
Abstract
Background
The genome of the gastric pathogen Helicobacter pylori is characterised by considerable variation of both gene sequence and content, much of which is contained within three large genomic islands comprising the cag pathogenicity island (cagPAI) and two mobile integrative and conjugative elements (ICEs) termed tfs3 and tfs4. All three islands are implicated as virulence factors, although whereas the cagPAI is well characterised, understanding of how the tfs elements influence H. pylori interactions with different human hosts is significantly confounded by limited definition of their distribution, diversity and structural representation in the global H. pylori population.
Results
To gain a global perspective of tfs ICE population dynamics we established a bioinformatics workflow to extract and precisely define the full tfs pan-gene content contained within a global collection of 221 draft and complete H. pylori genome sequences. Complete (ca. 35-55kbp) and remnant tfs ICE clusters were reconstructed from a dataset comprising > 12,000 genes, from which orthologous gene complements and distinct alleles descriptive of different tfs ICE types were defined and classified in comparative analyses. The genetic variation within defined ICE modular segments was subsequently used to provide a complete description of tfs ICE diversity and a comprehensive assessment of their phylogeographic context. Our further examination of the apparent ICE modular types identified an ancient and complex history of ICE residence, mobility and interaction within particular H. pylori phylogeographic lineages and further, provided evidence of both contemporary inter-lineage and inter-species ICE transfer and displacement.
Conclusions
Our collective results establish a clear view of tfs ICE diversity and phylogeographic representation in the global H. pylori population, and provide a robust contextual framework for elucidating the functional role of the tfs ICEs particularly as it relates to the risk of gastric disease associated with different tfs ICE genotypes.
Similar content being viewed by others

Background
Both Helicobacter pylori and other non-pylori Helicobacters have the capacity to colonise the gastric mucosa of humans, increasing the risk of development of a range of gastrointestinal diseases [1, 2]. In respect of H. pylori, these include chronic gastritis, peptic ulcer and gastric adenocarcinoma which account for significant morbidity and mortality worldwide [3]. However, of H. pylori infected individuals, estimated to comprise one half of the world’s population, clinical disease manifests in a subset of only 15-20%, less than 1% of which develop gastric cancer [4,5,6]. This incidence is considered a consequence of the multifactorial nature of infection, in which disease risk and susceptibility is influenced by complex interplay between a variety of host, bacterial and environmental factors [7,8,9].
H. pylori is acquired in infancy, predominantly from within familial or close community groups [10] and invariably maintains a persistent infection throughout the lifetime of the host [4]. The consequent long association with genetically related ethnic groups as a result of such localised transmission has led to the selection of locally adapted H. pylori genotypes with distinct phylogeographic signals [11, 12]. These geographic patterns of diversity are concordant with human diversity and have been used in population genetic analyses to define different H. pylori populations that co-evolved with human hosts ancestrally native to particular global geographical regions [12,13,14,15,16].
Currently, seven major genetically and geographically distinct H. pylori populations (‘hp’) and five subpopulations (‘hsp’) have been described by multi locus sequence typing (MLST) and the STRUCTURE Bayesian population cluster method, defined as hpAfrica1 (subpopulations hspWAfrica, hspSAfrica), hpAfrica2, hpNEAfrica, hpEurope, hpAsia2, hpEAsia (subpopulations hspAmerind, hspEAsia and hspMaori) and hpSahul [13,14,15,16]. More recent methods such as fineSTRUCTURE have further resolved the genetic structure of these populations, expanding the number of subdivisions and providing detailed insight of inter-population admixture that contributes to the extreme genetic diversity of H. pylori strains [17]. Through such analyses, the association of H. pylori with humans is understood to be ancient, significantly pre-dating the migratory expansion of anatomically modern humans out of Africa ca. 60, 000 years ago (60kya) [13, 14, 16, 18, 19]. The long co-evolutionary association with the geographically distinct human populations that subsequently emerged during colonisation of the globe provides a possible explanation for the benign, if not beneficial, lifetime carriage of H. pylori by the majority of infected individuals [8, 9, 20, 21] since co-evolution and host adaptation of pathogens, particularly in the context of vertical transmission (parent to child), is proposed to select for reduced virulence and the promotion of commensalism [22]. Consistently, H. pylori indigenous to globally remote human populations have been demonstrated to encode an attenuated, low virulence form of the host-interactive CagA oncoprotein [23, 24]. In contrast, events that disrupt the co-evolved complementarity between host and pathogen genotypes, such as horizontal transmission to non-ancestral host populations, may promote more adverse interactions which have consequences for clinical disease [22]. In the absence of discernible correlations with established virulence genotypes, these theories may account for the differing susceptibilities to gastric cancer in populations with similarly high rates of H. pylori infection [25,26,27,28], and presents discordant host-pathogen co-ancestry as an important co-factor in the determination of disease risk [22].
In addition to the possible competitive displacement of native strains [29, 30], horizontal transmission also provides opportunity for inter-strain DNA transfer and exchange in mixed infection and is a principal mechanism for acquisition of novel genes and virulent genotypes of existing determinants [31, 32]. Both DNA transformation and conjugative processes are considered to contribute to the variability of gene content encoded by different H. pylori strains [33,34,35] with further genomic diversity mediated by high rates of recombination, mutation and phase variation [11, 36,37,38]. The variable and strain-specific subset of H. pylori genes comprises 20-25% of the genome [39, 40] many of which are encoded within three large (>35kbp) horizontally acquired low G + C content chromosomal regions. These are identified as the cag pathogenicity island (cagPAI) and two distinct ‘plasticity zone’ clusters of genes termed tfs3 and tfs4 which both have features common to mobile integrative and conjugative elements (ICEs). These include a subset of genes encoding DNA processing (XerT recombinase involved in ICE excision/integration) and transfer functions (VirD2 relaxase) and the characteristic sequence motifs that direct their activities [41,42,43,44,45], and a complement of type IV secretion system (T4SS) genes with homology to the prototypical virB/D T4SS genes of Agrobacterium tumefaciens [41], in addition to a variable subset of cargo/accessory genes of unknown function [46, 47]. Although lacking equivalent recombination genes for self-mobility, the cagPAI similarly encodes a T4SS known to function in host-cell interaction and translocation of the CagA effector protein. These activities are strongly associated with increased risk of all H. pylori-mediated gastroduodenal disease as a consequence of well-established roles in dysregulation of gastric epithelial cell signalling and promotion of sustained host inflammatory responses [5]. A fourth, minimal T4SS encoded by the comB locus is present in all H. pylori strains and is required for natural DNA transformation competence [33].
Components of the tfs4 ICE have also been found to associate with varying risk of particular gastroduodenal disease. Most notably, the presence of the gene encoding the putative T4SS VirB4 ATPase (also referred to as duodenal ulcer promoting gene A, or dupA), is reported to correlate with an increased risk for development of duodenal ulcer and conversely, reduced risk of atrophy/gastric cancer in some populations [48, 49]. However, that dupA disease associations are strengthened when considered in the context of the full tfs4 ICE is indicative of an important role for other tfs4-encoded products in determination of disease risk [50]. Indeed, several other tfs4 genes (homologues of H. pylori strain J99 genes jhp0947, 949, 950, 951 in particular) are similarly more frequently associated with strains isolated from either or both peptic ulcer disease or gastric cancer disease groups [40, 51,52,53,54,55]. These studies provide compelling evidence of a role for the tfs4 ICE in H. pylori-mediated gastric disease although the identity and functional activity of encoded products relevant to a virulence phenotype remain unknown.
The tfs3 ICE in contrast has been determined to variably encode the host-interacting pro-inflammatory cell translocating kinase (CtkA) protein [56]. CtkA-mediated stimulation of both host immune and epithelial cell pro-inflammatory signalling suggests it might potentiate gastric mucosal inflammation with consequences for inflammation-associated disease outcomes such as atrophy and gastric cancer [57,58,59]. Consistently, ctkA (encoded by jhp0940 in strain J99) has been identified as a marker for gastric cancer in some populations [52, 55]. More recently, the tfs3 ICE-encoded T4SS (Tfs3) has been implicated in promotion of proinflammatory signalling by CtkA [56], although the variable and often low prevalence of ctkA in the H. pylori (tfs3+) strain population [39, 52, 54,55,56,57] suggests that the full role of the Tfs3 T4SS in H. pylori host interaction remains to be determined.
Rearranged remnant fragments of the tfs ICEs were initially identified in a comparison of the first two genome sequenced H. pylori strains, 26695 and J99, as components of the strain-specific and variable gene subset [60]. Subsequent studies defined full gene complements and context for representative tfs3 [41, 42] and tfs4 ICEs [43] and tfs4 variation apparent within subsets of complete genome sequences [44, 61, 62]. However, public sequence repositories contain considerably more numerous unfinished, draft whole genome shotgun sequences (WGSs) which, due to their fragmentary nature, have remained refractory to the study of large contiguous chromosomal segments such as ICEs. As these draft assemblies represent an increasingly greater global diversity of H. pylori strains, we sought to develop a strategy for efficient data-mining of their constituent, often short read sequence contigs for tfs ICE content. Our subsequent analysis of the resulting data-rich resource provides a comprehensive account of tfs ICE structure, representation, prevalence and phylogeographic diversity within an extensive global collection of H. pylori strains. We show that tfs ICEs are modular in their organisation and identify disease-associated accessory modules that are preferentially maintained in strains independently of tfs ICE T4SS activity. We further show that population-specific allelic diversity of tfs3 ICE modules in particular is discriminatory for ICE admixture and inter-population ICE exchange and displacement, and additionally provide evidence for acquisition of an H. pylori tfs3 ICE by the emerging zoonoses, H. suis. These collective events, in addition to the particular modular architecture of different tfs ICE types have potential to impact upon different outcomes of persistent infection.
