
Abstract
Background
Maturity-onset diabetes of the young (MODY) is the most common type of monogenic diabetes, being characterized by beta-cell disfunction, early onset, and autosomal dominant inheritance. Despite the rapid evolution of molecular diagnosis methods, many MODY cases are misdiagnosed as type 1 or type 2 diabetes. High costs of genetic testing and limited knowledge of MODY as a relevant clinical entity are some of the obstacles that hinder correct MODY diagnosis and treatment. We present a broad review of clinical syndromes related to most common MODY subtypes, emphasizing the role of biomarkers that can help improving the accuracy of clinical selection of candidates for molecular diagnosis.
Main body
To date, MODY-related mutations have been reported in at least 14 different genes. Mutations in glucokinase (GCK), hepatocyte nuclear factor-1 homeobox A (HNF1A), and hepatocyte nuclear factor-4 homeobox A (HNF4A) are the most common causes of MODY. Accurate etiological diagnosis can be challenging. Many biomarkers such as apolipoprotein-M (ApoM), aminoaciduria, complement components, and glycosuria have been tested, but have not translated into helpful diagnostic tools. High-sensitivity C-reactive protein (hs-CRP) levels are lower in HNF1A-MODY and have been tested in some studies to discriminate HNF1A-MODY from other types of diabetes, although more data are needed. Overall, presence of pancreatic residual function and absence of islet autoimmunity seem the most promising clinical instruments to select patients for further investigation.
Conclusions
The selection of diabetic patients for genetic testing is an ongoing challenge. Metabolic profiling, diabetes onset age, pancreatic antibodies, and C-peptide seem to be useful tools to better select patients for genetic testing. Further studies are needed to define cut-off values in different populations.
Similar content being viewed by others
Background
Diabetes is a heterogeneous disease, most cases corresponding to type 1 and type 2 diabetes. Nevertheless, a considerable proportion of patients does not fit into this classification and is known to have hyperglycemia caused by a mutation in a single gene. Maturity-onset diabetes of the young (MODY) is a heterogeneous group of monogenic diseases, normally associated with a secretory beta-cell defect [1]. Although classically described as a clinical syndrome of early-onset autosomal dominant diabetes [2, 3], MODY is now known to present as distinct clinical syndromes.
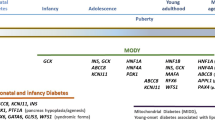
The most common causes are mutations in Glucokinase (GCK), presenting as mild non-progressive hyperglycemia since birth [4]; hepatocyte nuclear factor-1 homeobox A (HNF1A) and hepatocyte nuclear factor-4 homeobox A (HNF4A), presenting as familial symptomatic diabetes whereby hyperglycemia usually becomes evident during adolescence or early adulthood and deteriorates throughout life [5,6,7]; and hepatocyte nuclear factor-1 homeobox B (HNF1B), presenting mainly as renal alterations and diabetes [8]. Other rare forms of MODY can be caused by mutations in other genes (Table 1).
Despite the rapid evolution of molecular diagnosis methods, many MODY cases may be misdiagnosed as type 1 or type 2 diabetes [9]. Accurate etiological diagnosis of diabetes can be challenging, even within a single family [10]. In this context, there is a worldwide trend towards “Precision Medicine” (PM), an approach which aims to tailor prevention and treatment taking characteristics of individuals and/or subpopulations into account. PM is a possible approach to enhance treatment of patients with diabetes and has been successfully applied in monogenic diabetes, especially in neonatal diabetes (ND), since a single clinical criterion is used (age of diagnosis < 6 months). ND is mainly caused by mutations in the genes encoding the pore-forming (Kir6.2, KCNJ11) and regulatory (SUR1, ABCC8) subunits of the K ATP channel. It is well-known that sulfonylurea treatment in potassium channel-linked ND have marked impact on endogenous insulin secretion and is now considered the treatment of choice [11,12,13,14]. Therefore, ND is an excellent prototype of how the understanding of pharmacogenomics helps in tailoring treatment according to a patient’s genetic profile.
In MODY, nonetheless, this approach is more complex. The lack of a single clinical criterion, cost of genetic testing, and specialists’ emphasis on treatment rather than diagnosis are the major barriers for the dissemination of precision medicine in MODY [15]. With the establishment of sulfonylureas as the treatment of choice for HNF1A-MODY [16], molecular diagnosis of monogenic diabetes has been solidified as a necessary clinical tool with important prognostic implications, being part of clinical routine for patients with a clinical suspicion of MODY. Evidence that most patients with GCK mutations generally do not require pharmacological treatment [7, 17, 18] and do not develop long-term complications [19, 20] has established the importance of classifying MODY in clinical syndromes as described below.
The use of criteria based on absolute cut-offs have shown poor sensibility, resulting in many MODY patients misdiagnosed as either type 1 or type 2 diabetes [9, 21, 22]. Despite more widespread availability of molecular diagnosis, better strategies for clinical screening of monogenic diabetes are necessary, in order to better select candidates for molecular diagnosis and therefore optimize cost-effectiveness.
This review aims to describe the clinical syndromes related to the most common genetic causes of MODY and biomarkers that can potentially improve accuracy of clinical selection candidates for molecular diagnosis.
Literature search strategy
Pubmed was searched for publications on the subject by employing search terms: MODY, Maturity Onset Diabetes of the Young, monogenic diabetes, HNF1A, HNF-1 alpha, GCK, glucokinase, HNF1B, HNF-1 beta, HNF4A, HNF-4 alpha, biomarkers. Search was performed on May 18th, 2020, so literature review is up to date at this point. We manually screened results for relevant and recent papers limited to the English language. References from selected publications were also used when necessary.
Clinical syndromes related to most common MODY subtypes
Clinical criteria for diagnosing MODY devised at the time of its original description, the classical triad of early onset, autosomal dominant inheritance, and predominant secretory defect, have reasonable positive predictive value (PPV). Nevertheless, sensitivity and hence negative predictive value (NPV), hallmarks of an adequate screening test, are low. This results in many false negative MODY cases intermixed in the vast heterogeneity of major types of diabetes [21].
With the advancement of molecular diagnosis technologies, clinical criteria for suspicion of MODY have been refined according to specific characteristics of different genes, so the classic criteria of autosomal dominant early-onset diabetes could be said to be more adequate for the screening of MODY caused by transcription factors. Nevertheless, given its low sensitivity, many publications have extended these criteria to individuals initiating diabetes at a later age (before 35 years old) and with at least one first-degree relative with diabetes instead of three full generations, since penetrance of MODY mutations is incomplete and varies with age. These criteria have been used in most large cohorts of patients with MODY and have yielded identification of thousands of individuals [23,24,25,26,27], but refining those criteria can improve detection of other specific subtypes of MODY. As an example, an Italian group designed and validated a 7-item flowchart (7-iF) to identify patients that have a high probability of carrying GCK mutations, taking into account aspects such as autoimmune diabetes antibodies, HbA1c levels, and heredity [28]. In order to assist clinicians in selecting candidates for molecular diagnosis, we describe below the most common clinical presentations of MODY according to the causative gene [7].
