
Abstract
Background
Nifedipine is a generic, well-known and commonly-prescribed dihydropyridine calcium channel blocker used in the treatment of hypertension and Prinzmetal’s angina. A known but very rare and serious adverse effect of nifedipine is clinically-apparent hepatitis which can take months to resolve.
Case presentation
Here we present a case of nifedipine-induced hepatitis in a 78-year-old Caucasian female with no prior history of liver or autoimmune disease. We discuss our investigative and management approach, and present a review of prior cases. We offer an approach for patients who present with signs of acute liver injury with jaundice and high elevations in serum transaminases.
Conclusion
Not much is known about nifedipine-induced hepatitis due to its rare occurrence. Its prevalence is unknown. The disease appears to afflict older men and women. It can present acutely (within days) or subacutely (within 4–8 weeks after medication start) and in an idiosyncratic manner. Chronic or latent cases have also been described, some diagnosed as late as 3 years after medication start. Common symptoms include jaundice, nausea, chills, rigors, diaphoresis, fatigue, and abdominal pain. Laboratory investigations often reveal profound elevations in AST, ALT, GGT, and conjugated bilirubin. Peripheral blood smear may demonstrate eosinophilia. Histology from liver biopsy typically demonstrates infiltration of immune cells, cholestasis, and a picture of steatohepatitis. Treatment involves immediate discontinuation of the drug with supportive care. Thus far, all published instances of nifedipine-induced hepatitis were self-limiting without mortality due to fulminant liver failure. However, this disease can take months to resolve. There is no randomized evidence for other treatments such as corticosteroids.
Similar content being viewed by others

Background
Nifedipine-induced hepatitis is a rare type of drug-induced liver injury (DILI), which has been a subject of increased scrutiny in the medical community over the last several years. Broadly speaking, DILI is a serious complication of medication use, with a prevalence of up to 14 out of 100,000 people [1]. It is implicated in up to 33% of patients who present with acute liver injury [2], and has been cited as the most common cause of acute liver failure—about 52% of all cases according to one study [3].
The majority of DILI—up to 75% in one prospective study—is due to acetaminophen ingestion [3]. Other notable causes include antibiotics—especially amoxicillin-clavulanate [2] and fluoroquinolones [4] —as well as medications from other classes such as anti-epileptics (Table 1). DILI is typically dose-dependent, and occasionally idiosyncratic. Dose-dependent DILI is most commonly caused by large and often supratherapeutic doses of hepatotoxic drugs, in particular acetaminophen. Conversely, idiosyncratic DILI is not dose-dependent. DILI has a broad range of clinical presentations, from subclinical (or chronic hepatitis) to fulminant liver failure requiring transplantation or even death [5]. Histologically it is a diverse phenomenon, with findings that can resemble autoimmune hepatitis, cholestasis, steatosis, fibrosis, phospholipidosis, hepatic vein thrombosis, biliary sclerosis, granulomatous hepatitis, peliosis hepatis and cirrhosis [6,7,8,9,10]. Rarely, DILI is also associated with hepatic neoplasms, in particular adenomas, angiosarcomas, and hepatocellular carcinomas [11, 12]. For these reasons, DILI can be thought of as a “great imitator” of liver disease and should be suspected in any patient with acute or chronic hepatitis of unclear etiology.
Case presentation
A 78-year-old Caucasian female presented to a community hospital in Canada with jaundice, scleral icterus, and mild epigastric discomfort. She reported a 4-day history of progressive fatigue with intermittent nausea and vomiting, diminished appetite, as well as loose, lightly coloured, and foul-smelling stools. She had no evidence of coagulopathy or encephalopathy. Her past medical history included essential hypertension, dyslipidemia, and chronic obstructive pulmonary disease. She had no history of liver disease, intravenous drug use, excessive alcohol consumption, recent travel, infectious symptoms, or constitutional symptoms. She had no exposure to poisonous mushrooms or other common hepatotoxic agents. Her medications were atorvastatin 20 mg daily which she had been taking for many years, a course of clarithromycin for community-acquired pneumonia completed 28 days prior to admission, and nifedipine 10 mg daily for hypertension which was started 14 days prior to admission. She did not take any alternative or complementary therapies.