Results
Representation of tfs ICEs in a global H. pylori strain population
Previous descriptions of the tfs ICEs of H. pylori have predominantly resulted from examination of contiguous ICE clusters in a limited number of available complete genome sequences [44, 61, 62]. However, data-mining of substantially more numerous draft genome sequences in this context has been largely overlooked, not least because of difficulties in establishing full representation and context of large ca. 35-55 kb genomic segments which are invariably distributed over a variable number of non-sequential, often short read contigs. That tfs ICEs may also occur in a fragmented state or concurrent with additional tfs ICEs or additional partial segments further confounds contextual analysis. To address this, we developed a robust bioinformatics workflow to enable identification and contextual reconstruction of tfs ICE clusters encoded within draft WGS sequences. Initially, selected reference tfs ICE clusters were manually re-annotated to determine the full complement of tfs genes and resolve disparities in the often variable definition of coding sequence arising from automated annotation. A subset of 59 distinct tfs gene sequences resulting from this analysis were translated and subsequently used in a sequential BLASTp interrogation of the PATRIC RefSeqProt database, then search hits, comprising > 12,000 tfs genes extracted from 221 complete and draft genomes, manually compiled into an ordered dataset (Additional file 1). Sequences were tagged as being tfs3, tfs4 or com-encoded by identification of conserved sequence motifs, then full tfs representation determined sequentially for each strain genome. Complete and remnant tfs clusters were finally assembled by reference to the original re-annotated reference ICE clusters as necessary (Additional file 2). The resulting dataset provides a comprehensive assessment of the global prevalence, structure and allelic frequency of the tfs elements both in the context of each other and cag status in an extensive collection of H. pylori strains, representing 7 phylogeographic populations isolated from 23 different countries.
Comparison of reconstructed tfs ICE clusters identifies a core subset of 21 putative genes broadly conserved between both tfs3 and tfs4 ICEs (denoted t3/t4_C1-C21) and a further 19 genes unique to each (denoted t4_V1-V19 and t3_V20-V38 for tfs4 and tfs3 respectively) (Table 1). Within both core and variable gene subsets, two distinct variants of multiple tfs4 genes can be identified distributed along the entire length of the tfs4 ICE, broadly contained within each of two left, central and right segments, referred to here for brevity as L1/L2, C1/C2 and R1/R2 respectively (Table 1, Additional file 2). The tfs3 ICE in contrast, comprises a more modest subset of variable genes clustered within the left segment of the ICE. Accounting for these, the tfs4 and tfs3 pangene complements are determined to comprise 54 and 42 distinct genes/orthologues respectively.
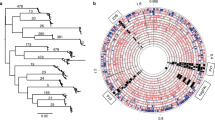
Hierarchical clustering of the tfs and cag datasets based on gene content contained within particular segments (Fig. 1a) highlights the relative ubiquity of the tfs4 ICE, either as a complete (96 strains) or fragmented (74 strains) cluster compared to either tfs3 or cag which, although broadly prevalent in the population, are entirely absent in 60 and 37 strains respectively and concurrently so in 11. Notably, tfs3 and tfs4 are maintained together in substantially intact form in 18 strains although commonly, the right end segment of the tfs3 ICE encoding the majority of putative T4SS genes is absent (53 strains with partial clusters compared with 43 containing intact tfs3 ICEs).

Prevalence of tfs ICE and selected cag genes in 187 H. pylori genomes (for a high resolution version of this figure, please see Additional file 6). a Clustering defines the strain representation and integrity of tfs ICEs relative to each other and cagPAI status. Distinct clusters (1–4) identify strains with similar representation of tfs and cag gene complements. Colouring of tfs4 gene subsets corresponds to different complements of orthologous genes contained within each of two left (L1 and L2), central (C1 and C2) or right (R1 and R2) ICE segment modules which may be contiguous with, isolated from or additional to the tri-module complement of a particular tfs4 ICE type. The tfs3 ICE lacks an equivalent pattern of modular variation and is more broadly defined by left and right segments. b tfs ICE and cag gene content data was transformed to reflect phylogeographic lineage of the originating strain as determined by MLST relative to a subset of 347 reference sequences obtained from the MLST database (Additional file 3: Figure S1). tfs and cag gene complements were coloured according to the phylogeographic population assigned to each strain, as indicated in the inset key. Hierarchical clustering identifies extensive erosion of the tfs ICEs in hpEurope strain populations
Transformation of the data to enable clustering on the basis of phylogeographic population highlights an apparent increase in fragmentation of both tfs ICEs in strains of European (hpEurope), and to a lesser extent, African (hpAfrica1) descent (Fig. 1b) suggesting that the ICEs may be less stable and/or more dispensable in these backgrounds. No correlation between presence, absence or co-occurrence of intact tfs and cag clusters could be determined suggesting that the clusters are independent of each other. Although tfs3/tfs4 co-carriage appeared less common in the hspEAsia population (Fig. 1b), there was a similar lack of correlation between the presence of intact/remnant tfs3 or tfs4 ICEs suggesting that neither ICE presents a strict barrier to acquisition or stable maintenance of the other. In view of this, it might therefore be considered that the encoded functions of the tfs3/4 ICEs are also unlikely to be antagonistic.
Modular disposition of the tfs4 ICE and hybrid tfs4 ICE types
Up to three tfs4 ICE types have been previously described from comparative analysis of complete genome sequences based on the presence of particular orthologous gene subsets, [42, 62], most recently referred to as ICEHptfs4a-c [44]. Hierarchical clustering of tfs4 ICE gene content similarly identifies these types, in which distinct orthologous gene complements are precisely contained within L2-C1-R2 (ICEHptfs4a), L1-C1-R1 (ICEHptfs4a) and L1/L2-C2-R2 (ICEHptfs4c) ICE segments (Table 1, Additional file 3: Figure S2, clusters 1 and 3). This assessment reveals that tfs4 ICEs have a distinct modular organisation in which each of two variant L-C-R modules may be interchangeable in the generation of a particular tfs4 type. Interestingly, L1 and L2 left flank modules are occasionally observed in addition to and in isolation from both complete and remnant ICE clusters (Additional file 3: Figure S2, cluster 4). This is similarly observed, albeit to a lesser extent, with R1 and R2 right flank modules, although only the R2 module is apparent in isolation from other sections of the tfs4 ICE (Additional file 3: Figure S2, cluster 3). Similar to the R2 module, the C2 central module is present in only a small subset of strains, invariably concurrent with the R2 flank.
Scrutiny of L1/L2-C1/C2-R1/R2 module representation in the context of contiguous gene content identifies the L1C1R1 modular configuration to be predominant in the H. pylori population, with 37 genomes harbouring a complete L1C1R1 ICE (Table 2). A second, previously undescribed configuration, L2C2R2 can be discerned intact in a further 9 genomes. Two rare configurations L2C1R2 (ICEhptfs4a in [44], 2 strains) and L2C1R1 (2 strains) can also be identified in addition to several others which incorporate either a truncated R1 module fragment (‘R1f’), or a mosaic left flank (‘Lm’) module comprising five and two L2 and L1 genes respectively flanking the L1/L2 conserved gene t4_C4 (Table 2), equivalent to ICEHptfs4c [44]. As illustrated by comparative global sequence alignment of each defined tfs4 ICE type (Fig. 2), both Lm and R1f flanks can associate with any modular organisation suggesting that they both represent stable modular types rather than constructions arising repeatedly through occasional recombination or aberrant ICE integration. A pairwise sequence comparison between the modular complements of encoded orthologous tfs T4SS proteins, and that of cag and com, further emphasises the distinctiveness of the different elements (Additional file 4: Table S1a and [44]).

Global pairwise sequence alignment of distinct tfs4 ICE modular subtypes. The ‘vir’ gene labelled tfs4 ICE gene cluster (top) is representative of the tfs4 ICE from strain G27 (L1C1R1, including genes 0959–0986) used as a reference scaffold sequence for each ICE alignment and closely approximates the position of genes in the alignment graphs. L1, C1 and R1 module gene complements are coloured blue, whereas orthologous genes characteristic of L2, C2, R2 modules are shown coloured red in the schematic of the L2C2R2 ICE type from strain R036d (bottom). Sequence conserved genes common to all tfs4 ICEs are coloured cyan. Global pairwise sequence alignments of different tfs4 ICEs was performed using the Shuffle-LAGAN alignment program in mVISTA. Individual mVISTA graphs depict sequence similarity of tfs4 ICEs from strains Shi470 (L1C1R1, genes 4480–4640), R036d (L2C2R2, genes 0976–1010), P12 (L2C1R2, genes 0437–0473), NQ4053 (L2C1R1, genes 1511–1596), P-25 (L2C1R1f, genes 1401–1438), A-8 (L1C1R1f, genes 0913–0945), A-11 (LmC1R1, genes 0721–0756), PeCan4 (LmC1R2, genes 7670–7835) and SouthAfrica7 (LmC2R2, genes 7540–7710) relative to the G27 tfs4 ICE using a 50 bp calculation window and 70% sequence identity cut-off. The midline in individual alignment graphs corresponds to 50% sequence identity to the G27 reference tfs4 ICE sequence and regions with sequence identity ≥ 70% are indicated by blue (coding sequence) or red (non-coding intergenic sequence) shading
Since L1C1R1 and L2C2R2 modular organisations are predominant in the H. pylori population and account for all orthologous and unique tfs4 genes, they likely represent two principal ancestral tfs4 ICE types. Other tfs4 module configurations are therefore presumably hybrid derivatives which conceivably arose as a consequence of partial integration/exchange during heterologous ICE transfer or genome rearrangement and recombination between two resident ICEs.
