
Abstract
Background
Intestinal fibrosis is a frequent complication of Crohn’s disease. However, the factors that cause chronicity and promote fibrogenesis are not yet understood.
Aims
In the present study, we evaluated the profibrotic effects of adherent-invasive Escherichia coli (AIEC) LF82 strain and Candida albicans in the gut.
Methods
Colonic fibrosis was induced in C57BL/6 mice by administration of three cycles of 2.5% (w/v) dextran sulfate sodium (DSS) for 5 weeks. LF82 and C. albicans were administered orally once at the start of each week or each cycle, respectively. Expression of markers of myofibroblast activation was determined in TGF-β1-stimulated human intestinal epithelial cells (IECs).
Results
LF82 administration exacerbated fibrosis in DSS-treated mice, revealed by increased colonic collagen deposition and expression of the profibrotic genes Col1a1, Col3a1, Fn1 and Vim. This was accompanied by enhanced gene expression of proinflammatory cytokines and chemokines, as well as more recruited inflammatory cells into the intestine. LF82 also potentiated TGF-β1-stimulated epithelial–mesenchymal transition and myofibroblast activation in IECs, by further inducing gene expression of the main mesenchymal cell markers FN1 and VIM and downregulating the IEC marker OCLN. Proinflammatory cytokines were overexpressed with LF82 in TGF-β1-stimulated IECs. Conversely, C. albicans did not affect intestinal fibrosis progression in DSS-treated mice or myofibroblast activation in TGF-β1-stimulated IECs.
Conclusions
These results demonstrate that AIEC strain LF82, but not C. albicans, may play a major profibrogenic role in the gut.
Similar content being viewed by others


Introduction
Crohn’s disease (CD) is a worldwide chronic inflammatory bowel disease (IBD) whose incidence is increasing across Europe [1,2,3]. Although the precise aetiology of CD is unknown, it is well accepted that this disease is the consequence of immune-mediated injury to the gut mucosa inflicted by an overactive immune response towards environmental factors in a genetically predisposed host [4]. Sustained inflammation and chronic wound healing response often lead to intestinal fibrosis, a condition defined by an excessive accumulation of extracellular matrix (ECM) proteins produced by activated myofibroblasts, which are alpha smooth muscle actin (α-SMA)-expressing cells. These cells not only derive from resident mesenchymal cells (fibroblasts and smooth muscle cells) but can also originate from epithelial and endothelial cells via epithelial/endothelial transition, as well as from stellate cells, pericytes and bone marrow stem cells [5]. Activation of myofibroblasts occurs in response to different stimuli, including growth factors such as transforming growth factor beta (TGF-β) and platelet-derived growth factor (PDGF), pro-inflammatory cytokines such as interleukin 1 (IL-1), IL-13 and IL-17, as well as CC chemokines like CCL2, CCL3 and CCL4, and lipid mediators released by immune and non-immune cells [6]. As a result, the formation of fibrotic scars in the intestinal wall results in a narrowing of the intestinal lumen and generates strictures and fistulae, or stenosis, in approximately 50% of CD patients. Despite the availability of treatments that target inflammation, no effective anti-fibrotic therapies exist, with surgical intervention being the only curative option although inflammation and stenosis may reoccur [5, 7, 8]. There is therefore an urgent need for a better understanding of the pathophysiology and identification of potential therapeutic targets in intestinal fibrosis.