Mild non-progressive hyperglycemia due to GCK mutations
GCK-MODY is characterized by mild non-progressive hyperglycemia. It was first suggested by Cammidge in 1928 [29] and it was first recognized as a monogenic disease in 1992, when Froguel et al. first observed a tight linkage between the glucokinase locus on chromosome 7p and diabetes in 16 French families with MODY [4]. Subsequently, in the same year, a nonsense mutation in the gene encoding glucokinase and its linkage with MODY in one family was reported [30].
GCK-MODY can be described as disturbed beta-cell glucose sensing. Decreased levels of glucokinase activity in beta-cells are predicted to lead to a defect in glucose-stimulated insulin secretion, and, therefore, a rightward shift of the dose–response curve of insulin secretion [31]. GCK-MODY has been reported in 40–50% of cases of incidental or asymptomatic hyperglycemia in the pediatric population [32]. One British study, employing a systematic population screening approach, tested C-peptide in 808 individuals with diabetes and age younger than 20 years old and, in those with detectable pancreatic residual function, pancreatic autoantibodies were assessed. Those without evidence of pancreatic autoimmunity were tested for monogenic diabetes, demonstrating 2.5% of the 808 individuals to have MODY, being 1% of the total caused by GCK [25]. This low prevalence reinforces the importance of clinical screening. Diagnosis of GCK mutations is suggested by the clinical characteristics depicted in Table 2. Measuring fasting glucose in apparently unaffected parents is important when considering a diagnosis of a GCK mutation in a proband, since mutations have complete penetrance [7, 33,34,35]. Due to the mild non-progressive hyperglycemia, HbA1c can have a role in differential diagnosis with other types of diabetes [18, 36]. Another British study showed 123 individuals carrying GCK mutations to have HbA1c between 5.6 and 7.3% in the subgroup with age below 40 years old, and between 5.9 and 7.6% in the subgroup aged 40 years or older [35].
In contrast to other forms of dysglycemia, insulin secretion continues to be regulated. Pharmacological treatment is not usually recommended since hyperglycemia in GCK-MODY is resistant to oral medication due to a set point alteration of glucose homeostasis. Therefore, treatment does not generally alter glycemic control or prognosis, with the exception of pregnancy, where treatment of an affected mother is needed due the possibility of in utero accelerated growth when the fetus is unaffected [36].
GCK-MODY is rarely associated with diabetes-related complications. A British study evaluated the association between chronic, mild hyperglycemia and complication prevalence in patients with GCK mutations. Despite a median duration of 48.6 years of hyperglycemia, prevalence of microvascular and macrovascular complications was low [20].
Presence of GCK mutations probably do not affect the risk of developing type 2 diabetes and obesity later in life [36], Therefore, diagnosis of GCK mutations in older individuals can be challenging due to the possibility of overlap with other metabolic conditions [37].
Early-onset autosomal dominant diabetes due to HNF1A and HNF4A mutations
Hepatocyte nuclear factors (HNFs), despite being first described in the liver of animal models, are transcription factors expressed in different tissues that play a substantive role in the normal development and function of pancreatic beta-cells. Reduction of insulin secretion in response to glucose occurs in patients with heterozygous HNF1A mutations. This secretory defect worsens over time due to progressive beta-cell dysfunction [17]. Diabetes mellitus manifests usually at the age of 6–25 years with mild osmotic symptoms (polyuria, polydipsia) or as asymptomatic postprandial hyperglycemia without ketosis or ketoacidosis [38]. In contrast to GCK-MODY, there is impairment of first and second-phase insulin secretion in individuals with HNF1A mutations [39, 40], resulting more often in overt diabetes as opposed to the mild hyperglycemia seen in the first.
Clinical expression of HNF1A-MODY varies considerably. Environmental and genetic characteristics contribute to this heterogeneity [41,42,43]. Therefore, some patients with HNF1A-MODY may not fulfill the classical diagnostic criteria. Nevertheless, in general, diagnosis of HNF1A mutations may be suggested by the presence of clinical characteristics described on Table 3 [7, 40, 44].
In HNF1A-MODY the frequency of cardiovascular and microvascular complications is high and similar to that of patients with type 1 and type 2 diabetes [45, 46]. Treatment depends on the age and HbA1c levels. Patients can initially be managed with diet but most patients will require pharmacological treatment. They are very sensitive to sulfonylureas which are usually more effective than insulin, particularly in children and young adults [47]. This treatment is usually effective for several decades, but in a case of severe decrease in beta-cell insulin production, insulin therapy is needed [48]. A study demonstrated that 80% of patients with HNF1A-MODY treated with sulfonylurea therapy remained insulin independent at 84 months of follow-up [49].
Diabetes caused by mutations in the HNF4A gene is considerably less common than HNF1A (5% to 10% of the cases) [9], but should be considered whenever clinical characteristics of HNF1A are present and genetic analysis does not detect a mutation in this gene. Some discriminatory factors between HNF4A from HNF1A are later age at diagnosis and the lack of pronounced glycosuria in the former (see Table 3). Patients are also often sensitive to sulfonylureas [50]. Differently from HNF1A, HNF4A mutations are associated with macrosomia (approximately 56% of mutation carriers) and transient neonatal hypoglycemia (approximately 15% of mutation carriers) [51]. A family history of marked macrosomia or diazoxide responsive neonatal hyperinsulinism in the context of familial diabetes should raise the hypothesis of HNF4A-MODY [51].
Renal/urogenital alterations and diabetes due to HNF1B mutations
HNF1B gene plays a role in the tissue-specific regulation of gene expression in liver, kidney, intestines, and pancreatic islets, therefore influencing their embryonic development [52]. Diabetes in HNF1B-MODY develops during adolescence or early adulthood. Median age of onset of diabetes was 28 years old in a multicenter cohort of 201 individuals. Patients present some degree of hepatic insulin resistance, explaining why approximately half of them do not respond to sulfonylureas, needing early insulin therapy [7, 53]. A recent study evaluated 35 patients with HNF1B mutations, 65.7% of whom were treated with insulin and, in 40%, extrapancreatic symptoms were reported [54]. Diabetes complications (especially microvascular disease) and cardiovascular risk factors are highly frequent and renal impairment can be a major issue (chronic kidney disease stages 3–4 was present in 44% of individuals and end-stage renal disease in 21%) [55, 56].
This subtype of MODY was thought to be rare when initially described, but since the diabetes phenotype is frequently associated with renal and urogenital malformations [57], search for MODY in patients with those features has demonstrated HNF1B to be more frequent, with a proportion of affected patients similar to HNF4A depending on the studied population [44]. Although HNF1B-MODY is often described as a syndrome of renal cysts and diabetes (RCAD), several different renal and urogenital phenotypes are now known to be associated with mutations in this gene. Presence of urogenital tract malformations, renal failure not explainable by diabetes progression, renal cysts, renal dysplasia, or hypoplastic glomerulocystic kidney disease in association with diabetes may prompt direct investigation of HNF1B, without necessarily investigating more common types of MODY beforehand [8]. It is important to point out in this context that spontaneous de novo mutations are relatively frequent and the absence of a familial history of renal disease should not discourage testing for HNF1B mutations [58]. Renal and other alterations associated with HNF1B mutations are described in Table 4. In addition to the marked heterogeneity of the urogenital phenotype observed in association with diabetes, many cases of HNF1B without diabetes have been described [59].