Phylogeographic distribution of tfs4 ICEs and component modules
On the basis of MLST, H. pylori strains can be assigned to different phylogeographic populations which are informative of their geographical and ancestral origins. To determine if phylogeographic origins of different tfs4 ICE types could be discerned, hierarchical clustering was performed on the tfs4 gene dataset in the context of host strain lineage. The analysis clearly demonstrated the presence of the L1C1R1 ICE in all phylogeographic groups (Additional file 3: Figure S3), of which, a significantly increased prevalence was apparent for hpAfrica2, hpAfrica1 and hspAmerind populations (Additional file 4: Table S2). By contrast, the L2C1R1f type appeared to be exclusive to either hpAfrica1 (p < 0.0001) and to a lesser extent, hpEurope strain lineages. Notably, all hybrid tfs4 ICEs with the single exception of LmC2R2, were only found in hpAfrica1/hpEurope strain populations in which the L2C2R2 ICE was also most prevalent (Additional file 4: Table S2). Hybrid ICEs are therefore indicated to have arisen from module exchange between the two predominant tfs4 ICEs following co-infection with these particular strain populations.
Given the apparent propensity of tfs ICEs for modular exchange and the relative abundance of individual L-C-R modules in the global strain population compared to intact ICEs, we also considered the distribution of modules independently of a particular ICE architecture. This analysis was more informative of previous ICE carriage and the population skew of L/R modules in particular. Consistent with findings for the L2C1R1f ICE, L2 and R1f modules were most commonly associated with the hpAfrica1 lineage (p < 0.0001). Similarly, findings for the R1 module reciprocated the association of the L1C1R1 ICE with hpAfrica2/hspAmerind lineages, whereas the R2 module was significantly more prevalent in hspEAsia strains (Table 3). As with L1C1R1 type ICEs in general, the L1 module was found to be widely distributed, but particularly prevalent in hpAfrica1/2 and hpAsia2 populations. This widespread but variable distribution of tfs4 ICEs and component modules is further emphasised by an assessment of module co-occurrence in strains, revealing a complex history of tfs ICE acquisition, exchange and erosion (Fig. 3a and b) which is likely to make a notable contribution to strain diversity.

Venn diagrams showing co-occurrence of tfs ICE modules/types with each other and the cagPAI. a Non-T4SS accessory/cargo tfs genes located within tfs4 L1/L2 modules and the tfs3 left segment. b tfs4 R1/R2 modules and the tfs3 right segment comprising the majority complement of tfs T4SS transfer genes. c Intact tfs3 and L1C1R1 and L2C1R1f tfs4 ICE types. Plots a. and b. highlight a complex history of tfs ICE/module acquisition in which cargo/accessory modules (a) are more commonly maintained in strain genomes compared with transfer modules (b). Erosion of transfer modules is further reflected by the low co-occurrence of intact tfs3 with either of the most prevalent tfs4 types, L1C1R1 (dupA+) or L2C1R1f (partial dupA), co-resident with tfs3 in 22 and 26% of strains respectively (c). Although most commonly co-resident with the cagPAI, all tfs modules and ICE types were also apparent in strains which lacked it. Representation of intact elements was determined in 181, 168 and 164 H. pylori genomes (a-c respectively). tfs4 ‘Lm’ hybrid left flanks were included with L2 counts in a. and L2C1R1f counts in c
With further respect to the R1f module, examination of the L2C1R1f genomic context identified the ICE to be invariantly integrated adjacent to 23 s-5 s RNA genes at the L2 left flank end and proximal to ftsZ-ftsA and a gene encoding a mechanosensitive ion channel domain-containing protein at the truncated R1f right flank. Characteristically, a gene homologous to jhp0914 from strain J99 is also uniquely present in this context in hpAfrica1 strains [39, 44]. This strict conservation of integration site, apparent in 57/58 genomes with an R1f module in this study (Additional file 2), restricted distribution and deficit of T4SS assembly genes presumably employed in conjugative ICE transfer, suggests that the L2C1R1f tfs4 subtype is likely to be immobile and therefore fixed in the hpAfrica1 strain population, conceivably arising from a single ancestral strain in which the R1f truncation originally occurred. Further support for this notion is provided by the observation that the great majority of strains with the L2C1R1f ICE (94%) are also cagPAI+ (Fig. 3c) in marked contrast to the prototypical L1C1R1 and L2C2R2 types (68 and 55% co-resident with the cagPAI respectively) which can be presumed to mobilise between more genetically diverse host strain populations. Unlike the observed disparity in cagPAI co-occurrence however, both L1C1R1 and L2C1R1f tfs4 types are notably most frequently observed in the absence of a complete, T4SS-competent tfs3 ICE (22 and 26% co-occurrence with tfs3 respectively).
Interestingly, a remarkably conserved partial tfs4 C1 module comprising genes t4_C7/C8/V5 flanked by remnants of t4_V4 (methylase) and t4_C9.1 (virD2) is also present in H. acinonychis str. Sheeba (Additional file 2). H. acinonychis is estimated to have diverged from the H. pylori hpAfrica2 super-lineage following a single host jump from humans to large felines 43–56,000 years ago [16, 18]. This suggests that the association of tfs4 with H. pylori is ancient and further, that the C1 module was part of an ancestral tfs4 ICE, which would reasonably account for its ubiquity in the current global H. pylori population.
Architecture and allelic diversity of the tfs3 ICE
Although apparent in all H. pylori populations, except hpAfrica2 in which the cagPAI is also absent, the overall distribution of the tfs3 ICE in strains is notably reduced in comparison with tfs4 (Fig. 1, 123 vs 170 strains respectively with evidence of tfs acquisition). It is also more commonly present as an incomplete cluster (intact: fragmented ratio of 1:8 and 1:3 for tfs3 and tfs4 respectively) although fragmentation characteristically entails loss (right segment) and retention (left segment) of defined ICE segments (Fig. 1, Additional file 3: Figure S4). In the examined dataset, the left segment is present in 78% of all strains with some form of tfs3 ICE and further enriched in hspEAsia/hpAsia2 and hpAfrica1 H. pylori populations (Table 4). Notably, in all strains in which it occurs, tfs3 and its more highly represented left segment appear co-resident with the more ubiquitous tfs4 L modules and/or the cagPAI (Fig. 3a) suggesting potential differences in stability or temporal acquisition of tfs3 compared with these other genomic elements.
Although tfs3 lacks the overt modular disposition of tfs4 illustrated in Fig. 2, similar left, central and right segments of the ICE which comprise broadly equivalent gene subsets can be defined (Fig. 4a). With the exception of several variably present central genes, t3_V29/V30/V31/V32/V4, central and right flank regions encoding the majority of T4SS assembly genes, are well conserved in both gene content (Fig. 4a) and sequence identity (> 85% nucleotide sequence identity for the majority of genes examined, Additional file 4: Table S3). tfs3 left flanks by contrast, are hypervariable in these respects, markedly differing in composition (selective gain/loss of genes t3_V1/V20/V21/V22/V23 as previously highlighted [56]) and possession of multiple distinct variants of genes t3_V24/C3/C2/C5 and t3_V25 in particular (Additional file 4: Table S3 and Fig. 4b). Within this variable subset, variation of the t3_C5 genes is distinctive, comprising 5′, 3′ and central regions of conserved sequence interspersed with more highly variable segments differing both in size and sequence composition.

