
Abstract
Type 2 diabetes is a complex trait with both environmental and hereditary factors contributing to the overall pathogenesis. One link between genes, environment, and disease is epigenetics influencing gene transcription and, consequently, organ function. Genome-wide studies have shown altered DNA methylation in tissues important for glucose homeostasis including pancreas, liver, skeletal muscle, and adipose tissue from subjects with type 2 diabetes compared with nondiabetic controls. Factors predisposing for type 2 diabetes including an adverse intrauterine environment, increasing age, overweight, physical inactivity, a family history of the disease, and an unhealthy diet have all shown to affect the DNA methylation pattern in target tissues for insulin resistance in humans. Epigenetics including DNA methylation may therefore improve our understanding of the type 2 diabetes pathogenesis, contribute to development of novel treatments, and be a useful tool to identify individuals at risk for developing the disease.
Similar content being viewed by others

Background
Type 2 diabetes is one of the most common chronic metabolic diseases in developed countries [1]. This form of diabetes is a consequence of the target tissues becoming resistant to the effects of insulin and the failure of pancreatic β-cells to produce enough insulin. It is shown that type 2 diabetes develops with age, physical inactivity, and obesity in subjects with a genetic predisposition and/or in subjects who have experienced an adverse intrauterine environment. It is a complex multifactorial disease whose development is dependent on interactions in and between the predisposing factors.
One link between genes, environmental exposure, and disease development is epigenetics. It provides a molecular mechanism to explain how interactions between genetic and environmental factors may be involved in a disease process. The term epigenetics is typically described as heritable changes in gene function that occur without a change in the nucleotide sequence [2]. Epigenetic regulation includes DNA methylation, histone modifications, and non-coding RNA. DNA methylation is the most studied epigenetic mark so far and occurs mainly at the fifth position of the cytosine ring in CpG dinucleotides. DNA methylation is required to maintain cell-specific gene expression, plays an important role during embryonic development, and contributes to the establishment of imprinting and X-chromosome inactivation [3,4,5]. DNA methylation in promoter regions has been associated with transcriptional silencing. However, emerging data show that the effect of DNA methylation depends on the genomic location, and it may also affect alternative splicing, genomic stability, transcriptional elongation, and transcription of non-coding RNAs [3]. It may thereby also be associated with increased gene expression. The establishment and maintenance of epigenetic modifications are susceptible to environmental factors including dietary factors and changes in metabolism [6]. This, in addition to the fact that epigenetic changes accumulate in the living individual [7], led to the hypothesis that epigenetic modifications could be involved in age and lifestyle-related metabolic diseases such as type 2 diabetes. Indeed, we and others have shown altered epigenetic states in tissues important for glucose homeostasis including pancreas, liver, skeletal muscle, and adipose tissue from subjects with type 2 diabetes compared with nondiabetic controls [8,9,10,11,12,13,14]. These studies include differential DNA methylation of candidate genes that affect insulin secretion from pancreatic beta cells and thereby could play an important role in the pathogenesis of type 2 diabetes [8, 14].
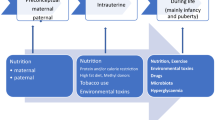
The aim of this review is to summarize studies supporting epigenetics as a link between genetics, the fetal environment, an unhealthy lifestyle and the development of type 2 diabetes. In particular, this review will focus on risk factors for type 2 diabetes and their impact on DNA methylation in target tissues for insulin resistance in humans, including skeletal muscle, liver and adipose tissue (Fig. 1).