Physical examination revealed a slightly distended abdomen with diffuse tenderness to palpation, particularly in the epigastric region, and jaundice with scleral icterus. There was no rebound tenderness, guarding, or hepatosplenomegaly. She did not have any other stigmata of liver disease and the rest of her physical examination was unremarkable.
Initial laboratory investigations revealed profound elevations in all liver enzymes, bilirubin, and LDH—all indicative of significant hepatocellular injury:
-
ALT 1912 U/L (29× upper limit of normal or “ULN”).
-
AST U/L (42× ULN).
-
ALP 439 U/L (3× ULN).
-
GGT 352 U/L (6× ULN).
-
Conjugated bilirubin 119 μmol/L (24× ULN).
-
Total bilirubin 134 μmol/L (7× ULN).
-
LDH 408 U/L (1.7× ULN).
Her INR was between 1.1 and 1.3 throughout the hospitalization, PTT was 27 s, and albumin was slightly low at 31 g/L (normal > 34 g/L). Lipase was within normal range at 288 U/L, and creatine kinase was normal at 31 U/L.
Her complete blood count was within normal limits. There was a mild monocytosis of 1.4 giga/L. Her electrolytes were within normal range. Creatinine was 65 μmol/L with an estimated glomerular filtration rate (eGFR) of 76 mL/min. Her lactate, anion gap, random blood glucose, and extended electrolytes were all also within normal limits.
Other laboratory investigations ruled out many infectious, metabolic, and autoimmune causes of acute liver injury. Serologies (IgM) for Epstein–Barr virus (EBV) and cytomegalovirus (CMV) were negative. Serologies for the human immunodeficiency virus (HIV) and hepatitis A, B, C, and E viruses (e.g. hepatitis A IgM, HBsAg, Anti-HBs, anti-HBc, anti-HBc IgM, anti-HCV, HBeAg) revealed no sign of current infection. Anti-liver kidney microsome type 1 (anti-LKM1) antibody, anti-mitochondrial antibody (AMA), anti-smooth muscle antibody (ASMA), ceruloplasmin, and immunoglobulins were negative. Alpha-1 antitrypsin was also negative. The ANA titre was 1:80, a non-specific finding in the context of the patient’s advanced age.
A computed tomography (CT) scan of the patient’s abdomen revealed that her hepatic parenchyma was unremarkable, with no evidence of hepatic obstruction, biliary duct dilatation, thrombosis, or malignancy (Fig. 1). Incidentally, the patient was discovered to have a prominent Riedel’s lobe of the liver and sigmoid diverticulosis. A follow-up abdominal ultrasound revealed that her liver had a heterogeneous echotexture. The gallbladder was markedly thickened and edematous but decompressed.

Findings on imaging. Computed tomography (CT) of the patient’s abdomen (A—abdominal view, and B—liver view) revealed a prominent Riedel lobe of the liver, with no evidence of obstruction, thrombosis, or malignancy. Mild changes of sigmoid diverticulosis were seen, with no evidence of acute diverticulitis. A repeat CT revealed mild ascites localized to the right upper quadrant, and the liver edge appeared to have a slightly nodular contour, a non-specific finding in this particular case, but which in general may suggest cirrhosis. Abdominal ultrasound (C, D) revealed a heterogeneous echotexture of the liver with regions of increased echogenicity in the right lobe, a non-specific finding which may be seen in hepatitis
The patient was admitted to hospital. Suspicious that her liver injury may be statin-induced, her atorvastatin was held on admission. Nifedipine was also held on admission.
Although her liver enzymes continued to steadily improve during the admission, the patient’s clinical condition deteriorated during the first 14 days of hospitalization with worsening fatigue, jaundice, mild hypotension and development of a generalized, pruritic urticarial eruption that partially responded to antihistamines. The patient then developed an acute COPD exacerbation treated with a 5-day course of prednisone, oral moxifloxacin, nebulized ipratropium and salbutamol, and supplemental oxygen.
Given the ongoing elevation in liver enzymes and jaundice, an ultrasound-guided core liver biopsy was performed. The biopsy demonstrated portal tract and central zone inflammation with necrosis and plasma cell infiltrates, which was consistent with a hypersensitivity-mediated drug-induced hepatitis (see Fig. 2).