Allelic diversity of the tfs3 ICE. a The ‘vir’ gene labelled tfs3 ICE gene cluster (top) is representative of the complete tfs3 ICE from strain Gambia94/24 (genes 7340–7535) used as reference for the ICE alignments and closely approximates the position of genes in the alignment schematic. Individual mVISTA graphs depict sequence similarity of tfs3 ICEs from strains P-41 (genes 0159–0204), A-5 (genes 1146–1181), PeCan18 (genes 4840–5050), UM114 (genes 1250–1445), CPY6311 (genes 0113–0153), India7 (genes 3720–3900), Shi112 (genes 1185–1415), Shi417 (genes 7490–7655), Aklavik117 (genes 2160–2315) and H. cetorum strain MIT 00–7128 (genes 8040–8260) relative to the Gambia94/24 tfs3 ICE in global pairwise sequence alignments using a 50 bp calculation window and 70% sequence identity cut-off. Labelled brackets indicate broad positional conservation of similar gene groupings as the tfs4 ICE. b Extent of sequence and gene content variability within the left flank of the tfs3 ICE. (Top) Schematic shows the complement of 20 genes in the left flank of the tfs3 ICE (virD2 [C9] to topA [C16] inclusive) annotated according to the nomenclature described in Table 1. Highly variable genes are block highlighted in orange. Relative position of genes closely approximates corresponding positions in the alignment graphs (bottom). Individual mVISTA graphs depict sequence similarity of tfs3 ICEs from 16 strains including strains P-41, H-29, SJM180, UM114, CPY6311, PeCan18, India7, OK310, Puno135, Shi112, Shi417, Aklavik117, H-3, P-13 and H. cetorum strain MIT 00–7128 relative to the Gambia94/24 tfs3 ICE (genes 7340–7425) using a 50 bp calculation window and 70% sequence identity cut-off. For both a. and b., the mid-line in individual alignment curves corresponds to 50% sequence identity to the Gambia94/24 reference tfs3 ICE sequence and regions with sequence identity ≥ 70% are indicated by blue (coding sequence) or red (non-coding intergenic sequence) shading. Gaps indicate absent genes
With the exception of t3_C5, Neighbour-Joining phylogenetic trees identify 2–5 distinct clades for each of these genes (illustrated in Additional file 3: Figure S5). However, divergence of the distinct tfs3 alleles is more modest compared with the substantial separation observed for the corresponding tfs4 orthologues. Phylogeographic origins can be clearly discerned for the two most prevalent alleles of each gene in the tfs3 variable subset (annotated as ‘1’ and ‘2’), invariantly corresponding to hpAfrica1/hpEurope or hpAsia2/hspEAsia populations respectively. Remaining distinct clades (‘3-5’), more distantly related to either of the two main clades, comprise more mixed populations but characteristically include sequences from hspAmerind isolates (Table 5). These latter include strains Shi112 and Shi417 isolated from residents of the remote Peruvian Amazonian village of Shimaa, which together with other Shimaa isolates are known to fall within a unique phylogenetic cluster more distantly related to other H. pylori populations [30]. The population heterogeneity of these ostensibly hspAmerind clades may therefore reflect historical interactions between different H. pylori strain populations which promoted the inter-population exchange of particular hspAmerind tfs3 alleles.
Heterogeneous distribution of the hspAmerind tfs3 ICE
Since tfs ICEs are indicated to be mobile it is reasonable to consider that the heterogeneity of strain background in which hspAmerind tfs3 left flank alleles are found could be readily explained by transfer of native hspAmerind ICEs to other phylogeographic H. pylori populations. To investigate this, nucleotide sequences of the four most variable tfs3 genes t3_C3, C2, C5 and V25 from 75 strains were concatenated then used for construction of a Neighbour-Joining phylogenetic tree as before. Sequences were assigned the MLST population of the host strain and further annotated with an allelic profile derived from the collective Neighbour-joining analysis of the individual variable genes.
The majority of sequences could be identified within one of four major clades, two with homogeneous populations corresponding to either hpAfrica1/hpEurope (allelic profile, ‘1111’) or hspEAsia (allelic profile, ‘2222’) (Fig. 5 and Additional file 4: Table S4) and as before, two clades more heterogeneous in population structure (‘3443’ and ‘4533’) presumably corresponding to distinct hspAmerind populations. Consistent with this, multiple distinct subdivisions of hspAmerind populations have been resolved previously [17]. That the subset of variable genes that these ostensibly hspAmerind profiles represent appear conserved in other phylogeographic H. pylori populations suggests that the encoding left flank segment of the tfs3 ICE was indeed acquired into these backgrounds as a single large block, most likely as a consequence of transfer of the entire tfs3 ICE. A diversity of other unclustered allelic profiles representing unique combinations of variant alleles were also identified (Fig. 5). However, as these comprised more discrete substitutions of genetic material they might equally have resulted as a consequence of transformation and recombinational exchange of individual genes or gene fragments rather than ICE mobilisation.

Neighbour-joining analysis of the tfs3 variable gene subset. The phylogenetic tree was calculated from concatenated sequence of four tfs3 variable subset genes comprising in order, t3_V24, t3_C3, t3_C2 and t3_V25 from 76 genomes. All positions in the initial sequence alignment containing gaps and missing data were eliminated prior to analysis. The four-digit number codes correspond to different alleles of each gene determined from previous phylogenetic analysis of individual genes (Additional file 3: Figure S5c, d). Two principal clades correspond to sequences from strains of hpAfrica1/hpEurope (clade 1) and hpEAsia (clade 2) lineages. The two remaining clades demonstrate characteristic high divergence of hspAmerind sequences but notably also include sequences from other H. pylori phylogeographic groups. A diversity of other unclustered allelic profiles arising from the inter-population exchange of variable subset tfs3 genes or gene fragments are also apparent. Coloured circles representing gene sequences are coloured according to phylogeographic lineage of the originating strain as indicated. u; unassigned (phylogeny could not be determined due to substantially admixed sequence). Uncoloured circles indicate sequences from strains which could not be definitively assigned to a particular phylogeographic population
To more fully resolve the genetic character of a selection of ICEs, we performed global pairwise sequence alignments, applying a stringent sequence identity cut-off (≥ 98%) to highlight distinct ICE regions homologous to each of three reference ICEs used as alignment scaffolds (hpAfrica1 or hspAmerind). These clearly showed admixture in the sequence of all hspAmerind-type tfs3 ICEs resident in a heterologous strain background, suggesting some level of inter-ICE recombination prior to full or partial displacement of the native ICE-type (Additional file 3: Figure S6).
Preservation of a tfs3-like ICE in other Helicobacter species
In addition to a variety of other species-specific clusters of T4SS genes, several other Helicobacter species harbor complete or remnant H. pylori-like tfs ICEs (Additional file 2). In particular, a large ca. 55 kb tfs3-like ICE is apparent in H. cetorum strain MIT-00-7128 [63], isolated from a captive Beluga whale, which, with the exception of a three gene insertion, is homologous to the corresponding H. pylori tfs3 along its entire length (Fig. 4a). Strikingly, this homology also extends to the definition of an allelic profile from the subset of variable left segment genes, most closely resembling that of the admixed H. pylori sequence from strain A-27 (Fig. 5). The variable segment of the tfs3 ICE therefore appears to have remained remarkably stable in these diverse backgrounds and seemingly subject to minimal host-adaptive changes. The prevalence and diversity of tfs3-like ICEs in the H. cetorum population is presently unknown, however, the presence of an H. pylori-homologous tfs3 left segment cluster of genes (t3_C9-V3 inclusive) in a second H. cetorum strain, MIT 99–5656 [63], isolated from a wild dolphin, suggests that these ICEs may indeed be widespread and, by implication, long-associated with H. cetorum strains.
Apparent remnants of tfs3 ICEs are also evident in two draft genomes of the porcine pathogen H. suis (Additional file 2). The tfs3 fragment in H. suis strain HS1 [64] spans genes t3_C9 to t3_C10 inclusive on one WGS contig (00063), continuing to t3_C14 on a second (00062), although several large deletions are apparent relative to H. pylori tfs3 ICEs, conceivably consequent with adaptive changes following initial acquisition [18]. However, whereas the H. cetorum tfs3 is somewhat sequence divergent from H. pylori tfs3, consistent with the sequence divergence of both genomes in general [63] (and also indicated by moderate sequence similarity of concatenated MLST sequences, Additional file 4: Table S5), the H. suis tfs3 sequences, in marked contrast, remain highly similar to the H. pylori tfs3 ICE, demonstrating 91-98% identity over the three gene clusters compared and ≥98% sequence identity over much of the t3_C9 to t3_C10 fragment when directly compared with the equivalent tfs3 segment from hpAfrica1 H. pylori strain Gambia94/24 (Additional file 3: Figure S7). This is particularly surprising given the substantial separation of H. suis and H. pylori species (68% identity between MLST concatenated sequence, Additional file 4: Table S5) and suggests a recent acquisition of H. pylori tfs3 by the H. suis HS1 strain.
Discussion
Difficulties in obtaining contextual information for large contiguous clusters of genes from draft genome sequences has undoubtedly presented a bottleneck in the study of large genomic elements such as ICEs, not least the tfs ICEs of H. pylori. In addressing this issue we have significantly extended observations from analysis of limited numbers of complete genome sequences, enabling detailed assessment of tfs ICE structure, variation and prevalence within the global H. pylori population. From this foundation, we have provided a novel classification of the tfs ICEs which has facilitated study of their historical association and mobility within and between geographically diverse human populations.
Distribution and distinctive variation of the tfs ICEs
H. pylori is considered to have infected anatomically modern humans since their emergence in Africa ~100kya, continuing to co-evolve with different human populations following their migratory expansion from Africa, beginning ~60kya [13, 14, 16]. Genetically distinct populations of H. pylori can be discerned as a consequence which reflect human colonisation of particular geographic regions and their subsequent interactions in both pre- and contemporary history. Currently, seven main H. pylori lineages are apparent, of which hpAfrica2 is considered to be ancestral, predating human migration out of Africa [13, 14, 16]. The ubiquitous presence of the tfs4 ICE in all phylogeographic H. pylori populations, including hpAfrica2 is therefore particularly striking, and alludes to the association of tfs4, and more particularly the L1C1R1 subtype, with an ancestral population of H. pylori potentially before spatial separation. That a fragment of the tfs4 C1 module is also apparent in H. acinonychis, which is part of the same super-lineage as hpAfrica2 [18] supports such an ancient origin.