A growing body of evidence suggests that an imbalance in the gut microbiota, or dysbiosis, is highly associated with CD pathogenesis, where it modulates the inflammatory status [9]. The bacterial microbiome has also been linked to intestinal fibrosis; however, the direct correlation between specific microbial species and fibrogenesis is still uncertain. CD patients are characterized by having fewer bacterial phyla, Firmicutes and Bacteroidetes, with anti-inflammatory properties, and more Actinobacteria and Proteobacteria, with proinflammatory roles [9, 10]. Among the Gram-negative Proteobacteria, pathogenic adherent-invasive Escherichia coli (AIEC) has been preferentially observed in ileal CD [11, 12]. The prototype AIEC strain, LF82, colonizes the intestinal mucosa and induces proinflammatory cytokines in a number of acute dextran sulfate sodium (DSS)-induced colitis mouse models [13,14,15]. Adhesion to intestinal epithelial cells (IECs) has been shown to be mediated by the interaction of LF82 type 1 pili with the abnormally expressed human carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) [14], the binding of flagella to TLR5 and IPAF flagellin receptors [13], or via chitin-binding domains, encoded by bacterial chitinase ChiA, that interact with human chitinase CHI3L1 expressed on IECs under inflammatory conditions [15].
In addition to bacteria, fungal microbiota dysbiosis is also considered a possible cause of CD. A number of studies have shown a decrease in levels of non-pathogenic Saccharomyces cerevisiae yeasts [16, 17] and an increase in abundance of Candida species in CD patients [18]. Candida albicans is the most prevalent opportunistic fungal pathogen in the intestine. Our group has previously demonstrated that colonic inflammation induced by DSS promotes C. albicans colonization in mice. In turn, C. albicans, via the β-galactoside-binding lectin receptor galectin-3, augments inflammation, as revealed by increased expression of TLR-2 and TNF-α, and triggers antibody generation directed against C. albicans antigens and also anti-S. cerevisiae antibodies (ASCA), which are serological markers of CD [19, 20]. Together, these findings indicate that microorganisms and their products can be profibrogenic in the gut.
In the current study, we investigated the effects of the AIEC strain LF82 and C. albicans on chronic intestinal inflammation and fibrosis progression.
Results
LF82, but not C. albicans, exacerbated intestinal fibrosis in DSS-induced chronic colitis in mice
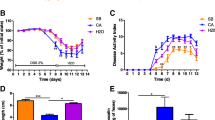
To examine the profibrotic role of the AIEC strain LF82 and C. albicans on the intestinal tract, chronic colitis was induced in mice exposed to 3 cycles of DSS as described previously [21]. The experimental design is illustrated in Fig. 1a. A previous study reported that wild-type (WT) mice challenged orally with LF82 showed neither colonization nor gut inflammation after 7 days [14]. Based on those results, LF82 was administered once per week to DSS-treated mice; E. coli counts in the stools increased in LF82-colonized DSS mice and persisted for 36 days (Fig. 1c). C. albicans administration was initiated once per cycle in DSS-treated mice according to a study done by our group showing colonization and persistence of C. albicans in the intestine of mice after acute DSS exposure for 7 days [20]. Our study demonstrated evidence of C. albicans colonization in the first cycle of DSS administration which persisted until the end of the experiment (Fig. 1c). As expected, mice treated with DSS showed decreased body weight during all cycles compared to control mice (H2O treated animals) (Fig. 1b). DSS-exposed mice colonized with LF82 showed significantly greater weight loss during the last DSS cycle when compared to DSS-treated mice (Fig. 1b).