A suggested model of type 2 diabetes development and focus of this review article. Risk factors for type 2 diabetes affect expression of genes involved in glucose homeostasis regulation via epigenetic modifications including DNA methylation
DNA methylation and risk factors for type 2 diabetes
Intrauterine environment
Fetal programming describes the process by which different environmental conditions during development can have long lasting effects on metabolic pathways influencing disease susceptibility. The first evidence for the importance of the intrauterine environment in relation to type 2 diabetes came in the early 1990s [15, 16]. Since then, numerous studies have reported a relationship between low or high birth weight and risk of developing type 2 diabetes later in life [17,18,19,20]. Already young adult individuals born with low birth weight show signs of metabolic impairment related to insulin resistance including an altered fat distribution, increased lipolysis, and reduced expression of key insulin signaling proteins in insulin target tissues [21,22,23,24]. The increased risk of insulin resistance and type 2 diabetes in these individuals has long been speculated to originate at the epigenetic level, since epigenetic marks are thought to respond to the surrounding environment. Prenatal factors including mode of conception, maternal smoking, and maternal diet have indeed shown to effect the DNA methylation pattern [25,26,27]. Differential methylation of IGF2, as well as other type 2 diabetes-related genes, have been found in children born to mothers experiencing famine, further pointing to epigenetics as a mechanism connecting prenatal nutrition and the onset of type 2 diabetes later in life [28,29,30]. Also, periconceptual folic acid supplementation has been shown to alter the methylation in IGF2 in children from the folic acid-supplemented mothers [31]. Godfrey et al. reported that epigenetic marks measured in umbilical cord tissue at birth can predict phenotypic outcomes such as obesity in later childhood [32]. We have shown that young men who had a low birth weight have differential DNA methylation in their adipose tissue compared with men born with a normal birth weight [33]. A recent study also suggests that altered epigenetic and transcriptional regulation of adipose-derived stem cells could play a role in programming adipose tissue dysfunction in individuals born with low birth weight [34].
The phenotypic effects of epigenetic modifications during development may not manifest until later in life in response to environmental challenges such as energy dense diets. As an example of this, we found that 5 days high-fat overfeeding unmasked a decreased plasticity in subjects born with low birth weight compared with subjects born with normal birth weight at the DNA methylation level in the skeletal muscle [35, 36]. Also, fasting induces DNA methylation changes in LEP and ADIPOQ promoters in adipose tissue among normal birth weight but not among low birth weight subjects. The altered epigenetic flexibility in low birth weight subjects might contribute to their differential response to fasting and increased risk of metabolic disease [37].
Genes
The genetic contribution to type 2 diabetes has been demonstrated with linkage analyses and twin and adoption studies [38]. However, our knowledge about the impact of environmental factors is so far greater than the understanding of the underlying genetic factors [39]. Association studies have until now identified > 120 genomic loci influencing type 2 diabetes susceptibility [40]. A large number of the identified single nucleotide polymorphisms (SNPs) are located in intergenic regions, in introns, and/or are not predicted to result in functional changes at the protein level. The molecular mechanisms through which these SNPs influence gene function and disease pathology are largely unknown but may include epigenetic mechanisms. The introduction or removal of a CpG site may be a molecular mechanism through which some of the type 2 diabetes SNPs affect gene function. Indeed, a study investigated the 40 SNPs associated with type 2 diabetes at that time and found that 19 of 40 (48%) SNPs introduce or remove a CpG site [41]. In adipose tissue, the degree of methylation seems to mediate the impact of SNPs on metabolic traits, including insulin sensitivity [42]. In the skeletal muscle, we further showed that interactions between genetic (SNP), epigenetic (DNA methylation), and non-genetic (age) factors influence gene expression and metabolism [43].
We have observed differential DNA methylation in the skeletal muscle from individuals with a family history of type 2 diabetes compared with individuals without such family history [11]. Although the epigenetic differences we find between these individuals may be due to genetic factors, they could of course also be due to a shared environment within families. However, based on heritability estimates in twin and family studies, several studies have found that DNA methylation levels are under genetic control [12, 44,45,46,47]. We also found support for a genetic impact on DNA methylation in a study where we found a stronger correlation of genome-wide adipose tissue DNA methylation levels in monozygotic twin pairs compared with same-sex dizygotic twins or unrelated subjects [9].
Diet
Methyl groups for all biological methylation reactions are primarily supplied from dietary methyl donors and co-factors such as folate, choline, methionine, vitamin B2, vitamin B6, and vitamin B12. These dietary components can affect the one-carbon metabolism that determines the amount of S-adenosylmethionine (SAM), which is the methyl donor for DNA and histone proteins. The reliance on dietary sources of methyl donors for DNA methylation reactions has led to the suggestion that nutrients may affect both the establishment and maintenance of DNA methylation patterns. Subjects with type 2 diabetes seem to have reduced folate levels in serum compared with healthy controls [10]. Interestingly, at the same time, the subjects with type 2 diabetes also had reduced DNA methylation of the majority (94%) of differentially methylated CpG sites in a genome-wide analysis in the human liver [10]. Folate levels correlated negatively with fasting glucose levels already in nondiabetic subjects, suggesting that reduced circulating folate levels may contribute to the development of type 2 diabetes.