In contrast, the lower abundance and population skew of L2C2R2 subtype modules suggests a different evolutionary history, perhaps reflecting later acquisition of this variant and/or adaptation to particular isolated H. pylori populations. With the exception of the notable predominance of the L2C1R1f tfs4 type in hpAfrica1 strains, L2/R2 flanks are otherwise most frequently associated with hpEAsia/hpEurope strains (Table 3) suggesting a possible ancestral association with early human inhabitants of the Eurasian continental landmass. Subsequent interactions between both human and H. pylori strain populations, enabling hybridisation of L1C1R1 and L2C2R2 subtypes, might then account for the introduction and stable inheritance of the L2 module within the hpAfrica1 strain population in the form of the transfer-deficient L2C1R1f hybrid. Although speculative, this general model is consistent with theories of human migrations out of Africa, in which waves of migration from North East Africa and Central Asia are considered to have converged in Western Asia 10-52kya, followed by human expansion throughout Europe, and ultimately, back migration to Africa from Europe and the Middle East ~10kya [16, 19].
Although apparent in the majority of H. pylori populations, the absence of the tfs3 ICE in hpAfrica2 strains, and indeed one third of all strains in this study (Figs. 1 and 3c, Table 4), is also suggestive of its acquisition contemporary to L1C1R1 tfs4. However, the clear association of individual tfs3 alleles with particular H. pylori lineages similarly alludes to significant co-evolution with geographically diverse H. pylori-infected human populations. Phylogeographic signals are particularly apparent for the subset of variable genes encoded within the left flank of tfs3, for which multiple distinct allelic forms can be discerned. Although the tfs3 variable subset comprises several genes unique to the tfs3 ICE, homologues of two, t3_c2 and t3_C3, are also apparent in both tfs4 ICE subtypes as one of two stably conserved, albeit highly divergent variants (Additional file 3: Figure S5). The reason for the difference in patterns of variation between these latter tfs genes is unclear, although it might presumably reflect the role of the encoded proteins relative to the overall function of each ICE within particular strain-host populations.
Functional considerations of modules and hybrid L-C-R modular configurations
ICEs are common in many bacterial species, invariably persisting within populations as a consequence of a particular fitness advantage they confer upon the host strain [65]. A modular structure is common, with particular modules conferring functions relevant to different ICE activities, including conjugation (encoding mating-pair formation [MPF] T4SS genes), transfer (encoding VirD2 relaxase and VirD4 coupling protein) and recombination (encoding relaxase and integrase/excisionase functions) [65]. These and other modules typically also harbour the adaptation/accessory genes that contribute to the evolutionary success of both ICE and host strain. A modular disposition of the tfs ICEs is also clearly apparent, most notably for tfs4 (Fig. 2) in which the C and R modules encode readily identifiable MPF and transfer functions [45], and the L modules a xer-mediated recombination function [44]. Common to other ICEs [66, 67], tfs4 L modules also include a putative VirB6–homologous MPF protein encoded independently from the main vir gene complement. All three L-C-R modules are therefore presumably required for intercellular transfer competence as they all encode functions relevant to this role.
Although module boundaries are less distinct, tfs3 in contrast appears to comprise two modules, broadly dividing the tfs3 ICE into left and right sections which harbour either recombination (plus VirD2) or MPF functions (plus VirD4) respectively (Fig. 4a). Both tfs4 and tfs3 ICEs commonly demonstrate loss and fragmentation of MPF modules and more frequent preservation of substantially intact L modules (Figs. 1 and 3a and b, Additional file 3: Figures S2-S4, Tables 3 and 4). However, mutational inactivation and erosion of mobility modules is common in ICEs that confer an adaptive advantage [65] suggesting that the selective maintenance of tfs L modules in the great majority of H. pylori strains might be attributable to a particular benefit their encoded protein products confer independently of the rest of the tfs ICE, including the encoded T4SS. It can therefore be considered that tfs L-C-R modules encode for different activities which are both dependent (ICE mobilisation) and independent (undetermined functions) of each other. This notion is further strongly supported by the high prevalence of the mobility-defective L2C1R1f tfs4 type (lacking essential virB4, B3, B2 T4SS assembly genes in the truncated R1 module) in the hpAfrica1 strain population, and also provides an explanation for the occasional stable generation of diverse hybrid tfs4 L-C-R modular configurations that may similarly be defective for T4SS assembly. With respect to the latter, it is unclear whether T4SS proteins encoded by orthologous, yet highly sequence divergent tfs4 gene subsets contained within the different modules (Additional file 4: Tables S1 and S3) have the capacity for cross-complementation in the assembly of a functional hybrid T4SS. Of the ten vir-homologous T4SS assembly proteins that are encoded by L-C-R modules, only one, VirB9, is highly conserved between both L1C1R1 and L2C2R2 tfs4 types (> 90% amino acid sequence identity, Additional file 4: Table S3). VirB9 is a core component in the T4SS assembly pathway, mediating interactions with multiple other Vir proteins in the formation of a stable secretion system complex [68]. It can therefore be assumed that there is at least some flexibility in the interactions of the conserved tfs4 VirB9 protein with both sets of orthologous Vir assembly proteins. Whether this is similarly the case for the other Vir proteins for which two distinct variants are apparent (43-80% sequence identity, Additional file 4: Table S4) remains to be determined, although if such heterologous interactions are not permissible then hybrid ICEs may also be defective for T4SS activity.
The potential for functional complementation by homologous components of tfs3 and tfs4 ICEs, and indeed tfs-homologous proteins encoded elsewhere in the genome (Additional file 2 and Additional file 4: Table S1b) similarly remains to be established. However, as ICE MPF functions are often exploited for the mobilisation of other unrelated genomic elements [65], it is reasonable to speculate that the tfs MPFs could at least each function in the mobilisation of any other ICE (and possibly also discrete ICE modules) that retains transfer and recombination activity.
Inter-population tfs ICE transfer and exchange
In support of previous observations from a more limited set of 36 CG sequences [44], Neighbour-joining analysis of tfs genes was frequently discordant with the distinct population divisions resolved by MLST, with predominant tfs clades more often comprising coalesced clusters of hpAfrica1/hpEurope (tfs4 L2 modules and tfs3 LF variable ‘1’ alleles) and hpAsia2/hspEAsia (tfs3 LF variable ‘2’ alleles) populations (Fig. 5, Additional file 3: Figure S5). The lack of fine population resolution for these alleles suggests a different evolutionary history relative to the core genome of the host strain which can be readily explained by ICE mobility, involving perhaps frequent inter-population transfer and exchange of tfs ICEs or component modules and a mechanism of replication-independent site-specific recombination mediating ICE excision and integration [69]. This contrasts with the more congruent phylogeny of the cagPAI with the core genome [70] which is indicative of its immobility within H. pylori genomes possibly from the time of its initial acquisition.
From the available data, it appears that tfs transfers occur preferentially between particular strain populations (such as hpAfrica and hpEurope) which may reflect either a history of extended interaction and population admixture or host-specific adaptations which potentially restrict the population range of particular ICE/module/allele types. Indeed, given the lack of a distinct hpEurope clade for any of the studied tfs genes/alleles and the common occurrence of hpEurope strains within clades predominated by other populations (hpAfrica1 and hspAmerind) it could be speculated that tfs’s do not have an appreciable evolutionary history with hpEurope populations, but rather, that these strains have more commonly been recipient to ICEs from other H. pylori lineages. The hpEurope population evolved through gradual admixture of two ancestral H. pylori populations, ancestral Europe 1 (AE1) and ancestral Europe 2 (AE2), following human migrations from North East Africa (AE2) and Central Asia (AE1) 10-52kya [13, 14, 16]. However, recent evidence suggests that the significant AE1/AE2 admixture characteristic of modern hpEurope only occurred after the Copper Age, ~ 5-6kya [71]. Association of tfs’s with modern hpEurope strains may therefore be much more recent than for other populations in which they have co-evolved over more significant periods of time, possibly accounting for the increased pseudogenisation of ICEs observed in these strains (Fig. 1b).
Our analyses of tfs3 ICEs in particular also provided evidence for inter-population transfer of hspAmerind ICEs to hpEurope and hpAfrica1 strains (Fig. 5, Additional file 3: Figures S5 and S6). No reciprocal transfers were observed suggesting that tfs transfers between these populations may be biased towards export rather than import, consistent with observations for DNA exchange within hspAmerind genomes as a whole [17]. In this context, hspAmerind strains have been shown to inefficiently transform with DNA from other H. pylori populations [30], potentially as a consequence of several strain-specific and functionally distinct restriction modification systems [72] which would similarly restrict the acquisition of foreign tfs ICEs. Such mechanisms may contribute to the low genetic diversity of hspAmerind strains and their consequent competitive displacement by other H. pylori populations [29, 30]. It is particularly noteworthy therefore that whereas hspAmerind strains may decline due to reduced fitness, their tfs ICEs by contrast, appear to have the capacity to survive the competitive interaction of mixed/transient infection to substantially displace the resident tfs ICE of competing foreign strains (Fig. 5, Additional file 3: Figure S6).
