Kratzsch J, Osgood D, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003;349(23):2211–22.
Woods KA, Camacho-Hubner C, Bergman RN, Barter D, Clark AJ, Savage MO. Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocrinol Metab. 2000;85(4):1407–11.
Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet. 2003;40(12):913–7.
Netchine I, Azzi S, Houang M, Seurin D, Perin L, Ricort JM, Daubas C, Legay C, Mester J, Herich R, et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab. 2009;94(10):3913–21.
Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37(9):1003–7.
Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Buttel HM, Schweizer R, van Workum W, Binder G, Eggermann T. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015;373(4):349–56.
Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104(38):15040–4.
Magnus P, Berg K, Bjerkedal T, Nance WE. Parental determinants of birth weight. Clin Genet. 1984;26(5):397–405.
Magnus P. Causes of variation in birth weight: a study of offspring of twins. Clin Genet. 1984;25(1):15–24.
Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, Feenstra B, van Zuydam NR, Gaulton KJ, Grarup N, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538(7624):248–52.
Little RE, Sing CF. Genetic and environmental influences on human birth weight. Am J Hum Genet. 1987;40(6):512–26.
Dunger DB, Ong KK, Huxtable SJ, Sherriff A, Woods KA, Ahmed ML, Golding J, Pembrey ME, Ring S, Bennett ST, et al. Association of the INS VNTR with size at birth. ALSPAC Study Team. Avon Longitudinal Study of Pregnancy and Childhood. Nat Genet. 1998;19(1):98–100.
Mitchell SM, Hattersley AT, Knight B, Turner T, Metcalf BS, Voss LD, Davies D, McCarthy A, Wilkin TJ, Smith GD, et al. Lack of support for a role of the insulin gene variable number of tandem repeats minisatellite (INS-VNTR) locus in fetal growth or type 2 diabetes-related intermediate traits in United Kingdom populations. J Clin Endocrinol Metab. 2004;89(1):310–7.
Johnston LB, Dahlgren J, Leger J, Gelander L, Savage MO, Czernichow P, Wikland KA, Clark AJ. Association between insulin-like growth factor I (IGF-I) polymorphisms, circulating IGF-I, and pre- and postnatal growth in two European small for gestational age populations. J Clin Endocrinol Metab. 2003;88(10):4805–10.
Frayling TM, Hattersley AT, Smith GD, Ben-Shlomo Y. Conflicting results on variation in the IGFI gene highlight methodological considerations in the design of genetic association studies. Diabetologia. 2002;45(11):1605–6.
Arends N, Johnston L, Hokken-Koelega A, van Duijn C, de Ridder M, Savage M, Clark A. Polymorphism in the IGF-I gene: clinical relevance for short children born small for gestational age (SGA). J Clin Endocrinol Metab. 2002;87(6):2720.
Lunde A, Melve KK, Gjessing HK, Skjaerven R, Irgens LM. Genetic and environmental influences on birth weight, birth length, head circumference, and gestational age by use of population-based parent-offspring data. Am J Epidemiol. 2007;165(7):734–41.
Mathews F, Yudkin P, Neil A. Influence of maternal nutrition on outcome of pregnancy: prospective cohort study. BMJ. 1999;319(7206):339–43.
Stein AD, Ravelli AC, Lumey LH. Famine, third-trimester pregnancy weight gain, and intrauterine growth: the Dutch Famine Birth Cohort Study. Hum Biol. 1995;67(1):135–50.
Engel SM, Joubert BR, Wu MC, Olshan AF, Haberg SE, Ueland PM, Nystad W, Nilsen RM, Vollset SE, Peddada SD, et al. Neonatal genome-wide methylation patterns in relation to birth weight in the Norwegian Mother and Child Cohort. Am J Epidemiol. 2014;179(7):834–42.
Turan N, Ghalwash MF, Katari S, Coutifaris C, Obradovic Z, Sapienza C. DNA methylation differences at growth related genes correlate with birth weight: a molecular signature linked to developmental origins of adult disease? BMC Med Genet. 2012;5:10.
Ouni M, Gunes Y, Belot MP, Castell AL, Fradin D, Bougneres P. The IGF1 P2 promoter is an epigenetic QTL for circulating IGF1 and human growth. Clin Epigenetics. 2015;7:22.
Ouni M, Castell AL, Rothenbuhler A, Linglart A, Bougneres P. Higher methylation of the IGF1 P2 promoter is associated with idiopathic short stature. Clin Endocrinol. 2016;84(2):216–21.
Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F, Mullen Y, Pfeifer GP, Ferreri K. Insulin gene expression is regulated by DNA methylation. PLoS One. 2009;4(9):e6953.
Fradin D, Le Fur S, Mille C, Naoui N, Groves C, Zelenika D, McCarthy MI, Lathrop M, Bougneres P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS One. 2012;7(5):e36278.