Considering the heterogenous phenotype of HNF1B, researchers have developed a score that could be used as a tool for clinicians to select patients with suspicion of HNF1B before genetic testing. One of these scores was developed based on the frequency of most typical findings considering clinical, biological, imaging, and familial characteristics. A score > 8 showed very good discriminatory power with high NPV (99%), and it would provide a useful aid for selecting patients for HNF1B testing [60]. A British study aimed at validating this score in a cohort of 686 individuals with a genetic diagnosis of a HNF1B mutation found the same cutoff point to have an NPV of 85% [61]. In a Brazilian cohort, 28 patients with clinical suspicion of HNF1B-MODY due to hyperglycemia and renal cysts were evaluated and two positive cases of HNF1B gene mutations were found. Both positive cases had a score higher than 8 [62].
Clinical heterogeneity, atypical diabetes, and discriminatory models of MODY
Clinical strategies to select patients for genetic test have benefited from inclusion of other clinical data in the past decade, with the added goal of excluding other common types of diabetes such as type 1 diabetes. Absence of pancreatic islet autoantibodies and presence of pancreatic residual function after 5 years of diabetes duration, when the honeymoon phase has unequivocally past (as demonstrated by low insulin requirements and/or detectable C-peptide) are now valuable clinical criteria in the selection of candidates to molecular diagnosis [7]. Superposition with type 2 diabetes has also to be considered, since monogenic diabetes does not usually show features of insulin resistance [63].
With typical MODY families identified, further exploration into the heterogeneity of type 1 and type 2 diabetes uncovered patients with MODY but not bearing typical clinical features, even when extended criteria have been used. This has drawn especial attention to atypical cases of the two most common types of diabetes. In 247 British individuals clinically labeled as type 1 diabetes, 20 had persistent residual beta-cell function outside the range usually expected for type 1 diabetes. There were no differences in GAD positivity, BMI, and parental diabetes history between C-peptide positive and negative patients. Among the 20 C-peptide positive individuals, two (10%) had mutations in HNF1A. Although the importance of diagnosing HNF1A diabetes correctly lies partly on the possibility of transferring patients from insulin to sulfonylureas, both patients couldn’t have their therapies successfully changed and were kept on insulin. Of note, both had positive GAD antibodies, opening the possibility of autoimmunity modulating MODY phenotype [63]. In the same series, among 322 patients clinically diagnosed as having type 2 diabetes, 80 met extended criteria for MODY. In this group, 10 individuals had HNF1A mutations and 2 had HNF4A mutations (only 5 of those 12 individuals had classical MODY criteria). Resequencing for GCK-MODY was carried out on 38 individuals and one previously reported mutation was found. This patient had a metabolic profile consistent with GCK-MODY [63].
In a French study, a model employing as predictors Euro-Caucasian origin, 3 or more affected generations, age, and BMI (only in patients without symptoms of hyperglycemia) yielded sensitivity 90% and specificity 49%. This model is useful to improve the low sensitivity of the classical criteria, although its own definition precludes its utilization in non-European populations [64].
Even with extended clinical criteria, most variables have been traditionally used in discrete form, resulting in poor sensitivity. To cope with this limitation, a mathematical model was developed for patients of white European origin, who have been diagnosed with diabetes at age equal or less than 35. Two-hundred and seventy-eight individuals with type 1 diabetes (clinically defined as requiring insulin within 6 months of diagnosis), 319 with type 2 diabetes (clinically defined as not requiring insulin within 6 months of diagnosis), and 594 probands with a genetic diagnosis of MODY (243 GCK, 296 HNF1A, and 55 HNF4A) have been compared regarding simple and widely available clinical variables [21].
When comparing type 2 diabetes with MODY, the following variables were associated with MODY: lower age at diagnosis, BMI, HbA1c, and current age; having one diabetic parent; not being treated with insulin or oral antidiabetic agents; female sex. Accuracy as measured by area under ROC curve was 0.95. Sensitivity was 92% and specificity 95%. Comparing type 1 diabetes with MODY, the latter was associated with: having one parent with diabetes; lower current age and HbA1c; higher age at diagnosis; female sex. Area under ROC curve was 0.95. Sensitivity was 87% and specificity 88%.
The mathematical model had better sensitivity than the classical criteria (72%). It is available online as a probability calculator at http://www.diabetesgenes.org. Individuals with type 2 diabetes should be tested for MODY if the probability provided is greater than 25%. In individuals with type 1 diabetes, probability should be greater than 10% [21]. Although promising, similar models need to be tested in other populations in order to gain wider acceptance in clinical use, since this specific model has been developed for European Caucasian individuals with diabetes diagnosed before 35 years old and has been validated only for the three most commons subtypes of MODY. Moreover, even when the shift from individual gene Sanger sequencing to targeted-NGS is completed in most centers around the world, algorithms to select candidates to genetic testing would still be necessary in order to improve cost-effectiveness [65,66,67].
Many stepwise algorithms of etiologic diagnoses of hyperglycemia have been proposed [68,69,70]. Recently, Urakami et al. suggested an algorithm used to identify candidates with diabetes who should undergo genetic testing considering age at onset of diabetes, pancreatic autoimmunity and residual function, obesity and insulin resistance and some nongenetic biomarkers [71].
Biomarkers employed in the clinical screening of monogenic diabetes
Nowadays, selection for molecular testing is based on nonspecific clinical characteristics such as age of onset, family background, and atypical presentation for the assumed etiology, although these criteria do not combine reasonable levels of specificity and sensitivity. In this context, many researches have been persistently looking for biomarkers to assist selection of individuals who deserve further investigation. Meanwhile, many candidates like apolipoprotein-M (ApoM), aminoaciduria, complement components, and glycosuria have been tested, but have not translated into useful biomarkers [72,73,74]. Biomarkers that have been studied as screening tools for MODY mutations are described on Table 5. An overview of the most studied biomarkers as well as its rationale and clinical limitations follow below.
High-sensitivity C-reactive protein (hsCRP) and HNF1A
Some studies have shown that common variants near the HNF1A gene are associated with small alterations in serum high-sensitive C-reactive protein (hsCPR) levels in healthy adults. Levels of hsCRP are lower in HNF1A-MODY than in other types of diabetes (including other types of MODY) and nondiabetic subjects. The rationale for associating hsCRP levels with HNF1A derives from two basic concepts. First, C-reactive protein is coded by the CRP gene. This gene bears binding sites specific for the HNF1A transcription factor. SNPs in HNF1A have been associated to CRP levels in various populations [75,76,77]. Second, although MODY can bear some clinical resemblance with type 2 diabetes, low-grade inflammatory process seen in type 2 diabetes, obesity, and cardiovascular disease does not participate in the pathophysiology of MODY.