Findings on liver core biopsy. Ultrasound-guided core needle biopsy of the liver demonstrated that the overall hepatic architecture was preserved. Abnormal features included the marked expansion and fibrosis of portal tracts. In A fibrous tongues can be seen extending outwards into the liver parenchyma. The centrilobular areas appear preserved. There is pericentral sinusoidal congestion. Within the portal tracts (B) there are heavy infiltration of acute and chronic inflammatory cells, mostly lymphocytes and neutrophils. This extends into the limiting plate and extensively involves the adjacent liver parenchyma. Many damaged bile ductules and necrotic hepatocytes can be seen in C, and isolated necrotic hepatocytes can also be seen in the liver parenchyma in D. These biopsy findings are consistent with severe subacute hepatitis with developing portal fibrosis. The presence of plasma cells within the inflammatory infiltrate suggests the likelihood of an immune mechanism (hypersensitivity) as a component of the damage. However, the overall pattern of liver damage is unlike typical autoimmune hepatitis. In the absence of anti-smooth muscle antibodies, the possibility of drug-induced hepatitis via a hypersensitivity or immune mechanism is favoured. In particular, bile ductular damage—which is commonly seen in drug-induced hepatitis—may in turn produce hypersensitivity-related liver damage
Plasma cell infiltration of the liver, seen in our patient’s liver biopsy, can be a sign of autoimmune hepatitis. However, in this case, we felt that the overall histologic pattern of liver damage was not consistent with what would be typically seen in autoimmune hepatitis. This, coupled with the absence of immunological markers associated with autoimmune hepatitis, made the diagnosis of autoimmune hepatitis less likely.
In addition, the patient’s presentation is very consistent with published cases of nifedipine-induced hepatitis, which have suggested that symptoms typically manifest within 2 weeks of initial exposure. This patient’s symptoms began 10 days after she started taking the drug.
During our patient’s third week of hospital stay, her bilirubin and liver enzymes began to normalize. Her jaundice, abdominal pain, and urticaria resolved, and her energy improved. She remained in hospital for 2 additional weeks for supportive care and rehabilitation. She was discharged home after 5 weeks of hospitalization with full resolution of her liver disease and return to baseline functioning.
The limitation of our diagnosis was that it largely relied on the exclusion of other diagnoses. While it remains possible that there was another etiology, we felt the odds were small. One way to confirm the diagnosis would be to re-challenge the patient with nifedipine after she has recovered from the illness, to see if a similar reaction recurred. However, given the severity of the patient’s liver injury, we did not have the option to do this.
The patient’s other medications—in particular atorvastatin and clarithromycin—have also been associated with drug-induced hepatitis. However, for various reasons discussed below, we felt that nifedipine was the most likely cause.
The possibility that the patient’s liver injury was caused by atorvastatin was strongly considered, however ultimately, we believe this to be less likely as the patient had been taking atorvastatin for years with no complications. Twelve weeks after the patient was discharged from hospital, the patient was re-challenged with atorvastatin at gradually increasing doses and experienced no liver injury.
Clarithromycin has been known to cause liver enzyme elevations in 1–3% of patients exposed to the drug, and rarely, it can also cause fulminant hepatitis similar to nifedipine. One key difference is that a number of deaths have been reported in the literature for clarithromycin, whereas none have been reported for nifedipine [13]. The histology of both statin and clarithromycin-induced hepatitis can be similar to the histology of nifedipine-induced hepatitis. All can exhibit signs of cholestasis [14] and infiltration of immune cells [15]. Despite these similarities, the diagnosis of clarithromycin-induced hepatitis was felt to be less likely for our patient as she has been exposed to clarithromycin on multiple occasions for COPD exacerbations over the past several years, and none of her previous exposures to clarithromycin has resulted in drug-induced liver injury. Furthermore, the onset and peak of liver injury in relation to the timing of clarithromycin exposure is not typical of clarithromycin-induced liver injury—at least for the ones that have been reported. A recent review of the literature on this topic demonstrated that the vast majority of fulminant liver injuries caused by clarithromycin occur within days of administration, usually within 1 week [13]. Onset of fulminant liver injury that occurs 3 weeks after discontinuation of clarithromycin has virtually been unreported.