Energy-dense food high in calories and fat is an important risk factor for obesity and type 2 diabetes [48]. A study investigating the effect of a short-term (5 days) high-fat overfeeding diet on the genome-wide DNA methylation pattern in the skeletal muscle from healthy young men found widespread DNA methylation changes affecting almost half of the investigated genes [49]. Due to the cross-over design of the overfeeding study, the reversibility of the induced DNA methylations could be investigated. The overfeeding-induced methylation changes were only partly reversed 6–8 weeks after returning to the control diet. The slow reversibility could have implications for build-up of CpG methylation over time. In a recent randomized control study, we investigated the impact of 7 weeks of saturated or polyunsaturated fat overfeeding on the DNA methylation pattern in human adipose tissue and observed distinct epigenetic changes induced by the two different overfeeding diets [50]. Both diets resulted in a similar weight gain and an increased mean degree of methylation in adipose tissue. However, the saturated fat diet resulted in elevated liver and visceral fat accumulations. Also, these data suggest that DNA methylation at baseline can predict weight gain in response to overfeeding. In addition, DNA methylation in adipose tissue seems to be able to predict weight loss in response to a low caloric diet as well [51, 52].
Obesity
Increased body fat stores resulting from an imbalance between energy intake and energy expenditure characterize obesity. The prevalence of overweight and obese individuals has increased dramatically in the last decades. Obesity constitutes a major risk factor for several serious diseases including type 2 diabetes. Studies focusing on epigenetic patterns in obesity have found altered DNA methylation levels in genes related to metabolic processes, for example, circadian clock system genes [53]. Because obese people are at increased risk of many age-related diseases, it is a possible hypothesis that obesity increases the biological age of some tissues. Horvath et al. reported a strong correlation between high body mass index (BMI) and the epigenetic age of liver tissue, suggesting that the accelerated epigenetic aging may play a role in liver-related comorbidities of obesity, such as insulin resistance [54]. BMI was related to DNA methylation in whole blood cells from 479 individuals and identified CpG sites annotated to HIF3A with increased DNA methylation associated with increased BMI. The association was further validated in adipose tissue from 635 females [55]. It has been proposed that the HIF system could play a role in mechanisms involved in adipose tissue inflammation, insulin resistance, and the etiology of obesity-related diseases [56, 57]. We analyzed the genome-wide DNA methylation pattern in human adipose tissue from 96 males and 94 females and found that DNA methylation of ~ 5000 CpG sites was associated to BMI [58]. The strong effect of increased BMI on the degree of DNA methylation in human adipose tissue indeed propose that obesity can mediate some of its effects via altering the epigenome. Interestingly, a large number of these CpG sites do also show differential DNA methylation in adipose tissue from subjects with type 2 diabetes compared with controls [9], suggesting that BMI-associated changes in DNA methylation may predispose to type 2 diabetes. We could also link BMI-associated DNA methylation to differential expression of almost 3000 genes [58]. Another epigenome-wide study showed that BMI is associated with widespread changes in DNA methylation in blood and that these changes correlated to methylation patterns in other tissues including subcutaneous and omental fat, liver, and muscle. They demonstrated that the DNA methylation alterations are a consequence rather than a cause of obesity. The methylation changes occurred in genes involved in lipid metabolism and inflammation and predicted future development of type 2 diabetes [59]. DNA methylation and gene expression patterns in adipose tissue have also been shown to differ significantly within young adult monozygotic BMI-discordant twin pairs [60]. We have in a recent study identified abnormal epigenetic changes during differentiation of skeletal muscle stem cells from obese compared with non-obese humans, proposing an altered epigenetic memory in muscle stem cells due to obesity [61]. Interestingly, there were many genes involved in epigenetic regulation and metabolic diseases among genes showing differential methylation and expression during differentiation in obese subjects only.