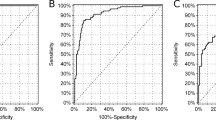
The use of hsCRP as a clinical screening tool for MODY has been first investigated in a British study, that showed patients with HNF1A-MODY to have significantly lower hsCRP levels when compared to autoimmune diabetes (both type 1 diabetes and LADA), young-onset type 2 diabetes, GCK-MODY, and non-diabetic individuals, even after correction for BMI and use of medications that could potentially lower hsCRP (aspirin and statins). Accuracy of hsCRP alone was 80% when discriminating HNF1A-MODY from type 2 diabetes and 75% when comparing HNF1A-MODY with all other types of diabetes. Analyzing various combinations of hsCRP with other criteria, utilizing CRP ≤ 0.2 mg/L or diagnosis of diabetes up to 30 years of age performed best, with 88% sensitivity and 75% specificity. This study, however, did not compare HNF1A diabetes with HNF4A, which bears many clinical similarities between each other [22].
These findings have been confirmed in a large multicenter trial involving 7 European countries, that showed hsCRP levels to be lower in HNF1A than every other type of diabetes, including HNF4A this time. Accuracy of hsCRP to distinguish between HNF1A-MODY patients and young adult- onset type 2 diabetes, as measured by ROC-derived C-statistic, ranged from 0.79 to 0.91, depending on the center [78].
Since the discriminating cutoff point occurs in very low levels of CRP, the utilization of a high-sensitivity assay is mandatory. In another British study, a cutoff point of 0.75 mg/L had a positive predictive value (PPV) of 2.7% and a negative predictive value (NPV) of 99.7% when comparing HNF1A-MODY to type 2 diabetes. When used to compare HNF1A with other MODY types, a 0.55 mg/L cutoff point may be useful to decide priority of sequencing in the context of using Sanger sequencing and testing genes separately [79].
The rise in obesity and type 2 diabetes have made the number of individuals with a family history of diabetes increase. Almost 30% of patients with HNF1A-MODY are overweight or obese, making differential diagnosis between HNF1A and familial young onset type 2 diabetes even more challenging [64]. A recent French study assessed the added value of hsCRP to distinguish between these two conditions. Area under ROC-curve was 0.82 with the clinical model (diabetes at age < 40 years, familial history of diabetes in at least two generations, and absence of obesity) and increased to 0.87 when hsCRP was included. These values were not satisfactory since the calculated probability of HNF1A-MODY diagnosis > 50% as a threshold for identifying patients for genetic screening would miss one-third of HNF1A-MODY cases [80].
Szopa et al. also evaluated the utility of hsCRP to improve diagnostic accuracy of MODY. In accordance with previous findings, the lowest levels of hsCRP were seen in HNF1A-MODY, but there was significant overlap of hsCRP distribution among individuals with HNF1A-MODY, GCK-MODY, and type 1 diabetes. Authors devised a three-step decision tree algorithm using C-peptide, BMI, 1,5-anhydroglucitol (1,5-AG), and hsCRP to identify patients with HNF1A mutations. Nevertheless, the model was not accurate enough to discriminate HNF1A patients without gene sequencing [68].
Although hsCRP is becoming a useful and promising marker for HNF1A, considering its extensive availability and low cost, clinicians should have in mind that it is a non-specific test, affected by several pathological conditions such as inflammation and acute infection, so caution should be exercised with its clinical significance until more data are available.
Pancreatic residual function
The measurement of C-peptide is used to assess endogenous insulin production in diabetic and non-diabetic individuals, despite treatment with insulin. Several methods of C-peptide measurement have been proposed. Venous blood C-peptide levels can be measured in the random, fasting or stimulated state [74]. Fasting and random non-fasting C-peptide are simple, quick to perform, and correlate with diabetes type. C-peptide is a small linear peptide, which is susceptible to enzyme proteolytic cleavage, consequently, quickly centrifuging and freezing sample is usually required. The 24-h urinary C-peptide (24 h UCP) sample collection is non-invasive and stable for 72 h in boric acid but is time-consuming and requires good patient compliance. Urinary C-peptide/creatinine ratio (UCPCR) is a reproducible measure that correlates well with 24-h UCP in nondiabetic subjects [81]. Second-void fasting UCPCR was suggested as the optimum approach for the assessment of baseline endogenous C-peptide production using a spot urine test [82]. Both tests are inaccurate in chronic kidney disease and affected by variations in creatinine [81, 82].
One important feature distinguishing type 1 diabetes from MODY is long-term evolution of residual pancreatic function. In type 1 diabetes, complete insulin deficiency ensues in most patients after 5 years of evolution [83, 84]. In MODY, since there is no direct destruction of beta-cells, residual endocrine pancreatic function may be observed after several years of evolution, therefore, detectable serum C-peptide outside the honeymoon period may indicate a diagnosis of MODY. This can be especially useful in transcription factor MODY, which presents frequently as a differential diagnosis to type 1 diabetes. In contrast, in type 2 diabetes, obesity-related insulin resistance may result in elevated levels of insulin and C-peptide [85].
C-peptide has been studied as a screening tool for HNF1A/4A MODY, specifically as post-prandial UCPCR, for the sake of methodological simplicity. Adults with diabetes duration equal or greater than 5 years were evaluated, including patients with monogenic diabetes, as well as clinically defined type 1 and type 2 diabetes. Individuals with type 1 diabetes had a median UCPCR < 0.02 nmol/mmol, compared to 1.72 nmol/mmol in HNF1A/4A patients. Area under ROC curve (ROC-AUC) showed good accuracy (0.98). Sensitivity was 97% and specificity 96% for discriminating HNF1A/4A MODY from type 1 diabetes, with an UCPCR cut-off point of 0.2 nmol/mmol. Accuracy levels were maintained after comparing only insulin-treated HNF1A/4A patients with type 1 diabetic subjects (area under ROC curve 0.96, 94% sensitivity, and 96% specificity) [86].
Considering that C-peptide is known to decline more rapidly in children than in adults, a study evaluated the use of UCPCR and its ability to discriminate pediatric diabetes subtypes even in short-duration diabetes. Two-hour postprandial UCPCR was measured in 264 patients with diabetes (MODY, type 1 diabetes, type 2 diabetes) aged < 21 years old. The UCPCR ≥ 0.7 nmol/mmol was effective in differentiating between type 1 and non-type 1 diabetes (type 2 diabetes and MODY), with a sensitivity of 100% and a specificity of 81%, independently of diabetes duration. If the duration of diabetes was greater than 2 years, a UCPCR ≥ 0.7 nmol/mmol was considered to be effective, with a sensitivity of 100% and a specificity of 97%. However, UCPCR was not able to discriminate MODY from type 2 diabetes (ROC-AUC 0.57) [87].
Another study compared UCPCR and fasting C-peptide together in patients with MODY and type 1 diabetes on the pediatric age group. UCPCR ≥ 0.22 nmol/mmol confirmed excellent differentiation between MODY and type 1 diabetes in children, yielding 96.3% sensitivity and 85.7% specificity. Fasting C-peptide levels in the type 1 diabetes group were lower than in MODY (p = 0.001). Fasting C-peptide cutoff determined by ROC curve analysis was 0.62 ng/ml, with a sensitivity of 93% and a specificity of 90% for discriminating between MODY and type 1 diabetes. All patients with type 1 diabetes had diabetes duration above 2 years, but a UCPCR level ≥ 0.7 nmol/mmol, as employed by the reference cited above, showed sensitivity of only 59% and specificity of 91% [88].