Discussion and conclusions
Nifedipine is a dihydropyridine calcium channel blocker, initially developed in Germany in the 1970s [16]. It remains a commonly prescribed medication for hypertension and Prinzmetal angina. Common side effects include facial flushing (up to 25%), peripheral edema (up to 30%), dizziness or lightheadedness (up to 27%), and gastrointestinal symptoms such as nausea and heartburn (up to 11%) [17]. A very rare but known adverse effect of nifedipine is drug-induced liver injury, which has been described in the literature in nine case reports and reviews that were published between 1979 and 1999 [14, 15, 17,18,19,20,21,22,23] [14, 15, 17, 19,20,21,22,23].
Characteristics of nifedipine-induced hepatitis
Not much is known about nifedipine-induced hepatitis due to its rare occurrence. The disease appears to afflict older men and women, with an age range of 59–80 years old as per published case reports [13, 14, 16,17,18,19,20,21] [13, 14, 17,18,19,20,21,22] [14, 15, 17,18,19,20,21,22] [14, 15, 17, 19,20,21,22,23].
Nifedipine-induced hepatitis is often acute and idiosyncratic, with symptoms occurring as rapidly as within 12 h to as late as 1 month after initial ingestion [14, 17]. Indolent cases have been described, diagnosed as late as 3 years after initial ingestion [14, 23]. Common presenting symptoms are jaundice, nausea, chills, rigors, diaphoresis, fatigue, and abdominal tenderness.
Biochemically, the initial pattern of liver injury is profound elevation of liver enzymes and conjugated bilirubin. Peripheral blood smear may reveal eosinophilia [15]. Data from published cases reflect an average elevation of AST of 17× ULN, ALT 7× ULN, GGT 16× ULN, and conjugated bilirubin of 14× ULN. Total bilirubin was also frequently elevated but to a lesser degree at 5× ULN, as were LDH (3× ULN) and ALP (3× ULN) (Table 2). Immunological markers of autoimmune hepatitis, ANA, ASMA, AMA, and anti-LKM1 are typically negative or inconclusive.
On liver biopsy, a multitude of findings have been described in the literature. There is often a general impression of “hypersensitivity” [20] or “subacute hepatitis” [17]. Signs of cholestasis and centrilobular necrosis have been observed [14]. Other features include an alcoholic-like picture with steatosis, hypertrophic hepatocytes and Mallory bodies [14], and an expansion of portal tracts with a mixed inflammatory infiltration of lymphocytes, histiocytes, neutrophils, and eosinophils [15]. In these latter findings, the parenchyma architecture may be preserved with mild infiltration of lymphocytes, neutrophils, and eosinophils [15]. The findings of nifedipine-induced hepatitis are similar to the findings of liver injury from verapamil, another calcium channel blocker [24].
In case reports where repeat liver biopsies were obtained following clinical and biochemical resolution of the liver injury, histological changes seen during the illness—in particular steatosis and Mallory bodies—typically resolve [14].
Diagnosis, management, and outcome of severe drug induced liver injury
DILIs are one of the few etiologies of liver injury that can result in very high elevations of liver enzymes that exceed 1000 U/L (Table 3). The main treatment for DILI is prompt discontinuation of the offending drug. Switching patients to another drug from the same class is not entirely safe as cross-reactions frequently occur. For instance, in one case of nifedipine-induced hepatitis, the patient’s jaundice recurred after he was started on amlodipine, another dihydropyridine calcium channel blocker [23].
While the majority of DILIs will resolve with prompt discontinuation of the offending drug, DILIs can worsen nonetheless and progress to liver failure that requires transplantation. Long-term sequelae of DILI include ongoing impaired liver function, progression to cirrhosis, and rarely, the development of hepatic malignancies. Mortality rates for DILI vary greatly depending on the offending drug and patient population, but has been reported as up to 60% in the published literature [4]. Fortunately, nifedipine-induced liver injury tends to have a favorable outcome, and no cases of mortality or liver transplantation have been reported in the literature [14].
Additional therapeutic options are available to clinicians in select situations. Acetaminophen toxicity can be treated with n-acetyl cysteine (NAC) [25]. Valproate-induced hepatitis can be managed with activated charcoal and emergent hemodialysis [26].