Inactivity
Physical inactivity contributes to lifestyle-related diseases, including obesity and type 2 diabetes. We have previously shown that 9 days of bed rest induce DNA methylation changes in PGC1α that are not totally reversed after a retraining period of 4 weeks in the skeletal muscle of healthy young men [62]. PGC1α is the master regulator of mitochondrial biogenesis, and PGC1α mRNA expression is reduced in the skeletal muscle from subjects with type 2 diabetes [63]. Interestingly, it has been shown that DNA methylation regulation of PGC1α in the skeletal muscle may explain interindividual variation in response to exercise training [64]. Although the benefits of regular exercise are well known, the underlying regulatory mechanisms are still not completely understood but may include epigenetics. Methylation changes in insulin target tissues have been reported after both acute and long-term physical exercise [11, 65,66,67,68]. A few years ago, it was shown that DNA methylation might be involved in muscle adaptation to regular exercise training in men with or without a family history of type 2 diabetes [11]. A 6-month exercise intervention changed both DNA methylation and mRNA expression of a number of genes involved in skeletal muscle metabolism. A study of fat biopsies from the same individuals showed that exercise induces genome-wide changes in DNA methylation also in human adipose tissue [66]. Here, a general global increase in DNA methylation in addition to changes in DNA methylation of 17,975 individual CpG sites including several genes associated with type 2 diabetes and obesity was observed. Two genes with differential methylation and expression in response to exercise, Hdac4 and Ncor2, were silenced in 3T3-L1 adipocytes which resulted in increased lipogenesis both in the basal and insulin-stimulated state. The epigenetic changes induced by bed rest or exercise may hence affect tissue function and predispose to or protect against disease risk (Table 1).
Aging
Aging is a complex multifactorial process affecting all living beings. It is associated with a progressive decline in physiological functions and an increased incidence of chronic metabolic diseases including type 2 diabetes. One possible explanation may be that aging causes epigenetic changes that effect expression of genes important for glucose homeostasis. A study of young and elderly monozygotic twin pairs showed that the young twin pairs exhibit a similar epigenetic pattern, whereas intrapair differences were substantial in elderly twin pairs, supporting the idea that epigenetic changes accumulate during life [7]. Studying the skeletal muscle from young and elderly twins, we could show age-related DNA methylation and expression changes of genes encoding proteins involved in the respiratory chain, i.e., NDUFB6 and COX7A1 [43, 69]. A prospective study has further shown that DNA methylation changes over time [45]. Epigenetic drift is suggested to be caused by environmental factors or spontaneous stochastic errors in the process of transmission of DNA methylation and leads to unpredictable differences in the methylome among aging individuals. The imperfect maintenance of epigenetic marks during the process of replication may be accentuated during aging as levels of DNA methyltransferase 1 (DNMT1), the maintenance DNMT, has been reported to decline in aging cells [70]. Several studies have investigated DNA methylation changes with age in target tissues for insulin resistance [58, 71,72,73,74,75]. Although many age-related changes depend on cell type [72], several studies from our group and others have shown that some DNA methylation changes occur at specific sites during aging in a highly reproducible way, independently of tissue type, sex, or disease state [58, 71, 76,77,78,79,80]. A study analyzing ~ 8000 samples representing 51 healthy human tissues and cell types identified that DNA methylation at 353 CpGs accurately predicted age [73]. Another study investigated the rate of DNA methylation change by building a predictive model of the aging methylome from the blood of individuals aged 19–101 years [81]. This study identified a set of 71 CpGs that have a high accuracy of age prediction and nearly all markers occur within or near genes associated with aging. Interestingly, a number of the blood-based epigenetic markers of age can also be found in tissues relevant for glucose homeostasis [58, 71, 76]. These include differential methylation of ELOVL2, KLF14, and FHL2 (Fig. 2). Although these studies clearly demonstrate that age affects the DNA methylation pattern, future studies need to address the mechanisms behind these specific age-related epigenetic changes taking place in multiple tissues.

A summary of some tissues and genes that are affected by age by altered DNA methylation
Conclusions
The prevalence of type 2 diabetes is rapidly increasing worldwide. This disorder causes suffering, deaths, and costs and is becoming a more and more severe problem for our society. It is important to better understand the underlying mechanisms in order to improve prediction, prevention, and treatment of the disease. As suggested in this review, type 2 diabetes develops due to an interplay between epigenetics, genetics, fetal environment, and lifestyle. Studies presented in this review show that DNA methylation is highly dynamic and responsive to the environment. Despite the facts that most of the findings in the reviewed papers are correlative and that most studies investigate only a small number of the existing CpG sites, these findings will be valuable for hypothesis development of future studies. Future work should focus on finding the optimal lifestyle (for example, type of diet and type/duration of exercise) for hindering the development of epigenetic-based diseases and how all these could be affected by genetic background. Epigenetics can either provide a biological mechanism for disease development, be targeted for therapy, or serve as a biomarker of disease or disease risk even if not directly involved in causing the disease. The epigenetic studies summarized in this review may improve our understanding of disease pathogenesis, contribute to development of novel therapies, and improve prediction of type 2 diabetes.