Chronic AIEC LF82, but not C. albicans, administration worsened intestinal fibrosis in DSS-induced chronic colitis in mice. a Illustration of the experimental design of DSS-induced chronic colitis. Wild-type mice (n = 5–10 mice/group) received 2.5% DSS in drinking water in three cycles (5 days of DSS followed by 7 days of drinking water) and were orally administered LF82 (3 × 108) or C. albicans (107) once at the beginning of each week or each cycle, respectively. b Body weight change (percentage of initial weight) of different groups (from week 0 to week 5). Data are means ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 for DSS-exposed mice vs. mice receiving drinking water; #P < 0.05, ####P < 0.0001 for colonized vs. non-colonized mice receiving DSS (two-way ANOVA test with a Bonferroni post hoc correction). c Quantification of E. coli or C. albicans (CFU/mg) in culture of faeces recovered from mouse groups on different days. d Representative images and quantification of Masson’s trichrome staining of colonic tissue sections. Orange arrows indicate collagen deposition (green) in the colonic submucosa and mucosa. e Colonic gene expression of Col1a1, Col3a1, Fn1 and Vim. Results correspond to fold-increase when compared to mice receiving drinking water and are expressed as means ± SEM; *P < 0.05; **P < 0.01 for colonized vs. non-colonized mice receiving DSS (Mann–Whitney U test)
Intestinal fibrosis was assessed by histological analysis using Masson trichrome staining of collagen I–III within the colon tissue. Chronic DSS exposure caused a statistically significant increase in collagen I–III deposition in the colon subepithelium and serosal areas, an effect that was further enhanced by the presence of LF82 only (Fig. 1d). Moreover, quantitative RT-PCR performed on entire colon samples, showed that the expression of the profibrotic genes Col1a1, Col3a1 and Fn1 was elevated with DSS exposure. LF82, but not C. albicans administration, was able to further amplify the expression of these three profibrotic genes, in addition to Vim, in DSS-treated mice (Fig. 1e). These results suggest that the severity of fibrogenesis was affected by LF82 but not by C. albicans presence in the DSS-induced model of chronic colitis.
LF82 and C. albicans differentially exhibit proinflammatory properties in DSS-induced chronic colitis in mice
Fibrosis is known to require chronic inflammation as a prerequisite. Thus, we analysed the impact of LF82 and C. albicans on the gene expression of colonic proinflammatory cytokines and chemokines. Colonic expression of the genes Il1b, Il6, Il12b, Il17, Ifng and Ccl4 was increased in the presence of DSS and overexpressed in the presence of LF82 (Fig. 2a). No significant differences in mRNA levels of most cytokines and chemokines were detected except for Il12b in C. albicans-colonized mice compared to uninfected controls after DSS exposure (Fig. 2a). We also examined the consequence of these two microorganisms on recruitment of different inflammatory cells into the intestine. DSS was able to induce the expression of the T-cell marker Cd3e and, in particular, Th1-specific Tbx21/Tbet, Th2-specific Gata3 and Treg-specific Foxp3 transcription factors (Fig. 2b). LF82 led to a significant further increase in gene expression of the macrophage/monocyte marker Adgre1-F4/80, the neutrophil marker Ly6g, Cd3e, Txb21, Gata3 and Foxp3 in DSS-treated groups, while C. albicans showed highly elevated mRNA levels of Ly6g, Cd3e, Txb21, Th17-specific Rorc/Rorγt transcription factor and Foxp3 in DSS-exposed mice (Fig. 2b). These data imply that whereas only LF82 worsened fibrogenesis, both LF82 and C. albicans were able to differently induce intestinal inflammation.

Chronic AIEC LF82 and C. albicans administration induced intestinal inflammation in DSS-induced chronic colitis in mice. Colonic gene expression of: a proinflammatory cytokines Il1b, Il6, Il12b, Il17, Ifng and chemokine Ccl4; b immune cell recruitment markers Adgre1, Ly6g and Cd3e, and T-cell transcription factors Tbx21, Gata3, Rorc and Foxp3. Results correspond to fold-increase when compared to mice receiving drinking water and are expressed as means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001 for colonized vs. non-colonized mice receiving DSS only (Mann–Whitney U test)
LF82, but not C. albicans, enhanced TGFβ-induced epithelial–mesenchymal transition (EMT) and myofibroblast activation of human epithelial cells
We then performed an in vitro experiment to determine the effect of LF82 and C. albicans on myofibroblast differentiation of monolayers of human IECs in the Caco-2 cell line in the presence of TGF-β1. Optimal TGF-β1 administration conditions inducing the differentiation of IECs to a myofibroblast phenotype were standardized. Four days of 20 ng/mL TGF-β1 exposure was able to significantly increase the expression of myofibroblast and activation markers, FN1 and VIM, without affecting the expression of the IEC marker OCLN or proinflammatory cytokines IL1B, IL6, IL12B and IL8 (Fig. 3). TGF-β1-stimulated Caco-2 cells challenged with LF82 presented significant overexpression of FN1 and VIM and reduced gene expression of OCLN, while there was no effect on expression of these genes when they were challenged with C. albicans (Fig. 3a). Moreover, only LF82 highly increased the gene expression of IL1b, IL6, IL12b and IL8 after TGF-β exposure (Fig. 3b). These results indicate that LF82, but not C. albicans, potentiated myofibroblast activation of IECs by EMT induced by TGF-β1.