Evaluation of the correlation between UCPCR and duration of diabetes demonstrated that UCPCR decreased as the duration of diabetes increased in both groups (type 1 diabetes and MODY). This seems contradictory at first glance, but in HNF1A/HNF4A-MODY there is actually a progressive decline in C-peptide related to reduction of beta-cell proliferation and increase of apoptosis. Conversely, GCK-MODY does not show decrease in C-peptide, as normal insulin secretion occurs, only at a higher glucose threshold. UCPCR values in the GCK-MODY group were higher than in type 1 diabetes, but the difference was not statistically significantly, and other studies confirmed this data [86, 88].
A recent study assessed random C-peptide measurements in patients with antibody-negative diabetes. A cut-off level of 0.15 nmol/L, obtained at 6 months or later after diabetes diagnosis, showed a negative predictive value of 96%. Thus, random C-peptide testing would be a potentially simple and affordable initial screening test for MODY in antibody-negative patients [89].
In conclusion, currently available evidence suggests that a UCPCR of ≥ 0.2 nmol/mmol indicates that a genetic test might be suitable. Biochemical parameters with autoimmune, demographic, physical characteristics, and the use of additional markers of pancreatic reserve may be critical to aid in the distinction between type 1 diabetes and MODY. Further studies in larger samples with a broader ethnical distribution of patients with specific MODY mutations are indicated.
Sulfonylurea sensitivity
Sulfonylurea sensitivity has been reported in MODY patients even before description of involved genes, including the first family described by Stephan Fajans [50]. Although better response of transcription factor MODY to sulfonylureas has been now solidly demonstrated, and many patients can be safely transferred back to oral medications even after many years of insulin therapy [16], it is frequent to encounter patients responding to sulfonylureas without a definite classification of diabetes, in clinical setting where molecular diagnosis is not readily available.
While transferring patients to sulfonylurea based solely in a clinical diagnosis of MODY is not a validated approach, patients without mutations in the known genes responding to oral medications suggest other undiscovered causes of MODY which also respond to sulfonylureas. The finding of 8% of ABCC8 mutations in 85 individuals with sulfonylurea-sensitive diabetes, negative for HNF1A and HNF4A mutation, and without neonatal onset illustrate this principle [90].
A British study showed that only 36% of individuals with HNF1A/HNF4A mutations achieved HbA1c ≤ 7.5% on sulfonylurea/diet alone. Shorter diabetes duration, lower HbA1c, and lower BMI at genetic diagnosis predicted successful treatment with sulfonylurea/diet alone. This study also suggested that sulfonylurea should be added to existing treatment, rather than replacing it, especially in those with longer duration of diabetes (> 11 years), overweight or obese and with a high HbA1c at the time of genetic diagnosis [91].
Sulfonylurea responsiveness is not endorsed as a valid criterion for patient selection for genetic testing. Nevertheless, marked sensitivity and long-term effectiveness to sulfonylureas among patients with long diabetes duration could be a useful clue to optimize the recruitment process [92].
Pancreatic autoimmunity
Type 1 diabetes is the most common form of diabetes in children and young adults. Approximately 80% of MODY patients are misdiagnosed [9]. This issue often leads to an inadvertent use of insulin, which has important implications on quality of life, side effects, level of acceptance of illness, and costs.
In this context, the assessment of islet antibodies to rule out type 1 diabetes gains importance. GAD and IA2 islet autoantibodies discriminate well between type 1 and MODY, with cross sectional studies showing they are present in 80% of patients with type 1 diabetes and in less than 1% of patients with MODY [93].
A recent study assessed the prevalence of MODY in a nationwide population-based registry of childhood diabetes. It used next-generation sequencing for the most common affected genes only in children negative for both GAD and IA-2 autoantibodies. The prevalence of MODY in antibody-negative childhood diabetes reached almost 6.5%. One-third of these MODY cases had not been recognized by clinical criteria alone [94, 95].
A Swedish study assessed the four islet autoantibodies: GAD (GADA), insulinoma antigen-2 (IA2A), zinc transporter 8 (ZnT8A), and insulin autoantibodies (IAA) at the time of diagnosis of diabetes in a pediatric population. This approach effectively resulted in more patients with type 1 diabetes being identified and reduced the number of patients needing consideration for MODY testing. Testing three islet autoantibody (GADA, IA-2A, and ZnT8A) seems to be the most cost-effective strategy, since testing IAA only reduced the number of patients who were autoantibody negative from 13% of pediatric diabetes to 12%. Testing 303 autoantibody-negative patients identified 46 patients with MODY (detection rate 15%). The detection rate rose to 49% when testing was limited to autoantibody-negative patients with HbA1c < 7.5% (58 mmol/mol) (36 out of 46 patients) [96]. Therefore, evaluation of autoantibodies can be a useful tool to select patients for further investigation.
Challenges and perspectives
The correct MODY diagnosis is crucial for proper treatment and improvement in quality of life. Recent advances in next-generation sequencing technology have enabled the maximization of diagnosis performance of monogenic diabetes. However, high costs of genetic testing and limited awareness of MODY as a relevant entity outside clinicians undermines accurate diagnosis. Moreover, paucity of studies in non-European populations (especially African and Latino), as well as access to molecular diagnosis in those populations, is also a challenge [97]. Likewise, refining the selection of patients to undergo genetic testing, using clinical criteria and inexpensive biomarkers, readily available and validated in various populations, could positively impact cost-effectiveness of diagnosis, follow-up, and treatment.
Conclusions
MODY is a heterogeneous group of monogenic forms of diabetes. Although it has been initially defined as a clinical syndrome of early-onset diabetes, subtypes of MODY caused by mutations in specific genes now stand on their own as separate pathological entities. Moreover, strict enforcement of the classical criteria to screen for MODY mutations yields poor sensitivity levels, detrimental to an adequate screening strategy. Clinical biomarkers have been studied to improve accuracy of recruitment for molecular diagnosis. Among them, models employing residual beta-cell function are the most promising, although they need to be further validated to other populations. This combined with advancements in molecular diagnosis technology and reduction of its costs may lead to more efficient detection of the great majority of undiagnosed MODY cases in the near future.