Abbreviations
- BMI:
-
Body mass index
- DNMT1:
-
DNA methyltransferase 1
- SAM:
-
S-adenosylmethionine
- SNPs:
-
Single nucleotide polymorphisms
References
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53.
Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science (New York, NY). 2001;293:1089–93.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9.
Dayeh T, Volkov P, Salo S, Hall E, Nilsson E, Olsson AH, Kirkpatrick CL, Wollheim CB, Eliasson L, Ronn T, Bacos K, Ling C. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014;10:e1004160.
Nilsson E, Jansson PA, Perfilyev A, Volkov P, Pedersen M, Svensson MK, Poulsen P, Ribel-Madsen R, Pedersen NL, Almgren P, Fadista J, Ronn T, Klarlund Pedersen B, Scheele C, Vaag A, Ling C. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes. 2014;63:2962–76.
Nilsson E, Matte A, Perfilyev A, de Mello VD, Kakela P, Pihlajamaki J, Ling C. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab. 2015;100:E1491–501.
Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, Yang BT, Lang S, Parikh H, Wessman Y, Weishaupt H, Attema J, Abels M, Wierup N, Almgren P, Jansson PA, Ronn T, Hansson O, Eriksson KF, Groop L, Ling C. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes. 2012;61:3322–32.
Ribel-Madsen R, Fraga MF, Jacobsen S, Bork-Jensen J, Lara E, Calvanese V, Fernandez AF, Friedrichsen M, Vind BF, Hojlund K, Beck-Nielsen H, Esteller M, Vaag A, Poulsen P. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS One. 2012;7:e51302.
Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, Calonne E, Volkmar U, Igoillo-Esteve M, Naamane N, Del Guerra S, Masini M, Bugliani M, Marchetti P, Cnop M, Eizirik DL, Fuks F. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012;31:1405–26.
Volkov P, Bacos K, Ofori JK, Esguerra JL, Eliasson L, Ronn T, Ling C. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes. 2017;
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–7.
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ (Clinical research ed). 1991;303:1019–22.
Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation. 1996;94:3246–50.
Harder T, Rodekamp E, Schellong K, Dudenhausen JW, Plagemann A. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. Am J Epidemiol. 2007;165:849–57.
Poulsen P, Vaag AA, Kyvik KO, Moller Jensen D, Beck-Nielsen H. Low birth weight is associated with NIDDM in discordant monozygotic and dizygotic twin pairs. Diabetologia. 1997;40:439–46.
Tian JY, Cheng Q, Song XM, Li G, Jiang GX, Gu YY, Luo M. Birth weight and risk of type 2 diabetes, abdominal obesity and hypertension among Chinese adults. Eur J Endocrinol. 2006;155:601–7.
Alibegovic AC, Hojbjerre L, Sonne MP, van Hall G, Alsted TJ, Kiens B, Stallknecht B, Dela F, Vaag A. Increased rate of whole body lipolysis before and after 9 days of bed rest in healthy young men born with low birth weight. Am J Phys Endocrinol Metab. 2010;298:E555–64.
Brons C, Jacobsen S, Hiscock N, White A, Nilsson E, Dunger D, Astrup A, Quistorff B, Vaag A. Effects of high-fat overfeeding on mitochondrial function, glucose and fat metabolism, and adipokine levels in low-birth-weight subjects. Am J Phys Endocrinol Metab. 2012;302:E43–51.
Ozanne SE, Jensen CB, Tingey KJ, Martin-Gronert MS, Grunnet L, Brons C, Storgaard H, Vaag AA. Decreased protein levels of key insulin signalling molecules in adipose tissue from young men with a low birthweight: potential link to increased risk of diabetes? Diabetologia. 2006;49:2993–9.