LF82, but not C. albicans, potentiated epithelial–mesenchymal transition (EMT) and myofibroblast activation of monolayers of human intestinal Caco-2 cells stimulated by TGF-β for 4 days. a Only LF82 induced overexpression of mesenchymal cell markers FN1 and VIM, and downregulation of the IEC marker OCLN in TGF-β-stimulated Caco-2 cells. b LF82 also increased the expression of the proinflammatory cytokines IL1B, IL6, IL12B and IL8 after TGF-β stimulation. Experiments were performed in triplicate and data correspond to fold-increase compared to control mice. Data are expressed as means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t-test). Results are representative of three independent experiments
Discussion
Intestinal fibrosis is the most common complication in patients with CD. It is the final outcome of the gut mucosal reaction to chronic inflammation and repair, which results in excessive deposition of extracellular matrix, leading eventually to intestinal dysfunction. Despite substantial efforts to identify the triggers for intestinal inflammation, the mechanisms underlying fibrosis remain poorly characterized and delay the development of effective anti-fibrotic therapies. Mounting evidence indicates that microorganisms can correlate directly with intestinal inflammation and could predict fibrosis. Here, we identified the AIEC strain LF82 as a new candidate organism affecting intestinal fibrogenesis, while the fungus C. albicans had no profibrogenic effect.
Most previous studies have shown that alterations in the bacterial microbiome are implicated in intestinal fibrosis. Antibiotic treatment in rats with chronic colitis significantly prevented TGFβ-1, collagen production and stricture formation [22], and intramural injection of faecal material or extracts from anaerobic bacteria into the intestinal wall induced chronic colitis with fibrosis and elevated levels of TGFβ-1 in colonic tissue [22, 23]. Additionally, a recent study showed that Tl1a-overexpressing mice had reduced colonic collagen deposition under pathogen-free conditions and interestingly proved causality by demonstrating that specific bacteria or bacterial consortia including groups of mucolytic bacteria such as Mucispirillum schaedleri, Ruminococcus, Anaeroplasma and members of the Streptococcus and Lactobacillus genera are directly correlated with the degree of fibrosis and fibroblast phenotype [24]. Severe and persistent intestinal fibrosis also occurred in mice infected chronically with Salmonella enterica [25].
AIEC is an E. coli pathotype that is present in high numbers in the inflamed gut of CD patients and its role in chronic colitis-associated fibrosis was first demonstrated in a chronic infection model of fibrosis using the AIEC strain NRG857c. NRG857c colonization persists for months in WT mice and is accompanied by chronic transmural inflammation and fibrosis [26]. This model was used in an attempt to avoid the use of LF82, which does not colonize conventional mice beyond 7 days and requires the expression of human CEACAM6 receptors to develop intestinal inflammation after acute DSS exposure [14]. In the present study, we observed the effects of LF82 on intestinal inflammation and fibrosis in WT mice in response to chronic DSS exposure by repeated LF82 challenges every 7 days. We demonstrated that LF82 worsened fibrosis, revealed by increased collagen deposition in the colon subepithelium and serosal areas and enhanced expression of the main fibrotic genes Col1a1, Col3a1, Fn1 and Vim.