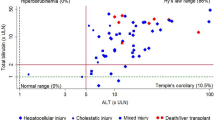
Analysis of the United States Network for Organ Sharing (UNOS) liver transplant database revealed that DILI accounted for 15% of liver transplants secondary to acute liver failure. Most (76%) of the recipients were female. Acetaminophen accounted for 49% of DILI-related liver transplants, followed by isoniazid (17.5%), propylthiouracil (9.5%), phenytoin (7.3%) and valproate (7.3%). A marker for increased severity and poorer prognosis was described by Hyman Zimmerman and recently validated by two different groups in studies involving a large number of patients with suspected DILI. Named Hy’s rule, it includes the presence of jaundice with a concomitant elevation in serum bilirubin (≥ 2× ULN) and serum transaminases, in particular ALT (≥ 3× ULN) [27].
Closing remarks
Hepatitis is a rare adverse effect of nifedipine. It typically affects older individuals who recently began taking the medication. Presenting symptoms include jaundice, nausea, chills, rigors, diaphoresis, fatigue, and right upper quadrant abdominal pain. Laboratory investigations reveal elevations in bilirubin and liver enzymes. Autoimmune serology are not expected to be positive. Imaging is usually benign or non-specific. Liver biopsy often shows a pattern of generalized inflammation, infiltration of immune cells in hepatic structures, and cholestasis. Patients will not improve unless the offending medication is discontinued. Other calcium channel blockers should be avoided if possible, as there have been reports of cross-reactivity. Treatment is supportive, as no treatments have been formally studied. Reassuringly, the nifedipine-induced DILI appears to be self-limiting although the elevations in liver enzymes can take months to resolve. No deaths from nifedipine-induced hepatitis have been reported in the literature.
In general, drug-induced hepatitis should be suspected in all patients with acute or chronic liver injury of unclear etiology. While most cases of drug-induced hepatitis resolve with prompt discontinuation of the offending agent, some may progress to fulminant and irreversible liver failure. Markers for increased severity and poorer prognosis include the presence of jaundice and elevation in bilirubin (≥ 2× ULN) and ALT ≥ 3× ULN [27]. This case serves as a reminder to clinicians to always consider the possibility of drug-induced hepatitis in patients with liver injury of unclear etiology.
Abbreviations
- ALP:
-
alkaline phosphatase
- ALT:
-
alanine aminotransferase or alanine transaminase
- AMA:
-
anti-mitochondrial antibody
- ANA:
-
anti-nuclear antibody
- ANCA:
-
anti-neutrophil cytoplasmic antibody
- Anti-LKM1:
-
anti-liver kidney microsomal type 1 antibody
- ASMA:
-
anti-smooth muscle antibody
- AST:
-
aspartate aminotransferase
- BPM:
-
beats per minute
- CMV:
-
cytomegalovirus
- COPD:
-
chronic obstructive pulmonary disease
- CT:
-
computed tomography
- DILI:
-
drug-induced liver injury
- EBV:
-
Epstein–Barr virus
- eGFR:
-
estimated glomerular filtration rate
- g/L:
-
grams per liter
- GGT:
-
gamma-glutamyl transpeptidase
- giga:
-
unit prefix in the metric system denoting a factor of billion
- HIV:
-
human immunodeficiency virus
- IADL:
-
instrumental activities of daily living
- INR:
-
international normalized ratio
- IV:
-
intravenous
- LDH:
-
lactate dehydrogenase
- mg:
-
milligram
- mL:
-
milliliter
- mL/minute:
-
milliliters per minute
- mmol/L:
-
millimoles per liter
- MRI:
-
magnetic resonance imaging
- NAC:
-
N-acetyl cysteine
- NSAID:
-
non-steroidal anti-inflammatory drug
- PTT:
-
partial thromboplastin time
- U/L:
-
units per liter
- ULN:
-
upper limit of normal
- umol/L:
-
micromole per liter
- UNOS:
-
United States Network for Organ Sharing
- WBC:
-
white blood cell
References
Sgro C, Clinard F, Ouazir K, Chanay H, Allard C, Guilleminet C, et al. Incidence of drug-induced hepatic injuries: a French population-based study. Hepatology. 2002;36:451–5.