Availability of data and materials
Not applicable
Abbreviations
- 1,5-AG:
-
1,5-Anhydroglucitol
- ABCC8:
-
ATP-binding Cassette C8
- ApoM:
-
Apolipoprotein-M
- APPL1:
-
Adaptor protein, phosphotyrosine interacting with PH domain and leucin zipper 1
- AUC:
-
Area under the curve
- BLK:
-
B-lymphoid tyrosine kinase
- BMI:
-
Body mass index
- C5:
-
Complement factor 5
- C8:
-
Complement factor 8
- CEL:
-
Cholesteryl-ester lipase
- CRP:
-
Creactive protein
- GAD:
-
Glutamic acid decarboxylase
- GCK:
-
Glucokinase
- GFR:
-
Glomerular filtration rate
- HbA1c:
-
Glycated hemoglobin
- HNF:
-
Hepatocyte nuclear factor
- HNF1A:
-
Hepatocyte nuclear factor-1 homeobox A
- HNF1B:
-
Hepatocyte nuclear factor-1 homeobox B
- HNF4A:
-
Hepatocyte nuclear factor-4 homeobox A
- hsCRP:
-
High-sensitivity C-reactive protein
- IA-2:
-
Insulinoma antigen-2
- IAA:
-
Insulin autoantibodies
- INS:
-
Insulin
- KCNJ11:
-
Potassium inwardly rectifying channel subfamily J member 11
- KLF11:
-
Krupell-like factor 11
- MODY:
-
Maturity-onset diabetes of the young
- ND:
-
Neonatal diabetes
- NEUROD1:
-
Neurogenic differentiation-1
- NGS:
-
Next-generation sequencing
- NPV:
-
Negative predictive value
- OGTT:
-
Oral glucose tolerance test
- PAX4:
-
Paired homeobox 4
- PDX:
-
Pancreas/duodenum homeobox 1
- PM:
-
Precision medicine
- PPV:
-
Positive predictive value
- RCAD:
-
Renal cysts and diabetes
- ROC:
-
Receiver operating characteristic
- SGLT2:
-
Sodium/glucose co-transporter 2
- SNP:
-
Single nucleotide polymorphism
- TTR:
-
Transthyretin
- UCP:
-
Urinary C-peptide
- UCPCR:
-
Urinary C-peptide/creatinine ratio
- ZnT8:
-
Zinc transporter 8
References
Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34:1878–84.
Tattersall RB. Mild familial diabetes with dominant inheritance. Q J Med. 1974;43:339–57.
Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24:44–53.
Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:162–4.
Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384:455–8.
Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature. 1996;384:458–60.
Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W, et al. ISPAD Clinical Practice Consensus Guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19(Suppl 27):47–63.
Bellanne-Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, et al. Large Genomic Rearrangements In The Hepatocyte Nuclear Factor-1 (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54:3126–32.
Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–8.
Franco LF, Peixoto-Barbosa R, Dotto RP, Vieira JGH, Dias-da-Silva MR, Reis LCF, et al. More than kin, less than kind: one family and the many faces of diabetes in youth. Arch Endocrinol Metabol. 2017;61:637–42.
Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New Engl j Med. 2006;355:467–77.
Greeley SAW, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal diabetes mellitus: a model for personalized medicine. TEM. 2010;21:464–72.
Ashcroft FM, Puljung MC, Vedovato N. Neonatal diabetes and the KATP Channel: from mutation to therapy. TEM. 2017;28:377–87.
Bowman P, Sulen Å, Barbetti F, Beltrand J, Svalastoga P, Codner E, et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6:637–46.
Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia. 2017;60:769–77.
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275–81.
Sanyoura M, Philipson LH, Naylor R. Monogenic diabetes in children and adolescents: recognition and treatment options. Curr Diab Rep. 2018;18:58.
Stride A, Shields B, Gill-Carey O, Chakera AJ, Colclough K, Ellard S, et al. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57:54–6.
Velho G, Blanché H, Vaxillaire M, Bellanné-Chantelot C, Pardini VC, Timsit J, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40:217–24.
Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311:279–86.
Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55:1265–72.
Owen KR, Thanabalasingham G, James TJ, Karpe F, Farmer AJ, Mccarthy MI, et al. Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care. 2010;33:1919–24.
Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metabol. 2013;98:4055–62.
Gandica RG, Chung WK, Deng L, Goland R, Gallagher MP. Identifying monogenic diabetes in a pediatric cohort with presumed type 1 diabetes. Pediatric Diabetes. 2015;16:227–33.
Shepherd M, Shields B, Hammersley S, Hudson M, McDonald TJ, Colclough K, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care. 2016;39:1879–88.
Chambers C, Fouts A, Dong F, Colclough K, Wang Z, Batish SD, et al. Characteristics of maturity onset diabetes of the young in a large diabetes center. Pediatric Diabetes. 2016;17:360–7.
Donath X, Saint-Martin C, Dubois-Laforgue D, Rajasingham R, Mifsud F, Ciangura C, et al. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis. BMC Med. 2019;17:132.
Pinelli M, Acquaviva F, Barbetti F, Caredda E, Cocozza S, Delvecchio M, et al. Identification of candidate children for maturity-onset diabetes of the young type 2 (MODY2) gene testing: a seven-item clinical flowchart (7-iF). PLoS ONE. 2013;8:e79933.
Cammidge PJ. Diabetes mellitus and heredity. BMJ. 1928;2:738–41.
Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:721–2.
Bell GI, Pilkis SJ, Weber IT, Polonsky KS. Glucokinase mutations, insulin secretion, and diabetes mellitus. Annu Rev Physiol. 1996;58:171–86.
Kavvoura FK, Owen KR. Maturity onset diabetes of the young: clinical characteristics, diagnosis and management. PER. 2012;10:234–42.
Feigerlová E, Pruhova S, Dittertová L, Lebl J, Pinterová D, Kološtová K, et al. Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr. 2006;165:446–52.
Lorini R, Klersy C, d’Annunzio G, Massa O, Minuto N, Iafusco D, et al. Maturity-onset diabetes of the young in children with incidental hyperglycemia: a multicenter Italian study of 172 families. Diabetes Care. 2009;32:1864–6.
Steele AM, Wensley KJ, Ellard S, Murphy R, Shepherd M, Colclough K, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS ONE. 2013;8:e65326.
Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38:1383–92.
Fendler W, Małachowska B, Baranowska-Jazwiecka A, Borowiec M, Wyka K, Malecki MT, et al. Population-based estimates for double diabetes amongst people with glucokinase monogenic diabetes, GCK-MODY. Diabetic Med. 2014;31:881–3.
Frayling TM, Evans JC, Bulman MP, Pearson E, Allen L, Owen K, et al. beta-cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes. 2001;50(Suppl 1):S94–100.
Vaxillaire M, Pueyo ME, Clément K, Fiet J, Timsit J, Philippe J, et al. Insulin secretion and insulin sensitivity in diabetic and non-diabetic subjects with hepatic nuclear factor-1alpha (maturity-onset diabetes of the young-3) mutations. Eur J Endocrinol. 1999;141:609–18.
Stride A, Vaxillaire M, Tuomi T, Barbetti F, Njølstad PR, Hansen T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45:427–35.
Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet. 2006;15:2216–24.
Bellanné-Chantelot C, Carette C, Riveline J-P, Valéro R, Gautier J-F, Larger E, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2007;57:503–8.
Allen HL, Johansson S, Ellard S, Shields B, Hertel JK, Raeder H, et al. Polygenic risk variants for type 2 diabetes susceptibility modify age at diagnosis in monogenic HNF1A diabetes. Diabetes. 2010;59:266–71.
Ellard S, Bellanné-Chantelot C, Hattersley AT, group EMGQNEM. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia. 2008;51:546–53.
Isomaa B, Henricsson M, Lehto M, Forsblom C, Karanko S, Sarelin L, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia. 1998;41:467–73.
Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27:157–61.
Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, et al. Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome 12. Diabetes. 1996;45:1503–10.