Rasmussen EL, Malis C, Jensen CB, Jensen JE, Storgaard H, Poulsen P, Pilgaard K, Schou JH, Madsbad S, Astrup A, Vaag A. Altered fat tissue distribution in young adult men who had low birth weight. Diabetes Care. 2005;28:151–3.
Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, Gaughan JP, Coutifaris C, Sapienza C. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum Mol Genet. 2009;18:3769–78.
Khulan B, Cooper WN, Skinner BM, Bauer J, Owens S, Prentice AM, Belteki G, Constancia M, Dunger D, Affara NA. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: a study of a unique resource in the Gambia. Hum Mol Genet. 2012;21:2086–101.
Murphy SK, Adigun A, Huang Z, Overcash F, Wang F, Jirtle RL, Schildkraut JM, Murtha AP, Iversen ES, Hoyo C. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494:36–43.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, Slieker RC, Stok AP, Thijssen PE, Muller F, van Zwet EW, Bock C, Meissner A, Lumey LH, Eline Slagboom P, Heijmans BT. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592.
Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53.
Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009;4:e7845.
Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, Emerald BS, Gale CR, Inskip HM, Cooper C, Hanson MA. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–34.
Gillberg L, Perfilyev A, Brons C, Thomasen M, Grunnet LG, Volkov P, Rosqvist F, Iggman D, Dahlman I, Riserus U, Ronn T, Nilsson E, Vaag A, Ling C. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: role of high-fat overfeeding. Diabetologia. 2016;59:799–812.
Broholm C, Olsson AH, Perfilyev A, Hansen NS, Schrolkamp M, Strasko KS, Scheele C, Ribel-Madsen R, Mortensen B, Jorgensen SW, Ling C, Vaag A. Epigenetic programming of adipose-derived stem cells in low birthweight individuals. Diabetologia. 2016;59:2664–73.
Brons C, Jacobsen S, Nilsson E, Ronn T, Jensen CB, Storgaard H, Poulsen P, Groop L, Ling C, Astrup A, Vaag A. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J Clin Endocrinol Metab. 2010;95:3048–56.
Jacobsen SC, Gillberg L, Bork-Jensen J, Ribel-Madsen R, Lara E, Calvanese V, Ling C, Fernandez AF, Fraga MF, Poulsen P, Brons C, Vaag A. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia. 2014;57:1154–8.
Hjort L, Jorgensen SW, Gillberg L, Hall E, Brons C, Frystyk J, Vaag AA, Ling C. 36 h fasting of young men influences adipose tissue DNA methylation of LEP and ADIPOQ in a birth weight-dependent manner. Clin Epigenetics. 2017;9:40.
Permutt MA, Wasson J, Cox N. Genetic epidemiology of diabetes. J Clin Invest. 2005;115:1431–9.
Groop L, Pociot F. Genetics of diabetes—are we missing the genes or the disease? Mol Cell Endocrinol. 2014;382:726–39.
Prasad RB, Groop L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes. 2015;6:87–123.
Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56:1036–46.
Volkov P, Olsson AH, Gillberg L, Jorgensen SW, Brons C, Eriksson KF, Groop L, Jansson PA, Nilsson E, Ronn T, Vaag A, Ling C. A genome-wide mQTL analysis in human adipose tissue identifies genetic variants associated with DNA methylation, Gene Expression and Metabolic Traits. PloS one. 2016;11:e0157776.
Ling C, Poulsen P, Simonsson S, Ronn T, Holmkvist J, Almgren P, Hagert P, Nilsson E, Mabey AG, Nilsson P, Vaag A, Groop L. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest. 2007;117:3427–35.
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10.
Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, Sapienza C, Gudnason V, Feinberg AP. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–83.
Fraser HB, Lam LL, Neumann SM, Kobor MS. Population-specificity of human DNA methylation. Genome Biol. 2012;13:R8.
Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, Wilk A, Barrett A, Small KS, Ge B, Caron M, Shin SY, Lathrop M, Dermitzakis ET, McCarthy MI, Spector TD, Bell JT, Deloukas P. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93:876–90.
Marshall JA, Bessesen DH. Dietary fat and the development of type 2 diabetes. Diabetes Care. 2002;25:620–2.