Disease implications of tfs ICE carriage
Component marker genes of both tfs ICEs have been found to associate with increased risk for H. pylori-related disease in some populations, indicating that the ICEs may confer as yet undefined virulence functions [40, 51, 52, 54, 55]. Such associations generally relate to genes encoded by tfs4 R1 (dupA+), L1 (jhp0947, jhp0949) or both L1/L2 (jhp0945) modules and the variably-encoded tfs3 left module ctkA gene (jhp0940). However, it is unclear from these studies whether these associations, particularly in the context of tfs4, relate to the activity of individual proteins, particular modules, tfs-types or are dependent or otherwise upon the function of the ICE-encoded T4SSs. In these contexts, our observations that 1) tfs4 L and R modules occur with high frequency in the global population, often in isolation from other ICE modules, and 2) the immobile L2C1R1f-type remains highly conserved without significant erosion in hpAfrica1 strains, both suggest that the L1/L2 modules may confer important function independently of Tfs4 T4SS activity. With respect to the latter, it is noteworthy that a recent study reported a remarkably low incidence of H. pylori-related disease in a Nigerian population infected with strains with the L2C1R1f tfs4-type but an otherwise highly virulent cagA/vacA genotype [27]. As the full R1 module (dupA+) is invariably associated with a C1 module (T4SS+) and defines an increased risk for duodenal ulcer and possibly reduced risk of gastric cancer [48], it is intriguing to speculate that in the absence of presumptive T4SS function, the L2C1R1f-type (or L2 module alone) might conceivably have a protective role in some settings; interactions of H. pylori with its human host which have a beneficial rather than pathogenic outcome have been suggested previously [9, 20, 22]. At the least, it can be considered that the prominence of the L2C1R1f tfs4-type in African strains, either alone or in addition to other factors, such as a concomitantly low co-occurrence of tfs3 (Fig. 3c), might contribute to overall reduced pathogenicity. With regard to the latter, as certain tfs3 genes have been reported to associate with increased risk of gastric cancer [40, 52, 55, 56], the low co-occurrence of T4SS-competent tfs3 and L1C1R1 tfs4 (Fig. 3c) might similarly be proposed as a factor in the observed inverse association of dupA+ H. pylori with this particular outcome of H. pylori infection [48].
In additional consideration of dupA, we also note that 29% of R1 (dupA+) modules are apparent in strains lacking a complete L1C1R1 tfs4 (Fig. 3b and c) suggesting that dupA presence alone is not a reliable marker for an intact tfs4 ICE and the functions it encodes.
tfs ICE transfer between populations and species
Whereas mutualistic interaction within co-evolved H. pylori-human populations is suggested to limit pathogenicity, discordant bacteria-host ancestry in contrast has been proposed to increase the risk of gastric disease [22, 26]. As tfs ICEs show evidence of evolution within particular strain populations (hpAfrica1/hspEAsia/hspAmerind in particular) it could be considered that inter-population heterologous exchange of ICEs (Fig. 5) might also change the virulence character of otherwise native host strains, possibly with detrimental consequences to the human host.
In addition to tfs ICE mobilisation between different H. pylori populations, the remarkable sequence similarity of the left segment remnant tfs3 ICE from the porcine H. suis strain HS1 with H. pylori tfs3 (Additional file 3: Figure S7) is intriguing and suggests contemporary interaction and ICE exchange between different Helicobacter species. H. suis is a recognised zoonoses and the most prevalent gastric non-H. pylori Helicobacter capable of causing gastric disease in humans [2, 73, 74]. Although the route of H. suis transmission from swine to humans is unresolved, transient co-colonisation of the human gastric niche with both H. suis and H. pylori would present reasonable opportunity for tfs ICE exchange. However, the isolation of the H. suis HS1 strain from the gastric mucosa of an infected swine [64] invokes more speculative models of tfs3 ICE acquisition, requiring either transient infection of the source animal with H. pylori, or human to animal (re)transmission of the HS1 strain. These models of anthroponotic infection remain unexplored, although are plausible as H. pylori has proven capacity to infect other animal species [75, 76] and is the likely ancestral origin of H. acinonychis [18]. Important clarity is therefore required regarding contemporary tfs3 gene flow between H. pylori and H. suis, since a human adapted H. pylori tfs3 could conceivably confer attributes which influence colonisation and/or virulence by H. suis within a human or porcine host.
Conclusions
In conclusion, the tfs ICE environment of individual H. pylori strains is shown to be complex and highly variable, reflecting both ancient and contemporary accretion, erosion and exchange of different ICE types and their genetically distinct component modules. That tfs modules might encode for activities which are both dependent and independent of each other suggests that inclusion of full ICE modular representation in the definition of a strain tfs genotype will aid understanding of the function and disease risk potential of particular ICE modular types. Finally, further knowledge of tfs gene flow within and between different Helicobacter populations and species will provide important context in assessing the beneficial or detrimental impact of different ICE types on both strain fitness and health of the infected human host.
Methods
Bioinformatics analyses and generation of datasets
tfs ICE gene clusters were identified within complete H. pylori genome sequences obtained from the NCBI [77, 78] or PATRIC [79, 80]. Sequences were compared in pairwise and multiple sequence alignments as appropriate (Needle/Stretcher and ClustalOmega/KAlign [81] respectively, accessible from [82]), then manually re-annotated to establish precise definition of coding sequence and full gene content using BioEdit [83, 84] and the suite of analysis tools available through the EXPASY resource [85, 86]. This comprehensive analysis resolved disparities arising from automated database annotation of frameshifted genes (not annotated or annotated as multiple individual genes) and provided clear discrimination of variant alleles.
Reference sequences selected from these comparative analyses, representative of the entire tfs gene pool (59 genes) were translated and subsequently used in sequential BLASTp (NCBI BLAST version 2.2.22, BLOSUM62 matrix) [77] interrogation of the RefSeqProt database (contemporary to June 2015) within the PATRIC bioinformatics platform [79] to obtain tfs homologous sequences from available draft WGS sequences. Significant hits were collated into an ordered searchable dataset comprising ~ 56,000 nucleotide and amino acid sequences (Additional file 1). The latter were initially identified and broadly tagged as either tfs3 or tfs4 ICE-encoded components by the presence of conserved sequence motifs characteristic of each (determined from previous multiple sequence alignment of reference sequences). This facilitated rapid sequential determination of full tfs gene representation and context relative to intact reference tfs3 and tfs4 ICEs (from strains PeCan18 and G27/P12 respectively) for each of 221 H. pylori genomes, comprising 53 complete (including 8 pairs/replicates) and 168 WGS (including 26 pairs/replicates) genome sequences (Additional file 2). Representation of homologous genes separate from the tfs ICEs and those present in 11 non-pylori Helicobacters were also included in the dataset in addition to the complement of com genes and a subset of nine cag genes selected at intervals along the entire length of the cagPAI (ca. 30 kbp). These latter were included to assess the utility of the overall approach for effective determination of complete gene subsets which may be encoded in both multiple separate genomic locations (com, akin to fragmented tfs ICEs) or in a single large contiguous cluster (cagPAI comprising 28–30 conserved genes, akin to complete intact tfs ICEs). Selected subsets for both com and cag genes were identified in their entirety for the majority of strains with isolated exceptions due to bona fide deletion of the complete cagPAI/individual genes or lack of automated database annotation for specific genes due to multiple frameshift mutation (Additional file 2). This was similarly found to be the case for tfs3 and tfs4 ICEs following validation of the complements of BLASTp tfs hits against NCBI database entries of complete genomes and cross-reference to the initial manual re-annotation of the tfs clusters.
Percentage sequence identity matrices for selected genes were generated from multiple sequence alignments in ClustalOmega [82]. Allelic variants with sequence identity < 70% were subsequently assigned to an arbitrarily numbered allelic group on the basis of evolutionary distance inferred using the Neighbour-joining method in MEGA 6.0 [87] as described below. Representation and definition of tfs ICE modules and subtypes was deduced from contiguous gene content and clusters annotated accordingly for all strains. A facile nomenclature based on tfs4 gene order and accounting for all conserved and variable tfs-homologous genes was additionally developed for standardised description and ease of reference to genes in the final datafile (Additional file 2). Additional meta-data for individual H. pylori strains was obtained from a survey of genome sequence entries in Genbank [78] and associated publications.
MLST and phylogenetic analyses
All H. pylori strains interrogated for tfs gene content were assigned to previously established populations or subpopulations [13] using multilocus sequence typing (MLST). Briefly, partial sequence of seven housekeeping genes, atpA, efp, mutY, ppa, trpC, ureI and yphC for each strain was obtained as described above, concatenated, then sequences aligned together with a selection of 347 reference sequences obtained from the MLST database [88, 89] using the MUSCLE alignment program in MEGA 6.0 [87]. Alignments were subsequently used for generation of phylogenetic trees in MEGA 6.0 using the Neighbour-joining method and Kimura 2-parameter model of nucleotide substitution with 1000 bootstrap replicates (Additional file 4: Figure S1). Evolutionary relationships of both individual and concatenated sequence derived from a subset of variable tfs3 genes from 76 complete and draft genomes (Additional file 5) was inferred using a similar approach. All positions in the alignment containing gaps and missing data were eliminated prior to analysis leaving 2474 positions in the final dataset. Phylogenetic trees were also constructed for individual tfs genes using CLUSTALW codon alignments, but otherwise identical parameters in MEGA 6.0.
Comparative alignment of tfs ICE sequences
Comparative analyses of selected contiguous tfs ICE sequences contained within individual FASTA sequence files was done in mVISTA using the Shuffle-LAGAN alignment program [90]. Similarity between aligned sequences was depicted using a 50 bp calculation window and sequence identity cut-off ranging from 70 to 98%.
Analyses in the R programming environment
To illustrate the representation of tfs and cag genes in Helicobacter complete and draft genomes, gene content matrices were built in the R (v3.2.2) programming environment [91, 92] using the ‘gplots’ package [93]. Loci were hierarchical clustered by similarity of content using the ‘hclust’ function (ward.D2 method and Euclidean distance measure) to generate a sidelong dendrogram. The R environment was also used for representation of tfs and cag gene co-occurrence in relevant H. pylori genomes using the VennDiagram package [94].