Galan MV, Potts JA, Silverman AL, Gordon SC. The burden of acute nonfulminant drug-induced hepatitis in a United States tertiary referral center. J Clin Gastroenterol. 2005;39:64–7.
Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SHB, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–54.
Paterson JM, Mamdani MM, Manno M, Juurlink DN, Canadian Drug Safety and Effectiveness Research Network. Fluoroquinolone therapy and idiosyncratic acute liver injury: a population-based study. Cancer Med Assoc J. 2012;184:1565–70.
Russo MW, Galanko JA, Shrestha R, Fried MW, Watkins P. Liver transplantation for acute liver failure from drug induced liver injury in the United States. Liver Transplant. 2004;10:1018–23.
Chang CC, Petrelli M, Tomashefski JF, McCullough AJ. Severe intrahepatic cholestasis caused by amiodarone toxicity after withdrawal of the drug: a case report and review of the literature. Arch Pathol Lab Med. 1999;123:251–6.
Perarnau J-M, Bacq Y. Hepatic vascular involvement related to pregnancy, oral contraceptives, and estrogen replacement therapy. Semin Liver Dis. 2008;28:315–27.
Tsirigotis P, Sella T, Shapira MY, Bitan M, Bloom A, Kiselgoff D, et al. Peliosis hepatis following treatment with androgen-steroids in patients with bone marrow failure syndromes. Haematologica. 2007;92:e106–10.
Ramachandran R, Kakar S. Histological patterns in drug-induced liver disease. J Clin Pathol. 2009;62:481–92.
Harrison RF, Elias E. Amiodarone-associated cirrhosis with hepatic and lymph node granulomas. Histopathology. 1993;22:80–2.
Tazawa K, Yasuda M, Ohtani Y, Makuuchi H, Osamura RY. Multiple hepatocellular adenomas associated with long-term carbamazepine. Histopathology. 1999;35:92–4.
Zimmerman HJ, Lewis JH. Chemical-and toxin-induced hepatotoxicity. Gastroenterol Clin N Am. 1995;24:1027–45.
Babany G, Uzzan F, Larrey D, Degott C, Bourgeois P, René E, et al. Alcoholic-like liver lesions induced by nifedipine. J Hepatol. 1989;9:252–5.
Shaw DR, Misan GM, Johnson RD. Nifedipine hepatitis. Aust N Z J Med. 1987;17:447–8.
Vater W, Kroneberg G, Hoffmeister F, Saller H, Meng K, Oberdorf A. Pharmacology of 4-(2′-nitrophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylic acid dimethyl ester (Nifedipine, BAY a 1040). Arzneimittelforschung. 1972;22:1–14.
Rotmensch HH, Roth A, Liron M, Rubinstein A, Gefel A, Livni E. Lymphocyte sensitisation in nifedipine-induced hepatitis. Br Med J. 1980;281:976–7.
Rotmensch HH, Weintraub M, Sofferman G, Livni E, Klejman A, Liron M. Experience with immunological tests in drug-induced hepatitis. Z Gastroenterol. 1981;19:691–7.
Abramson M, Littlejohn GO. Hepatic reactions to nifedipine. Med J Aust. 1985;142:47–8.
Welch HG, Lazar B, Gresser J, McMahon BJ. Nifedipine-induced hepatitis. Alaska Med. 1986;28:11–2.
Sawaya GF, Robertson PA. Hepatotoxicity with the administration of nifedipine for treatment of preterm labor. Am J Obstet Gynecol. 1992;167:512–3.
Basile C, Mascia E. Dihydropyridine calcium channel blockers: a rare and reversible cause of hepatotoxicity with cholestasis in a CAPD patient. Nephrol Dial Transplant. 1999;14:2776–7.
Guarascio P, D’Amato C, Sette P, Conte A, Visco G. Liver damage from verapamil. Br Med J (Clin Res Ed). 1984;288:362–3.
Saritas A, Kandis H, Baltaci D, Yildirim U, Kaya H, Karakus A, et al. N-Acetyl cysteine and erdosteine treatment in acetaminophen-induced liver damage. Toxicol Ind Health. 2014;30:670–8.
Tank JE, Palmer BF. Simultaneous “in series” hemodialysis and hemoperfusion in the management of valproic acid overdose. Am J Kidney Dis. 1993;22:341–4.