Valkovicova T, Skopkova M, Stanik J, Gasperikova D. Novel insights into genetics and clinics of the HNF1A-MODY. Endocr Regul. 2019;53:110–34.
Bacon S, Kyithar MP, Rizvi SR, Donnelly E, McCarthy A, Burke M, et al. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A–MODY cohort. Diabetic Med. 2016;33:976–84.
Fajans SS, Brown MB. Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care. 1993;16:1254–61.
Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Medicine. 2007;4:e118.
Coffinier C, Thépot D, Babinet C, Yaniv M, Barra J. Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development (Cambridge, England). 1999;126:4785–94.
Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200–13.
Warncke K, Kummer S, Raile K, Grulich-Henn J, Woelfle J, Steichen E, et al. Frequency and Characteristics of MODY 1 (HNF4A Mutation) and MODY 5 (HNF1B Mutation): analysis From the DPV Database. J Clin Endocrinol Metabol. 2018;104:845–55.
Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–5.
Bingham C, Bulman MP, Ellard S, Allen LI, Lipkin GW, Hoff WG, et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219–24.
Dubois-Laforgue D, Cornu E, Saint-Martin C, Coste J, Bellanné-Chantelot C, Timsit J, et al. Diabetes, associated clinical spectrum, long-term prognosis and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1 B (HNF1B) molecular defects. Diabetes Care. 2017;40:1436–43.
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. JASN. 2006;17:497–503.
Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. 2006;43:84–90.
Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, Casemayou A, et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 2014;86:1007–15.
Clissold R, Shields B, Ellard S, Hattersley A, Bingham C. Assessment of the HNF1B Score as a Tool to Select Patients for HNF1B Genetic Testing. Nephron. 2015;130:134–40.
Dotto RP, de Santana LS, Lindsey SC, Caetano LA, Franco LF, Moisés RCMS, et al. Searching for mutations in the HNF1B gene in a Brazilian cohort with renal cysts and hyperglycemia. Arch Endocrinol Metab. 2019;63:250–7.
Thanabalasingham G, Pal A, Selwood MP, Dudley C, Fisher K, Bingley PJ, et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 2012;35:1206–12.
Bellanné-Chantelot C, Lévy DJ, Carette C, Saint-Martin C, Riveline J-P, Larger E, et al. Clinical characteristics and diagnostic criteria of maturity-onset diabetes of the young (MODY) due to molecular anomalies of the HNF1A gene. J Clin Endocrinol Metabol. 2011;96:E1346–51.
Naylor RN, John PM, Winn AN, Carmody D, Greeley SAW, Philipson LH, et al. Cost-effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care. 2014;37:202–9.
Szopa M, Ludwig-Gałęzowska A, Radkowski P, Skupien J, Zapała B, Płatek T, et al. Genetic testing for monogenic diabetes using targeted next-generation sequencing in patients with maturity-onset diabetes of the young. Pol Arch Med Wewn. 2015;125:845–51.
Ang SF, Lim SC, Tan CS, Fong JC, Kon WY, Lian JX, et al. A preliminary study to evaluate the strategy of combining clinical criteria and next generation sequencing (NGS) for the identification of monogenic diabetes among multi-ethnic Asians. Diabetes Res Clin Pract. 2016;119:13–22.
Szopa M, Klupa T, Kapusta M, Matejko B, Ucieklak D, Glodzik W, et al. A decision algorithm to identify patients with high probability of monogenic diabetes due to HNF1A mutations. Endocrine. 2019;64:75–81.
Nguyen HV, Finkelstein EA, Mital S, Gardner DSL. Incremental cost-effectiveness of algorithm-driven genetic testing versus no testing for maturity onset diabetes of the young (MODY) in Singapore. J Med Genet. 2017;54:747.
Delvecchio M, Mozzillo E, Salzano G, Iafusco D, Frontino G, Patera PI, et al. Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metabolism. 2007;2017(102):1826–34.
Urakami T. Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metabol Syndrome Obes Targets Ther. 2019;12:1047–56.
Cervin C, Axler O, Holmkvist J, Almgren P, Rantala E, Tuomi T, et al. An investigation of serum concentration of apoM as a potential MODY3 marker using a novel ELISA. J Intern Med. 2010;267:316–21.
Richter S, Shih DQ, Pearson ER, Wolfrum C, Fajans SS, Hattersley AT, et al. Regulation of apolipoprotein M gene expression by MODY3 gene hepatocyte nuclear factor-1alpha: haploinsufficiency is associated with reduced serum apolipoprotein M levels. Diabetes. 2003;52:2989–95.
Karlsson E, Shaat N, Groop L. Can complement factors 5 and 8 and transthyretin be used as biomarkers for MODY 1 (HNF4A-MODY) and MODY 3 (HNF1A-MODY)? Diabet Med. 2008;25:788–91.
Curocichin G, Wu Y, McDade TW, Kuzawa CW, Borja JB, Qin L, et al. Single-nucleotide polymorphisms at five loci are associated with C-reactive protein levels in a cohort of Filipino young adults. J Hum Genet. 2011;56:823–7.
Naitza S, Porcu E, Steri M, Taub DD, Mulas A, Xiao X, et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet. 2012;8:e1002480.
Reiner AP, Barber MJ, Guan Y, Ridker PM, Lange LA, Chasman DI, et al. Polymorphisms of the HNF1A gene encoding hepatocyte nuclear factor-1 alpha are associated with C-reactive protein. Am J Hum Genet. 2008;82:1193–201.
Thanabalasingham G, Shah N, Vaxillaire M, Hansen T, Tuomi T, Gašperíková D, et al. A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia. 2011;54:2801–10.
McDonald TJ, Shields BM, Lawry J, Owen KR, Gloyn AL, Ellard S, et al. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care. 2011;34:1860–2.
Bellanné-Chantelot C, Coste J, Ciangura C, Fonfrède M, Saint-Martin C, Bouché C, et al. High-sensitivity C-reactive protein does not improve the differential diagnosis of HNF1A-MODY and familial young-onset type 2 diabetes: a grey zone analysis. Diabetes Metab. 2016;42:33–7.
Leighton E, Sainsbury CA, Jones GC. A practical review of C-peptide testing in diabetes. Diabetes Ther. 2017;8:475–87.
McDonald TJ, Knight BA, Shields BM, Bowman P, Salzmann MB, Hattersley AT. Stability and reproducibility of a single-sample urinary C-peptide/creatinine ratio and its correlation with 24-h urinary C-peptide. Clin Chem. 2009;55:2035–9.
The Diabetes Control and Complication Trial Research Group. Effect of intensive therapy on residual β-cell function in patients with type 1 diabetes in the diabetes control and complications trial. Ann Intern Med. 1998;128:517.
Palmer JP, Fleming GA, Greenbaum CJ, Herold KC, Jansa LD, Kolb H, et al. C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21-22 October 2001. Diabetes. 2004;53:250–64.
Tfayli H, Bacha F, Gungor N, Arslanian S. Phenotypic type 2 diabetes in obese youth: insulin sensitivity and secretion in islet cell antibody-negative versus -positive patients. Diabetes. 2009;58:738–44.