Niklasson A, Albertsson-Wikland K. Continuous growth reference from 24th week of gestation to 24 months by gender. BMC Pediatr. 2008;8:8.
Le Stunff C, Fallin D, Schork NJ, Bougneres P. The insulin gene VNTR is associated with fasting insulin levels and development of juvenile obesity. Nat Genet. 2000;26(4):444–6.
Nawathe AR, Christian M, Kim SH, Johnson M, Savvidou MD, Terzidou V. Insulin-like growth factor axis in pregnancies affected by fetal growth disorders. Clin Epigenetics. 2016;8:11.
Iniguez G, Gonzalez CA, Argandona F, Kakarieka E, Johnson MC, Cassorla F. Expression and protein content of IGF-I and IGF-I receptor in placentas from small, adequate and large for gestational age newborns. Horm Res Paediatr. 2010;73(5):320–7.
Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK, et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res. 2012;22(8):1395–406.
Armstrong DA, Lesseur C, Conradt E, Lester BM, Marsit CJ. Global and gene-specific DNA methylation across multiple tissues in early infancy: implications for children's health research. FASEB J. 2014;28(5):2088–97.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–31.
Caffrey A, McNulty H, Walsh H, Irwin R, Pentieva K. Effect of folic acid supplementation in pregnancy on gene specific DNA methylation in the child: evidence from a randomised controlled trial. Proc Nutr Soc. 2016;75(OCE3):E132.
Acknowledgements
Fanny Lachaux did the monitoring of Epichild cohort of mothers and children. We thank the nurses and midwives of the Antoine Béclère Maternity for their motivation and help.
Funding
The study was supported by the INSERM-CEA-Paris Saclay U1169 Research Unit and the EPICHILD ANR R13107KK.
Availability of data and materials
Data are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
PB designed the study, interpreted the data, and wrote the manuscript. CLS did the experiments and interpreted the results. NT did the statistical analyses. NF prepared the data bases for final analysis. ALC interpreted the results. CM and MPB did the experiments. SBT did insulin and IGF1 measurements. AB and DF recruited subjects and discussed clinical aspects. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We obtained informed consent from all participating mothers. All protocols were agreed by French ethic boards (CODECOH DC-2013-2017, CPP CO-14-001, CCTIRS no. 14-124bis, CNIL no. 914253).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised. The title and a number of typos have been corrected.
Additional files
Additional file 1:
Figure S1. Schematic representation of the IGF1 and INS loci. (A) The two IGF1 gene promoters are figured. CpGs are indicated as lollypops (studied CpGs in white and non-studied CpG in black). rs35767 is indicated by a black arrow. TSS are shown as broken arrows. (B) INS promoter is figured. CpGs are indicated as lollypops (studied CpGs in white and non-studied CpG in black). TSS is shown as broken arrow. Location of primers are indicated by arrow for CpG methylation and genotyping. Sequences of primers and location on chromosome are provided in Additional file 2: Table S1. (PPTX 67 kb)
Additional file 2:
Table S1. List of primers and location used in our study. Sequences are given from 5′ to 3′. (DOCX 15 kb)
Additional file 3:
Figure S2. Correlation matrix of methylation values (%) at the CpG located in the P1 and P2 promoters of the IGF1 gene in newborns patients. Pearson correlation coefficient is indicated in bold, and P value below. (PPTX 88 kb)
Additional file 4:
Figure S3. Relationship between promoter CpG methylation and genotypes. (A) Methylation at CpGs-137 of the IGF1 P2 promoter is independent from the rs35767 genotypes. (B) Methylation at CpGs-206 and CpG-180 in insulin promoter is closely dependent on rs689 alleles. (PPTX 242 kb)
Additional file 5:
Figure S4. Relation between CpG methylation and birth length (SDS) at the IGF1 promoter 1 and 2. (A) at IGF1 P1 promoter, we observed no significant correlation of birth length with the studied CpGs, (B) at IGF1 P2 promoter, only two CpGs other that CpG-137 showed a weak correlation with birth length. The correlation between CpG-224 (%) and birth length (SDS) is described by the equation: Birth length = − 0.014*[CpG-224 methylation] + 0.43 (r = 0.17, P = 0.03). The correlation between CpG-218 (%) and birth length (SDS) is described by the equation: Birth length = − 0.016*[CpG-218 methylation] + 0.58 (r = 0.2, P = 0.02). (PPTX 752 kb)
Additional file 6:
Figure S5. Relationship between insulin rs689 genotype and birth weight (sds) and birth length (sds). Birth weight (sds) and birth length (sds) are independent from the rs 689 genotypes. (PPTX 161 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Le Stunff, C., Castell, AL., Todd, N. et al. Fetal growth is associated with CpG methylation in the P2 promoter of the IGF1 gene. Clin Epigenet 10, 57 (2018). https://doi.org/10.1186/s13148-018-0489-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-018-0489-9