Jacobsen SC, Brons C, Bork-Jensen J, Ribel-Madsen R, Yang B, Lara E, Hall E, Calvanese V, Nilsson E, Jorgensen SW, Mandrup S, Ling C, Fernandez AF, Fraga MF, Poulsen P, Vaag A. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia. 2012;55:3341–9.
Perfilyev A, Dahlman I, Gillberg L, Rosqvist F, Iggman D, Volkov P, Nilsson E, Riserus U, Ling C. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: a randomized controlled trial. Am J Clin Nutr. 2017;105:991–1000.
Cordero P, Campion J, Milagro FI, Goyenechea E, Steemburgo T, Javierre BM, Martinez JA. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J Physiol Biochem. 2011;67:463–70.
Bouchard L, Rabasa-Lhoret R, Faraj M, Lavoie ME, Mill J, Perusse L, Vohl MC. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am J Clin Nutr. 2010;91:309–20.
Milagro FI, Gomez-Abellan P, Campion J, Martinez JA, Ordovas JM, Garaulet M. CLOCK, PER2 and BMAL1 DNA methylation: association with obesity and metabolic syndrome characteristics and monounsaturated fat intake. Chronobiol Int. 2012;29:1180–94.
Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, Becker T, Rocken C, Schafmayer C, Hampe J. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A. 2014;111:15538–43.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, Meduri E, Morange PE, Gagnon F, Grallert H, Waldenberger M, Peters A, Erdmann J, Hengstenberg C, Cambien F, Goodall AH, Ouwehand WH, Schunkert H, Thompson JR, Spector TD, Gieger C, Tregouet DA, Deloukas P, Samani NJ. DNA methylation and body-mass index: a genome-wide analysis. Lancet (London, England). 2014;383:1990–8.
Main AM, Gillberg L, Jacobsen AL, Nilsson E, Gjesing AP, Hansen T, Pedersen O, Ribel-Madsen R, Vaag A. DNA methylation and gene expression of HIF3A: cross-tissue validation and associations with BMI and insulin resistance. Clin Epigenetics. 2016;8:89.
Pfeiffer S, Kruger J, Maierhofer A, Bottcher Y, Kloting N, El Hajj N, Schleinitz D, Schon MR, Dietrich A, Fasshauer M, Lohmann T, Dressler M, Stumvoll M, Haaf T, Bluher M, Kovacs P. Hypoxia-inducible factor 3A gene expression and methylation in adipose tissue is related to adipose tissue dysfunction. Sci Rep. 2016;6:27969.
Ronn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL, Jorgensen SW, Brons C, Jansson PA, Eriksson KF, Pedersen O, Hansen T, Groop L, Stener-Victorin E, Vaag A, Nilsson E, Ling C. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015;24:3792–813.
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, Tsai PC, Ried JS, Zhang W, Yang Y, Tan S, Fiorito G, Franke L, Guarrera S, Kasela S, Kriebel J, Richmond RC, Adamo M, Afzal U, Ala-Korpela M, Albetti B, Ammerpohl O, Apperley JF, Beekman M, Bertazzi PA, Black SL, Blancher C, Bonder MJ, Brosch M, Carstensen-Kirberg M, de Craen AJ, de Lusignan S, Dehghan A, Elkalaawy M, Fischer K, Franco OH, Gaunt TR, Hampe J, Hashemi M, Isaacs A, Jenkinson A, Jha S, Kato N, Krogh V, Laffan M, Meisinger C, Meitinger T, Mok ZY, Motta V, Ng HK, Nikolakopoulou Z, Nteliopoulos G, Panico S, Pervjakova N, Prokisch H, Rathmann W, Roden M, Rota F, Rozario MA, Sandling JK, Schafmayer C, Schramm K, Siebert R, Slagboom PE, Soininen P, Stolk L, Strauch K, Tai ES, Tarantini L, Thorand B, Tigchelaar EF, Tumino R, Uitterlinden AG, van Duijn C, van Meurs JB, Vineis P, Wickremasinghe AR, Wijmenga C, Yang TP, Yuan W, Zhernakova A, Batterham RL, Smith GD, Deloukas P, Heijmans BT, Herder C, Hofman A, Lindgren CM, Milani L, van der Harst P, Peters A, Illig T, Relton CL, Waldenberger M, Jarvelin MR, Bollati V, Soong R, Spector TD, Scott J, McCarthy MI, Elliott P, Bell JT, Matullo G, Gieger C, Kooner JS, Grallert H, Chambers JC. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–6.