Inflammation in CD has been shown to be driven predominantly by Th1 and Th17 responses [27]. We observed high gene expression of Il17, as well as of Ifng, which was consistent with elevated mRNA expression of the Th1 transcription factor Tbet, suggesting a Th1 response in our model. Other experimental colitis models have shown that both Th1- and Th2-mediated pathways contribute to the pathogenesis of CD, where they participate in different stages of chronic colitis development and affect diverse components of the inflammatory response [28, 29]. This may explain the increase in the Th2 transcription factor Gata3 in LF82-challenged mice after DSS exposure. Mucosal Treg cells and activated macrophages are also increased in paediatric and adult CD patients [30]. In our system, the Treg transcription factor Foxp3 was expressed at increased levels. Furthermore, there was also an upregulation in gene expression of the monocyte/macrophage marker Adgre1 and the neutrophil marker Ly6g, as well as higher expression levels of the proinflammatory cytokines related to them such as Il1b, Il6 and Il12b. Our results are similar to those observed with chronic NRG857c inflammation involving Th1 and Th17 responses and a significant role for macrophages and Treg cells [26].
Since EMT has been identified as a key contributor to the pool of activated fibroblasts associated with fibrosis in a mouse model of chronic colitis and to fistula formation in CD patients [31, 32], we evaluated the in vitro effect of LF82 on EMT and myofibroblast activation in TGF-β1-stimulated human IEC Caco-2 cells. LF82 aggravated TGF-β1-stimulated myofibroblast activation of these cells by EMT, as revealed by highly increased gene expression of mesenchymal cell markers FN1 and VIM and downregulated expression of the IEC marker OCLN. TGF-β signalling in epithelial cells appears to play an anti-inflammatory role [33]; however, we did not observe any effect of TGF-β on expression of the proinflammatory cytokines IL1B, IL6, IL12B and IL8 in our model. LF82 had strong proinflammatory properties and thus was able to overexpress all of these genes in IECs in the presence of TGF-β. Overall, these findings indicate that LF82 is associated with severe intestinal inflammation and fibrosis, and can affect fibroblast function directly or possibly via its products. Future studies should identify the mechanism for LF82-stimulated intestinal inflammation and fibrosis.
The importance of the fungal microbiome has received little attention in the context of intestinal fibrosis. Here, we evaluated whether C. albicans, the most prevalent fungal species in CD patients, is positively or negatively correlated with fibrosis severity. Despite the pro-inflammatory effects observed, we found that C. albicans did not affect fibrosis severity in DSS-treated mice. This could be due to certain products of this fungus that may have opposing effects on intestinal inflammation compared to fibrosis and this requires further investigation. In vitro studies showed no impact of C. albicans on myofibroblast activation and proinflammatory properties of TGF-β1-stimulated IECs, thus confirming the lack of a profibrogenic effect of C. albicans and that its effect on inflammation depends on different cell types.
Epithelial integrity is compromised in DSS-induced colitis suggesting penetration of microbes and diffusion of associated antigens into the mucosa [34]. One could thus think that the observed effects in our experiments had nothing to do with LF82 itself but reflect a difference between yeasts and bacteria, or may be attributed to both LF82 and other microbes colonizing the gut including other E. coli strains. Pathogenic and commensal E. coli were both increased in CD [35], but there was no direct evidence that they could directly affect intestinal fibrosis. However, it has been shown that TLR4, which could be activated by Gram-negative derived LPS, mediates chronic intestinal inflammation and fibrosis by regulating cytokine expression on intestinal macrophages and myofibroblasts and inducing epithelial–mesenchymal transition [36]. These findings suggest a role for E. coli in inducing colitis and fibrosis. Additionally, it has been reported that stimulation of intestinal myofibroblasts with LPS can upregulate TLRs (2, 3, 4, 6, 7) and their accessory molecules (MyD88, TIRAP), activate the MAPK pathway and increase IL-8 secretion, thus indicating that intestinal myofibroblasts participate in the immune response in the intestine after activation by bacterial products and may play a role in CD-associated fibrosis [37]. In our study, the effects of DSS on gut epithelial injury, resulting in increased intestinal permeability, changes in the microbiota and immunological and fibrotic alterations, as observed in the DSS-treated group, were in the same range as in the LF82-treated and C. albicans-treated DSS-induced groups. On the other hand, LF82 enhanced fibrogenesis compared to the DSS-treated group, meaning that we had an additive or synergistic proinflammatory and profibrogenic role of LF82. In addition, in in vitro experiments, LF82 enhanced the effect of the fibrogenic cytokine TGFβ inducing EMT and myofibroblast activation.