Björnsson E. Drug-induced liver injury: Hy’s rule revisited. Clin Pharmacol Ther. 2006;79:521–8.
Maddrey WC. Isoniazid-induced liver disease. Semin Liver Dis. 1981;1:129–33.
Karthik SV, Casson D. Erythromycin-associated cholestatic hepatitis and liver dysfunction in children: the British experience. J Clin Gastroenterol. 2005;39:743–4.
Iverson SL, Uetrecht JP. Identification of a reactive metabolite of terbinafine: insights into terbinafine-induced hepatotoxicity. Chem Res Toxicol. 2001;14:175–81.
Koch RO, Graziadei IW, Zangerle R, Romani N, Maier H, Vogel W. Acute hepatic failure and lactate acidosis associated with antiretroviral treatment for HIV. Wien Klin Wochenschr. 2003;115:135–40.
Miller KD, Cameron M, Wood LV, Dalakas MC, Kovacs JA. Lactic acidosis and hepatic steatosis associated with use of stavudine: report of four cases. Ann Intern Med. 2000;133:192–6.
O’Connor N, Dargan PI, Jones AL. Hepatocellular damage from non-steroidal anti-inflammatory drugs. QJM. 2003;96:787–91.
Andrade RJ, Lucena MI, Kaplowitz N, García-Muņoz B, Borraz Y, Pachkoria K, et al. Outcome of acute idiosyncratic drug-induced liver injury: long-term follow-up in a hepatotoxicity registry. Hepatology. 2006;44:1581–8.
Tolman KG. The liver and lovastatin. Am J Cardiol. 2002;89:1374–80.
Ben-Yehuda A, Bloom A, Lijovetzky G, Flusser D, Tur-Kaspa R. Chlorpromazine-induced liver and bone marrow granulomas associated with agranulocytosis. Isr J Med Sci. 1990;26:449–51.
Monteith DK, Emmerling MR, Garvin J, Theiss JC. Cytotoxicity study of tacrine, structurally and pharmacologically related compounds using rat hepatocytes. Drug Chem Toxicol. 1996;19:71–84.
Geubel AP, De Galocsy C, Alves N, Rahier J, Dive C. Liver damage caused by therapeutic vitamin A administration: estimate of dose-related toxicity in 41 cases. Gastroenterology. 1991;100:1701–9.
Davidson AR. Lymphocyte sensitisation in nifedipine-induced hepatitis. Br Med J. 1980;281(6251):1354.
Authors’ contributions
DY obtained consent from the patient and her family and drafted the article. DO assisted in diagnosing the case and manuscript preparation. JC and MH assisted in the preparation and publication of the manuscript. DL was the attending physician and provided much of the inspiration behind the article. MJF assisted in diagnosing the case and provided additional clinical guidance. All authors read and approved the final manuscript.
Authors’ information
DY is an internist and alumnus of the University of British Columbia Faculty of Medicine. JC is an alumnus of the Faculty of Land and Food Systems at the University of British Columbia and an MBA student at the Beedie School of Business. DO is a professor of pathology in the Department of Pathology and Laboratory Medicine at the University of British Columbia and consultant pathologist at Vancouver General Hospital. His major interest is the morphologic aspects of gastrointestinal and pancreatic disease including the influence of histopathologic appearances on the prognosis of malignant neoplasms. MH is a general internist and alumnus of the University of British Columbia Faculty of Medicine. DL is a family physician and clinical professor in the Department of Family Medicine at the University of British Columbia. MJF is a gastroenterologist and clinical professor in the Department of Medicine at the University of British Columbia.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
We thank the patient and her family for giving us their permission to study and publish this case for the advancement of medical knowledge and for the benefit of future patients.
Availability of data and materials
This article contains all of the data and materials that were available to us and utilized in the production of this article. No additional data or materials exist beyond what is presented in this article.
Consent for publication
Express written consent was provided to the authors by the patient for the publication of this case report.
Ethics approval and consent to participate
Not applicable.
Funding
This study was self-funded by the authors. No outside source of funding exists.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yusuf, D., Christy, J., Owen, D. et al. A case report of nifedipine-induced hepatitis with jaundice. BMC Res Notes 11, 228 (2018). https://doi.org/10.1186/s13104-018-3322-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-018-3322-9