Pietilainen KH, Ismail K, Jarvinen E, Heinonen S, Tummers M, Bollepalli S, Lyle R, Muniandy M, Moilanen E, Hakkarainen A, Lundbom J, Lundbom N, Rissanen A, Kaprio J, Ollikainen M. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int J Obesity (2005). 2016;40:654–61.
Davegardh C, Broholm C, Perfilyev A, Henriksen T, Garcia-Calzon S, Peijs L, Hansen NS, Volkov P, Kjobsted R, Wojtaszewski JF, Pedersen M, Pedersen BK, Ballak DB, Dinarello CA, Heinhuis B, Joosten LA, Nilsson E, Vaag A, Scheele C, Ling C. Abnormal epigenetic changes during differentiation of human skeletal muscle stem cells from obese subjects. BMC Med. 2017;15:39.
Alibegovic AC, Sonne MP, Hojbjerre L, Bork-Jensen J, Jacobsen S, Nilsson E, Faerch K, Hiscock N, Mortensen B, Friedrichsen M, Stallknecht B, Dela F, Vaag A. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am J Phys Endocrinol Metab. 2010;299:E752–63.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Bajpeyi S, Covington JD, Taylor EM, Stewart LK, Galgani JE, Henagan TM. Skeletal muscle PGC1alpha -1 nucleosome position and -260nt DNA methylation determine exercise response and prevent ectopic lipid accumulation in men. Endocrinology. 2017;158:2190–9.
Barres R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O'Gorman DJ, Zierath JR. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–11.
Ronn T, Volkov P, Davegardh C, Dayeh T, Hall E, Olsson AH, Nilsson E, Tornberg A, Dekker Nitert M, Eriksson KF, Jones HA, Groop L, Ling C. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013;9:e1003572.
Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekstrom TJ, Tegner J, Sundberg CJ. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9:1557–69.
Rowlands DS, Page RA, Sukala WR, Giri M, Ghimbovschi SD, Hayat I, Cheema BS, Lys I, Leikis M, Sheard PW, Wakefield SJ, Breier B, Hathout Y, Brown K, Marathi R, Orkunoglu-Suer FE, Devaney JM, Leiken B, Many G, Krebs J, Hopkins WG, Hoffman EP. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in type 2 diabetic obesity. Physiol Genomics. 2014;46:747–65.
Ronn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, Tuomi T, Isomaa B, Groop L, Vaag A, Ling C. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51:1159–68.
Casillas MA Jr, Lopatina N, Andrews LG, Tollefsbol TO. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol Cell Biochem. 2003;252:33–43.
Bysani M, Perfilyev A, de Mello VD, Ronn T, Nilsson E, Pihlajamaki J, Ling C. Epigenetic alterations in blood mirror age-associated DNA methylation and gene expression changes in human liver. Epigenomics. 2017;9:105–22.
Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013;14:R102.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Huse SM, Gruppuso PA, Boekelheide K, Sanders JA. Patterns of gene expression and DNA methylation in human fetal and adult liver. BMC Genomics. 2015;16:981.
Zykovich A, Hubbard A, Flynn JM, Tarnopolsky M, Fraga MF, Kerksick C, Ogborn D, MacNeil L, Mooney SD, Melov S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell. 2014;13:360–6.
Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, Gjesing AP, Eiberg H, Tuomi T, Almgren P, Groop L, Eliasson L, Vaag A, Dayeh T, Ling C. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. 2016;7:11089.
Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin SY, Dempster EL, Murray RM, Grundberg E, Hedman AK, Nica A, Small KS, Dermitzakis ET, McCarthy MI, Mill J, Spector TD, Deloukas P. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629.
Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97.
Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Swedish Research Council, Region Skåne (ALF), the Royal Physiographic Society in Lund, the Påhlsson Foundation, and the Swedish Diabetes Foundation.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
Both authors reviewed the literature and wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nilsson, E., Ling, C. DNA methylation links genetics, fetal environment, and an unhealthy lifestyle to the development of type 2 diabetes. Clin Epigenet 9, 105 (2017). https://doi.org/10.1186/s13148-017-0399-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-017-0399-2