Besser REJ, Shepherd MH, McDonald TJ, Shields BM, Knight BA, Ellard S, et al. Urinary C-peptide creatinine ratio is a practical outpatient tool for identifying hepatocyte nuclear factor 1-{alpha}/hepatocyte nuclear factor 4-{alpha} maturity-onset diabetes of the young from long-duration type 1 diabetes. Diabetes Care. 2011;34:286–91.
Besser REJ, Shields BM, Hammersley SE, Colclough K, McDonald TJ, Gray Z, et al. Home urine C-peptide creatinine ratio (UCPCR) testing can identify type 2 and MODY in pediatric diabetes. Pediatr Diabetes. 2013;14:181–8.
Agladioglu SY, Sagsak E, Aycan Z. Urinary C-peptide/creatinine ratio can distinguish maturity-onset diabetes of the young from type 1 diabetes in children and adolescents: a single-center experience. Hormone Res Paediatr. 2015;84:54–61.
Majidi S, Fouts A, Pyle L, Chambers C, Armstrong T, Wang Z, et al. Can biomarkers help target maturity-onset diabetes of the young genetic testing in antibody-negative diabetes? Diabetes Technol Therap. 2018;20:106–12.
Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55:123–7.
Shepherd MH, Shields BM, Hudson M, Pearson ER, Hyde C, Ellard S, et al. A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia. 2018;61:2520–7.
Urbanova J, Brunerova L, Broz J. How can maturity-onset diabetes of the young be identified among more common diabetes subtypes? Wien Klin Wochenschr. 2019;131:435–41.
McDonald TJ, Colclough K, Brown R, Shields B, Shepherd M, Bingley P, et al. Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from Type 1 diabetes. Diabetic Med. 2011;28:1028–33.
Johansson BB, Irgens HU, Molnes J, Sztromwasser P, Aukrust I, Juliusson PB, et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia. 2017;60:625–35.
Shields B, Colclough K. Towards a systematic nationwide screening strategy for MODY. Diabetologia. 2017;60:609–12.
Carlsson A, Shepherd M, Ellard S, Weedon M, Lernmark Å, Forsander G, et al. Absence of islet autoantibodies and modestly raised glucose values at diabetes diagnosis should lead to testing for MODY: lessons from a 5-year pediatric swedish national cohort study. Diabetes Care. 2020;43:82–9.
Kleinberger JW, Pollin TI. Undiagnosed MODY: time for Action. Curr Diab Rep. 2015;15:110.
Bonnefond A, Philippe J, Durand E, Dechaume A, Huyvaert M, Montagne L, et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE. 2012;7:e37423.
Molven A, Ringdal M, Nordbø AM, Raeder H, Støy J, Lipkind GM, et al. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57:1131–5.
Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest. 1999;104:R33–9.
Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–8.
Raeder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54–62.
Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA. 2005;102:4807–12.
Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92:2821–6.
Borowiec M, Liew CW, Thompson R, Boonyasrisawat W, Hu J, Mlynarski WM, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci USA. 2009;106:14460–5.
Prudente S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. 2015;97:177–85.
Bellanné-Chantelot C, Chauveau D, Gautier J-F, Dubois-Laforgue D, Clauin S, Beaufils S, et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med. 2004;140:510–7.
Iwasaki N, Okabe I, Momoi MY, Ohashi H, Ogata M, Iwamoto Y. Splice site mutation in the hepatocyte nuclear factor-1 beta gene, IVS2nt + 1G > A, associated with maturity-onset diabetes of the young, renal dysplasia and bicornuate uterus. Diabetologia. 2001;44:387–8.
Bingham C, Ellard S, Cole TRP, Jones KE, Allen LIS, Goodship JA, et al. Solitary functioning kidney and diverse genital tract malformations associated with hepatocyte nuclear factor-1beta mutations. Kidney Int. 2002;61:1243–51.
Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89:2905–8.
Kolatsi-Joannou M, Bingham C, Ellard S, Bulman MP, Allen LI, Hattersley AT, et al. Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. JASN. 2001;12:2175–80.
Furuta H, Furuta M, Sanke T, Ekawa K, Hanabusa T, Nishi M, et al. Nonsense and missense mutations in the human hepatocyte nuclear factor-1 beta gene (TCF2) and their relation to type 2 diabetes in Japanese. J Clin Endocrinol Metab. 2002;87:3859–63.
Carbone I, Cotellessa M, Barella C, Minetti C, Ghiggeri GM, Caridi G, et al. A novel hepatocyte nuclear factor-1beta (MODY-5) gene mutation in an Italian family with renal dysfunctions and early-onset diabetes. Diabetologia. 2002;45:153–4.
Bohn S, Thomas H, Turan G, Ellard S, Bingham C, Hattersley AT, et al. Distinct molecular and morphogenetic properties of mutations in the human HNF1beta gene that lead to defective kidney development. JASN. 2003;14:2033–41.
Bingham C, Ellard S, Hoff WG van’t, Simmonds HA, Marinaki AM, Badman MK, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int. 2003;63:1645–51.
Mache CJ, Preisegger K-H, Kopp S, Ratschek M, Ring E. De novo HNF-1 beta gene mutation in familial hypoplastic glomerulocystic kidney disease. Pediatr Nephrol (Berlin, Germany). 2002;17:1021–6.
Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transpl. 2004;19:2703–8.
Montoli A, Colussi G, Massa O, Caccia R, Rizzoni G, Civati G, et al. Renal cysts and diabetes syndrome linked to mutations of the hepatocyte nuclear factor-1 beta gene: description of a new family with associated liver involvement. Am J Kidney Dis. 2002;40:397–402.
Kitanaka S, Miki Y, Hayashi Y, Igarashi T. Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with Type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J Clin Endocrinol Metab. 2004;89:1369–78.
Lindner TH, Njølstad PR, Horikawa Y, Bostad L, Bell GI, Søvik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–8.
Nowak N, Szopa M, Thanabalasingham G, McDonald TJ, Colclough K, Skupien J, et al. Cystatin C is not a good candidate biomarker for HNF1A-MODY. Acta Diabetol. 2013;50:815–20.
Skupien J, Gorczynska-Kosiorz S, Klupa T, Wanic K, Button EA, Sieradzki J, et al. Clinical application of 1,5-anhydroglucitol measurements in patients with hepatocyte nuclear factor-1alpha maturity-onset diabetes of the young. Diabetes Care. 2008;31:1496–501.
Pal A, Farmer AJ, Dudley C, Selwood MP, Barrow BA, Klyne R, et al. Evaluation of serum 1,5 anhydroglucitol levels as a clinical test to differentiate subtypes of diabetes. Diabetes Care. 2010;33:252–7.
Acknowledgements
Not applicable.
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
RPB and FMAG conducted literature review and drafted the manuscript. FMAG and AFR provided scientific guidance, participated in editing the manuscript, and made critical revisions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
As this is a review article, there were no participants directly involved.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peixoto-Barbosa, R., Reis, A.F. & Giuffrida, F.M.A. Update on clinical screening of maturity-onset diabetes of the young (MODY). Diabetol Metab Syndr 12, 50 (2020). https://doi.org/10.1186/s13098-020-00557-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13098-020-00557-9