Conclusions
In conclusion, this study demonstrates a role for LF82 AIEC strains in the intestinal fibrotic process and suggests a possible role of the gut microbiome in the evolution of intestinal inflammation. To our knowledge, this is the first study to address the involvement of the fungal microbiome in intestinal fibrosis, ruling out a positive correlation between Candida species and fibrosis progression in our model.
Materials and methods
Strains and cultures of bacteria and yeasts
The AIEC strain LF82, derived from a chronic ileal lesion in a CD patient, was kindly provided by Christel Neut (Faculté de Médecine, Pôle Recherche, Laboratoire J&K, Lille, France) and maintained on MacConkey agar. C. albicans was isolated from the stool of a CD patient and maintained on Sabouraud dextrose agar containing amikacin. Prior to the experiments, LF82 and C. albicans were incubated overnight on a shaker at 37 °C in Luria–Bertani broth and yeast peptone dextrose broth, respectively, and harvested by centrifugation for 3 min at 3000×g for bacteria and 500×g for yeasts. The pellets were resuspended in 1× phosphate-buffered saline (PBS).
Animals
A total of 41, 7–8-week-old, male, C57/BL6 mice obtained from Janvier Laboratories (France) were used in the study and housed under specific pathogen-free conditions.
Mouse model of DSS-induced chronic colitis and fibrosis and infection
Chronic colitis and fibrosis were induced in mice by oral administration of 2.5% (w/v) DSS (MW: 36,000–44,000, purchased from TdB Consultancy, Uppsala, Sweden) in drinking water and administered ad libitum for three cycles (5 days of DSS followed by 7 days of tap water), as described previously [21]. Mice were challenged orally with 200 µL of PBS containing 3 × 108 LF82 live cells once per week, or with 200 µL of PBS containing 107 C. albicans live cells once per cycle. Mice were distributed into two control groups, including mice receiving drinking water (H2O) (n = 5) or DSS (n = 10) alone, and two experimental groups (n = 9–10/group), including LF82 + DSS and C. albicans (CA) + DSS. Animals underwent regular clinical follow-up (stool consistency and bleeding) and were weighed at the beginning of the study and every 2 days thereafter. Following oral administration, E. coli and C. albicans colonization in the intestinal tract was monitored by counting the number of colony-forming units (CFUs) in faeces collected from different animals. Faecal homogenates were plated on the corresponding agars (as mentioned above) and incubated at 37 °C for 24 h to 1 week. Blood and colon tissues were collected at sacrifice.
Cell culture and treatment
The human IEC line Caco-2 (ATCC HTB-37) was grown in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 10% foetal bovine serum (FBS). Cell cultures were maintained in a humidified atmosphere of 95% air/5% CO2 at 37 °C. Cells were seeded in 12-well plates at a density of 2.5 × 105 and incubated in medium without serum and supplemented with 20 ng/mL TGF-β1 for 4 days for myofibroblast activation. Cells were treated with LF82 and C. albicans at a multiplicity of infection = 1 during the last 24 h.
Masson trichrome collagen staining
Colon specimens from all animals were fixed overnight in 4% paraformaldehyde-acid, embedded in paraffin, cut to a thickness of 4 μm and stained with modified Masson’s trichrome counterstain using a Tissue-Tek Prisma Automated Slide Stainer (Sakura). The reagents for this staining technique include: haematoxylin, Mallory red (acid fuchsin stain and orange G), phosphotungstic–phosphomolybdic acid solution and light green, and when applied sequentially the resulting stain colours are: nuclei—dark blue, cytoplasm—pink, erythrocytes—bright red, muscles and collagen—green. The slides were scanned with a ZEISS Axio Scan and images were analysed using ImageJ software (NIH, USA).