Statistical analyses
Fisher’s Exact test was used for statistical analysis of contingency tables using Graphpad Prism 7.01 (GraphPad Software, California, USA).
Abbreviations
- ICE:
-
Integrative and conjugative elements
- MLST:
-
Multi locus sequence typing
- PAI:
-
Pathogenicity island
- T4SS:
-
Type IV secretion system
- WGS:
-
Whole genome shotgun (sequence)
References
Solnick JV, Schauer DB. Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin Microbiol Rev. 2001;14:59–97.
Haesebrouck F, Pasmans F, Flahou B, Chiers K, Baele M, Meyns T, Decostere A, Ducatelle R. Gastric Helicobacters in domestic animals and nonhuman primates and their significance for human health. Clin Microbiol Rev. 2009;22:202–23. Table of Contents
Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–73.
Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–33.
Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Houghton J, Wang TC. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology. 2005;128:1567–78.
Bridge DR, Merrell DS. Polymorphism in the Helicobacter pylori CagA and VacA toxins and disease. Gut Microbes. 2013;4:101–17.
Lin D, Koskella B. Friend and foe: factors influencing the movement of the bacterium Helicobacter pylori along the parasitism-mutualism continuum. Evol Appl. 2015;8:9–22.
Schwarz S, Morelli G, Kusecek B, Manica A, Balloux F, Owen RJ, Graham DY, van der Merwe S, Achtman M, Suerbaum S. Horizontal versus familial transmission of Helicobacter pylori. PLoS Pathog. 2008;4:e1000180.
Morelli G, Didelot X, Kusecek B, Schwarz S, Bahlawane C, Falush D, Suerbaum S, Achtman M. Microevolution of Helicobacter pylori during prolonged infection of single hosts and within families. PLoS Genet. 2010;6:e1001036.
Montano V, Didelot X, Foll M, Linz B, Reinhardt R, Suerbaum S, Moodley Y, Jensen JD. Worldwide population structure, long-term demography, and local adaptation of Helicobacter pylori. Genetics. 2015;200:947–63.
Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–5.
Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P, Falush D, Stamer C, Prugnolle F, van der Merwe SW, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–8.
Moodley Y, Linz B, Yamaoka Y, Windsor HM, Breurec S, Wu JY, Maady A, Bernhoft S, Thiberge JM, Phuanukoonnon S, et al. The peopling of the Pacific from a bacterial perspective. Science. 2009;323:527–30.
Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, Bernhoft S, Hale J, Suerbaum S, Mugisha L, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012;8:e1002693.
Yahara K, Furuta Y, Oshima K, Yoshida M, Azuma T, Hattori M, Uchiyama I, Kobayashi I. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol Biol Evol. 2013;30:1454–64.
Eppinger M, Baar C, Linz B, Raddatz G, Lanz C, Keller H, Morelli G, Gressmann H, Achtman M, Schuster SC. Who ate whom? Adaptive Helicobacter genomic changes that accompanied a host jump from early humans to large felines. PLoS Genet. 2006;2:e120.
Lopez S, van Dorp L, Hellenthal G. Human dispersal out of Africa. A lasting debate. Evol Bioinformatics Online. 2015;11:57–68.
Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57:561–7.
Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16:1077–84.
Kodaman N, Sobota RS, Mera R, Schneider BG, Williams SM. Disrupted human-pathogen co-evolution: a model for disease. Front Genet. 2014;5:290.
Suzuki M, Kiga K, Kersulyte D, Cok J, Hooper CC, Mimuro H, Sanada T, Suzuki S, Oyama M, Kozuka-Hata H, et al. Attenuated CagA oncoprotein in Helicobacter pylori from Amerindians in Peruvian Amazon. J Biol Chem. 2011;286:29964–72.
Camorlinga-Ponce M, Perez-Perez G, Gonzalez-Valencia G, Mendoza I, Penaloza-Espinosa R, Ramos I, Kersulyte D, Reyes-Leon A, Romo C, Granados J, et al. Helicobacter pylori genotyping from American indigenous groups shows novel Amerindian vacA and cagA alleles and Asian, African and European admixture. PLoS One. 2011;6:e27212.
de Sablet T, Piazuelo MB, Shaffer CL, Schneider BG, Asim M, Chaturvedi R, Bravo LE, Sicinschi LA, Delgado AG, Mera RM, et al. Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut. 2011;60:1189–95.
Kodaman N, Pazos A, Schneider BG, Piazuelo MB, Mera R, Sobota RS, Sicinschi LA, Shaffer CL, Romero-Gallo J, de Sablet T, et al. Human and Helicobacter pylori coevolution shapes the risk of gastric disease. Proc Natl Acad Sci U S A. 2014;111:1455–60.
Harrison U, Fowora MA, Seriki AT, Loell E, Mueller S, Ugo-Ijeh M, Onyekwere CA, Lesi OA, Otegbayo JA, Akere A, et al. Helicobacter pylori strains from a Nigerian cohort show divergent antibiotic resistance rates and a uniform pathogenicity profile. PLoS One. 2017;12:e0176454.
Campbell DI, Warren BF, Thomas JE, Figura N, Telford JL, Sullivan PB. The African enigma: low prevalence of gastric atrophy, high prevalence of chronic inflammation in west African adults and children. Helicobacter. 2001;6:263–7.
Dominguez-Bello MG, Perez ME, Bortolini MC, Salzano FM, Pericchi LR, Zambrano-Guzman O, Linz B. Amerindian Helicobacter pylori strains go extinct, as european strains expand their host range. PLoS One. 2008;3:e3307.
Kersulyte D, Kalia A, Gilman RH, Mendez M, Herrera P, Cabrera L, Velapatino B, Balqui J, de la Vega Paredes Puente F, Rodriguez Ulloa CA, et al. Helicobacter pylori from Peruvian amerindians: traces of human migrations in strains from remote Amazon, and genome sequence of an Amerind strain. PLoS One. 2010;5:e15076.
Ghose C, Perez-Perez GI, van Doorn LJ, Dominguez-Bello MG, Blaser MJ. High frequency of gastric colonization with multiple Helicobacter pylori strains in Venezuelan subjects. J Clin Microbiol. 2005;43:2635–41.
Morales-Espinosa R, Castillo-Rojas G, Gonzalez-Valencia G. Ponce de Leon S, Cravioto a, Atherton JC, Lopez-Vidal Y. Colonization of Mexican patients by multiple Helicobacter pylori strains with different vacA and cagA genotypes. J Clin Microbiol. 1999;37:3001–4.
Hofreuter D, Odenbreit S, Henke G, Haas R. Natural competence for DNA transformation in Helicobacter pylori: identification and genetic characterization of the comB locus. Mol Microbiol. 1998;28:1027–38.
Rohrer S, Holsten L, Weiss E, Benghezal M, Fischer W, Haas R. Multiple pathways of plasmid DNA transfer in Helicobacter pylori. PLoS One. 2012;7:e45623.
Backert S, Kwok T, Konig W. Conjugative plasmid DNA transfer in Helicobacter pylori mediated by chromosomally encoded relaxase and TraG-like proteins. Microbiology. 2005;151:3493–503.
Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. Free recombination within Helicobacter pylori. Proc Natl Acad Sci U S A. 1998;95:12619–24.
Falush D, Kraft C, Taylor NS, Correa P, Fox JG, Achtman M, Suerbaum S. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc Natl Acad Sci U S A. 2001;98:15056–61.
Kennemann L, Didelot X, Aebischer T, Kuhn S, Drescher B, Droege M, Reinhardt R, Correa P, Meyer TF, Josenhans C, et al. Helicobacter pylori genome evolution during human infection. Proc Natl Acad Sci U S A. 2011;108:5033–8.
Gressmann H, Linz B, Ghai R, Pleissner KP, Schlapbach R, Yamaoka Y, Kraft C, Suerbaum S, Meyer TF, Achtman M. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 2005;1:e43.
Romo-Gonzalez C, Salama NR, Burgeno-Ferreira J, Ponce-Castaneda V, Lazcano-Ponce E, Camorlinga-Ponce M, Torres J. Differences in genome content among Helicobacter pylori isolates from patients with gastritis, duodenal ulcer, or gastric cancer reveal novel disease-associated genes. Infect Immun. 2009;77:2201–11.
Kersulyte D, Velapatino B, Mukhopadhyay AK, Cahuayme L, Bussalleu A, Combe J, Gilman RH, Berg DE. Cluster of type IV secretion genes in Helicobacter pylori’s plasticity zone. J Bacteriol. 2003;185:3764–72.
Kersulyte D, Lee W, Subramaniam D, Anant S, Herrera P, Cabrera L, Balqui J, Barabas O, Kalia A, Gilman RH, Berg DE. Helicobacter pylori’s plasticity zones are novel transposable elements. PLoS One. 2009;4:e6859.
Fischer W, Windhager L, Rohrer S, Zeiller M, Karnholz A, Hoffmann R, Zimmer R, Haas R. Strain-specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 2010;38:6089–101.
Fischer W, Breithaupt U, Kern B, Smith SI, Spicher C, Haas R. A comprehensive analysis of Helicobacter pylori plasticity zones reveals that they are integrating conjugative elements with intermediate integration specificity. BMC Genomics. 2014;15:310.