RNA isolation and real time-PCR
Total RNA from entire mouse colon samples was isolated using Trizol reagent (Thermo Fisher Scientific) and RNA from human intestinal cell lines was extracted with a NucleoSpin RNA II kit (Macherey–Nagel) following the manufacturer’s instructions. Genomic DNA was removed using DNase I (RNase-free) according to the manufacturer’s protocol. RNA quantification was performed by spectrophotometry (Nanodrop). Reverse transcription was carried out on 1 µg RNA using a High Capacity cDNA reverse transcriptase kit (Thermo Fisher Scientific) and real time-PCR was performed with Fast SYBER Green Master Green (Thermo Fisher Scientific) according to the manufacturer's protocol. Gene expression from entire mouse colon samples and human IECs was normalized to Polr2a and GAPDH, respectively. The sequences and relative house-keeping genes are listed in Table 1.
Statistical analysis
Data are expressed as the mean ± SEM. The Student’s t-test was used to compare the mean values of two related groups. The Mann–Whitney U test was used for comparisons between two independent groups. Two-way ANOVA test with a Bonferroni post hoc correction was used for multiple intergroup comparisons. The results were considered statistically significant at P < 0.05. All statistical analyses were performed using Prism software (Graphpad, La Jolla, CA).
Availability of data and materials
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
References
Chouraki V, Savoye G, Dauchet L, Vernier-Massouille G, Dupas J-L, Merle V, Laberenne J-E, Salomez J-L, Lerebours E, Turck D, et al. The changing pattern of Crohn’s disease incidence in northern France: a continuing increase in the 10- to 19-year-old age bracket (1988–2007). Aliment Pharmacol Ther. 2011;33:1133–42.
Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. 2017;152:313.e2-321.e2.
Nerich V, Monnet E, Etienne A, Louafi S, Ramée C, Rican S, Weill A, Vallier N, Vanbockstael V, Auleley G-R, et al. Geographical variations of inflammatory bowel disease in France: a study based on national health insurance data. Inflamm Bowel Dis. 2006;12:218–26.
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78.
Latella G, Di Gregorio J, Flati V, Rieder F, Lawrance IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. 2015;50:53–65.
Rieder F, Fiocchi C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–35.
Latella G, Papi C. Crucial steps in the natural history of inflammatory bowel disease. World J Gastroenterol. 2012;18:3790–9.
Rieder F. The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci Transl Med. 2013;5:190ps10.
Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–84.
Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis. 2008;14:858–67.
Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–13.
Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15:872–82.
Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A. Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis. 2008;14:1051–60.
Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CHF, Stanners CP, Darfeuille-Michaud A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009;206:2179–89.
Low D, Tran HT, Lee I-A, Dreux N, Kamba A, Reinecker H-C, Darfeuille-Michaud A, Barnich N, Mizoguchi E. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology. 2013;145(602–612):e9.
Liguori G, Lamas B, Richard ML, Brandi G, da Costa G, Hoffmann TW, Di Simone MP, Calabrese C, Poggioli G, Langella P, et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. J Crohns Colitis. 2016;10:296–305.
Sokol H, Leducq V, Aschard H, Pham H-P, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–48.
Chehoud C, Albenberg LG, Judge C, Hoffmann C, Grunberg S, Bittinger K, Baldassano RN, Lewis JD, Bushman FD, Wu GD. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:1948–56.
Gerard R, Sendid B, Colombel J-F, Poulain D, Jouault T. An immunological link between Candida albicans colonization and Crohn’s disease. Crit Rev Microbiol. 2015;41:135–9.
Jawhara S, Thuru X, Standaert-Vitse A, Jouault T, Mordon S, Sendid B, Desreumaux P, Poulain D. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis. 2008;197:972–80.
Speca S, Rousseaux C, Dubuquoy C, Rieder F, Vetuschi A, Sferra R, Giusti I, Bertin B, Dubuquoy L, Gaudio E, et al. The novel PPARγ modulator GED-0507-34 Levo ameliorates inflammation-driven intestinal fibrosis. Inflamm Bowel Dis. 2016;22:279–92.
Mourelle M, Salas A, Guarner F, Crespo E, García-Lafuente A, Malagelada JR. Stimulation of transforming growth factor beta1 by enteric bacteria in the pathogenesis of rat intestinal fibrosis. Gastroenterology. 1998;114:519–26.
Medina C, Santos-Martinez MJ, Santana A, Paz-Cabrera MC, Johnston MJ, Mourelle M, Salas A, Guarner F. Transforming growth factor-beta type 1 receptor (ALK5) and Smad proteins mediate TIMP-1 and collagen synthesis in experimental intestinal fibrosis. J Pathol. 2011;224:461–72.
Jacob N, Jacobs JP, Kumagai K, Ha CWY, Kanazawa Y, Lagishetty V, Altmayer K, Hamill AM, Von Arx A, Sartor RB, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 2018;11:1466–76.
Grassl GA, Valdez Y, Bergstrom KSB, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. 2008;134:768–80.
Small CLN, Reid-Yu SA, McPhee JB, Coombes BK. Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun. 2013;4:1957.
Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1756–67.
Bamias G, Martin C, Mishina M, Ross WG, Rivera-Nieves J, Marini M, Cominelli F. Proinflammatory effects of TH2 cytokines in a murine model of chronic small intestinal inflammation. Gastroenterology. 2005;128:654–66.
Spencer DM, Veldman GM, Banerjee S, Willis J, Levine AD. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology. 2002;122:94–105.
Reikvam DH, Perminow G, Lyckander LG, Gran JM, Brandtzaeg P, Vatn M, Carlsen HS. Increase of regulatory T cells in ileal mucosa of untreated pediatric Crohn’s disease patients. Scand J Gastroenterol. 2011;46:550–60.
Bataille F, Rohrmeier C, Bates R, Weber A, Rieder F, Brenmoehl J, Strauch U, Farkas S, Fürst A, Hofstädter F, et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm Bowel Dis. 2008;14:1514–27.
Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285:20202–12.
Marincola Smith P, Means AL, Beauchamp RD. Immunomodulatory effects of TGF-β family signaling within intestinal epithelial cells and carcinomas. Gastrointest Disord. 2019;1:290–300.
Nagalingam NA, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. 2011;17:917–26.
Çekin AH. A microbial signature for Crohn’s disease. Turk J Gastroenterol. 2017;28:237–8.
Jun YK, Kwon SH, Yoon HT, Park H, Soh H, Lee HJ, Im JP, Kim JS, Kim JW, Koh S-J. Toll-like receptor 4 regulates intestinal fibrosis via cytokine expression and epithelial–mesenchymal transition. Sci Rep. 2020;10:19867.
Otte J-M, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–78.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm Unité 995) and Agence Nationale de la Recherche (ANR) in the setting of the project “InnateFun”, promotional reference ANR-16-IFEC-0003-05, in the “Infect-ERA” program.
Author information
Authors and Affiliations
Contributions
DC, MC, SS, BS conceived and designed the experiments; DC, MC, CB, RC, SS performed the experiments; DC, MC, CN, PD, BS analysed the data; DC, BS, MC wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All mouse experiments were conducted according to the protocol approved by the Subcommittee on Research Animal Care of Nord-Pas-de Calais, France and the French Ministry of Higher Education, Research and Innovation (201701408265726, 23/2/2018) and in accordance with European legal and institutional guidelines for the care and use of laboratory animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chokr, D., Cornu, M., Neut, C. et al. Adherent invasive Escherichia coli (AIEC) strain LF82, but not Candida albicans, plays a profibrogenic role in the intestine. Gut Pathog 13, 5 (2021). https://doi.org/10.1186/s13099-021-00401-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-021-00401-z