Grove JI, Alandiyjany MN, Delahay RM. Site-specific relaxase activity of a VirD2-like protein encoded within the tfs4 genomic island of Helicobacter pylori. J Biol Chem. 2013;288:26385–96.
Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A. 1996;93:14648–53.
Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–48.
Lu H, Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology. 2005;128:833–48.
Shiota S, Matsunari O, Watada M, Hanada K, Yamaoka Y. Systematic review and meta-analysis: the relationship between the Helicobacter pylori dupA gene and clinical outcomes. Gut Pathog. 2010;2:13.
Jung SW, Sugimoto M, Shiota S, Graham DY, Yamaoka Y. The intact dupA cluster is a more reliable Helicobacter pylori virulence marker than dupA alone. Infect Immun. 2012;80:381–7.
Sugimoto M, Watada M, Jung SW, Graham DY, Yamaoka Y. Role of Helicobacter pylori plasticity region genes in development of gastroduodenal diseases. J Clin Microbiol. 2012;50:441–8.
Occhialini A, Marais A, Alm R, Garcia F, Sierra R, Megraud F. Distribution of open reading frames of plasticity region of strain J99 in Helicobacter pylori strains isolated from gastric carcinoma and gastritis patients in Costa Rica. Infect Immun. 2000;68:6240–9.
de Jonge R, Kuipers EJ, Langeveld SC, Loffeld RJ, Stoof J, van Vliet AH, Kusters JG. The Helicobacter pylori plasticity region locus jhp0947-jhp0949 is associated with duodenal ulcer disease and interleukin-12 production in monocyte cells. FEMS Immunol Med Microbiol. 2004;41:161–7.
Santos A, Queiroz DM, Menard A, Marais A, Rocha GA, Oliveira CA, Nogueira AM, Uzeda M, Megraud F. New pathogenicity marker found in the plasticity region of the Helicobacter pylori genome. J Clin Microbiol. 2003;41:1651–5.
Yakoob J, Abbas Z, Naz S, Islam M, Abid S, Jafri W. Associations between the plasticity region genes of Helicobacter pylori and Gastroduodenal diseases in a high-prevalence area. Gut Liver. 2010;4:345–50.
Alandiyjany MN, Croxall NJ, Grove JI, Delahay RM. A role for the tfs3 ICE-encoded type IV secretion system in pro-inflammatory signalling by the Helicobacter pylori ser/Thr kinase. CtkA PLoS One. 2017;12:e0182144.
Rizwan M, Alvi A, Ahmed N. Novel protein antigen (JHP940) from the genomic plasticity region of Helicobacter pylori induces tumor necrosis factor alpha and interleukin-8 secretion by human macrophages. J Bacteriol. 2008;190:1146–51.
Kim DJ, Park KS, Kim JH, Yang SH, Yoon JY, Han BG, Kim HS, Lee SJ, Jang JY, Kim KH, et al. Helicobacter pylori proinflammatory protein up-regulates NF-kappaB as a cell-translocating ser/Thr kinase. Proc Natl Acad Sci U S A. 2010;107:21418–23.
Tenguria S, Ansari SA, Khan N, Ranjan A, Devi S, Tegtmeyer N, Lind J, Backert S, Ahmed N. Helicobacter pylori cell translocating kinase (CtkA/JHP0940) is pro-apoptotic in mouse macrophages and acts as auto-phosphorylating tyrosine kinase. Int J Med Microbiol. 2014;304:1066–76.
Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, de Jonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–80.
Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N, Takahashi N, Yoshida M, Azuma T, Hattori M, et al. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori east Asian genomes. BMC Microbiol. 2011;11:104.
Lu W, Wise MJ, Tay CY, Windsor HM, Marshall BJ, Peacock C, Perkins T. Comparative analysis of the full genome of Helicobacter pylori isolate Sahul64 identifies genes of high divergence. J Bacteriol. 2014;196:1073–83.
Kersulyte D, Rossi M, Berg DE. Sequence divergence and conservation in genomes of Helicobacter cetorum strains from a dolphin and a whale. PLoS One. 2013;8:e83177.
Vermoote M, Vandekerckhove TT, Flahou B, Pasmans F, Smet A, De Groote D, Van Criekinge W, Ducatelle R, Haesebrouck F. Genome sequence of Helicobacter suis supports its role in gastric pathology. Vet Res. 2011;42:51.
Bellanger X, Payot S, Leblond-Bourget N, Guedon G. Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol Rev. 2014;38:720–60.
Lavigne JP, Vergunst AC, Bourg G, O'Callaghan D. The IncP island in the genome of Brucella suis 1330 was acquired by site-specific integration. Infect Immun. 2005;73:7779–83.
Graindorge A, Menard A, Monnez C, Cournoyer B. Insertion sequence evolutionary patterns highlight convergent genetic inactivations and recent genomic island acquisitions among epidemic Burkholderia cenocepacia. J Med Microbiol. 2012;61:394–409.
Jakubowski SJ, Cascales E, Krishnamoorthy V, Christie PJ. Agrobacterium tumefaciens VirB9, an outer-membrane-associated component of a type IV secretion system, regulates substrate selection and T-pilus biogenesis. J Bacteriol. 2005;187:3486–95.
Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8:552–63.
Olbermann P, Josenhans C, Moodley Y, Uhr M, Stamer C, Vauterin M, Suerbaum S, Achtman M, Linz B. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010;6:e1001069.
Maixner F, Krause-Kyora B, Turaev D, Herbig A, Hoopmann MR, Hallows JL, Kusebauch U, Vigl EE, Malfertheiner P, Megraud F, et al. The 5300-year-old Helicobacter pylori genome of the iceman. Science. 2016;351:162–5.
Aras RA, Small AJ, Ando T, Blaser MJ. Helicobacter pylori interstrain restriction-modification diversity prevents genome subversion by chromosomal DNA from competing strains. Nucleic Acids Res. 2002;30:5391–7.
De Groote D, Van Doorn LJ, Van den Bulck K, Vandamme P, Vieth M, Stolte M, Debongnie JC, Burette A, Haesebrouck F, Ducatelle R. Detection of non-pylori Helicobacter species in “Helicobacter heilmannii”-infected humans. Helicobacter. 2005;10:398–406.
Van den Bulck K, Decostere A, Baele M, Driessen A, Debongnie JC, Burette A, Stolte M, Ducatelle R, Haesebrouck F. Identification of non-Helicobacter pylori spiral organisms in gastric samples from humans, dogs, and cats. J Clin Microbiol. 2005;43:2256–60.
Handt LK, Fox JG, Yan LL, Shen Z, Pouch WJ, Ngai D, Motzel SL, Nolan TE, Klein HJ. Diagnosis of Helicobacter pylori infection in a colony of rhesus monkeys (Macaca mulatta). J Clin Microbiol. 1997;35:165–8.
Doi SQ, Kimbason T, Reindel J, Dubois A. Molecular characterization of Helicobacter pylori strains isolated from cynomolgus monkeys (M. fascicularis). Vet Microbiol. 2005;108:133–9.
Coordinators NR. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2017;45:D12–7.
Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, Conrad N, Dietrich EM, Disz T, Gabbard JL, et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017;45:D535–42.
PATRIC: https://www.patricbrc.org/.
McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–603.
EXPASY: http://www.expasy.org/.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–9.
Team RC. R: a language and environment for statistical computing. R. Vienna: R Foundation for Statistical Computing; 2016.
Warnes GRB, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. Gplots: various R programming tools for plotting data. R package version 3.0.1. 2016.
Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35.
Acknowledgements
This work was supported by the Medical Research Council [grant number G0901104] (https://www.mrc.ac.uk/) to RMD. We are grateful to the numerous researchers in the field who have provided public access to their draft H. pylori genome sequences and to the anonymous reviewers of the manuscript for their constructive comments.
Funding
This work was supported by the Medical Research Council [grant number G0901104] (https://www.mrc.ac.uk/) to RMD.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional files.
Author information
Authors and Affiliations
Contributions
RMD conceived the project and study design. RMD, NJC, and ADS contributed to data collection, annotation and collation. RMD performed the analyses; RMD and ADS interpreted the results. RMD wrote the manuscript and all authors revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Dataset 1. This file contains compiled BLASTp search results for each of 59 tfs-encoded proteins. (XLSX 3851 kb)
Additional file 2:
Dataset 2. This file contains complete datasets for tfs, com and (partial) cag gene content extracted from 232 Helicobacter strains, additionally including gene/tfs ICE description, classification and nomenclature and relevant strain meta-data. (XLSX 249 kb)
Additional file 3: Figures S1 – S7.
This file contains Figures S1 – S7 and associated Figure legends. (PDF 9338 kb)
Additional file 4: Tables S1 – S5.
This file contains Tables S1 – S5. (PDF 52 kb)
Additional file 5:
Dataset 3. This file contains FASTA-formatted concatenated sequence of tfs3 ICE genes t3_V24, t3_C3, t3_C2 and t3_V25 from 76 H. pylori and H. cetorum genomes. (XLSX 50 kb)
Additional file 6:
High quality, 600dpi resolution version of Fig. 1. (TIF 14438 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Delahay, R.M., Croxall, N.J. & Stephens, A.D. Phylogeographic diversity and mosaicism of the Helicobacter pylori tfs integrative and conjugative elements. Mobile DNA 9, 5 (2018). https://doi.org/10.1186/s13100-018-0109-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-018-0109-4